A Splice Intervention Therapy for Autosomal Recessive Juvenile Parkinson’s Disease Arising from Parkin Mutations

 ,
,  , and
, and
Abstract
:
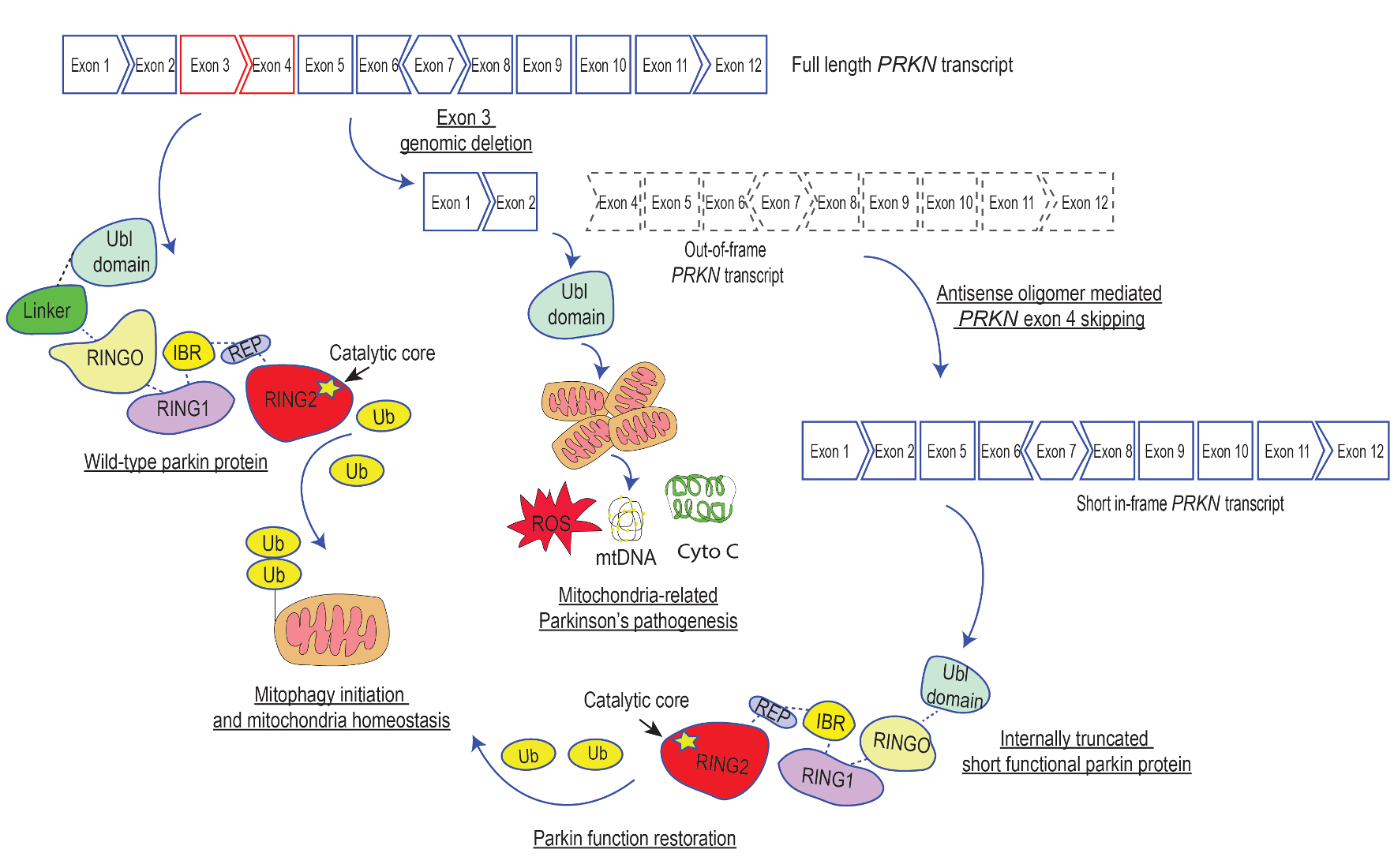
1. Introduction
2. Results
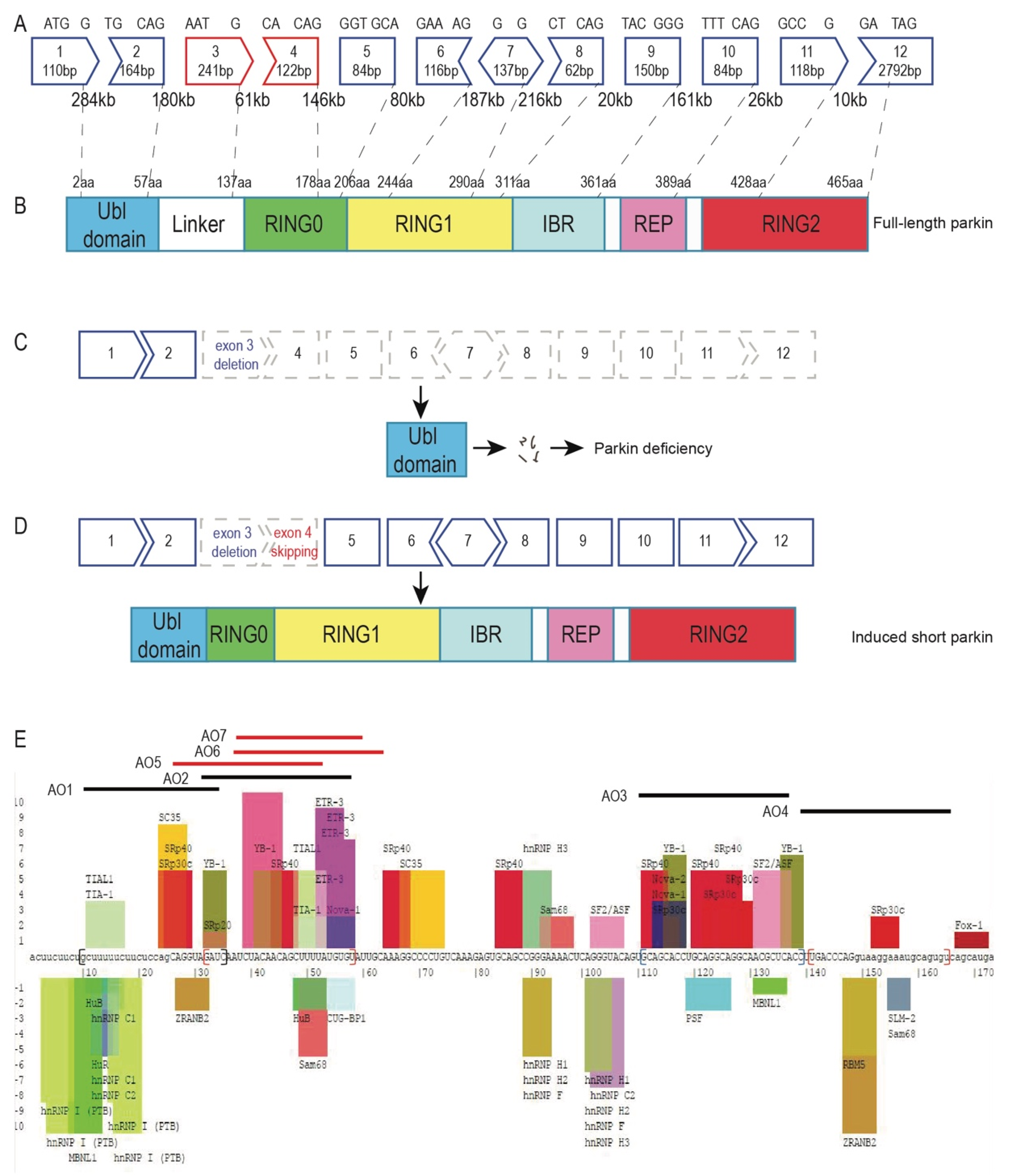
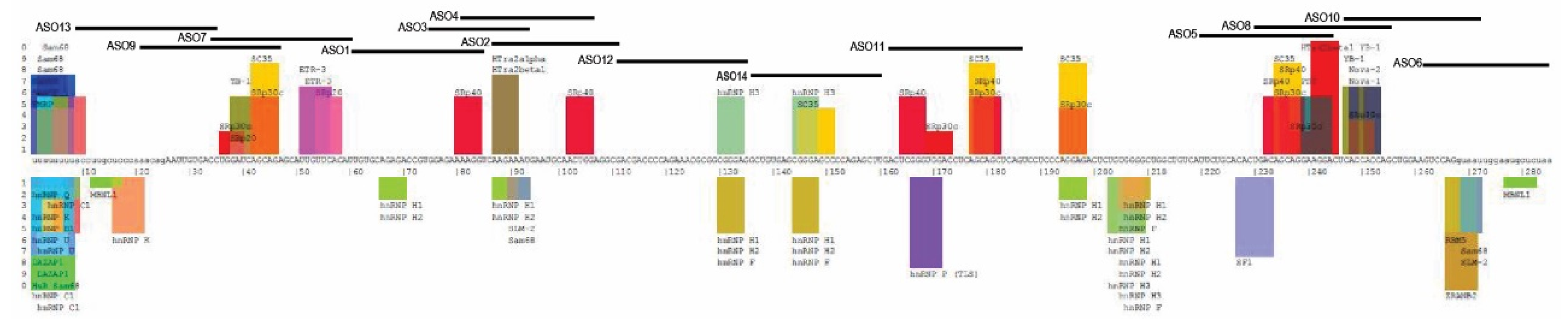
2.1. Design of Antisense Oligomers to Induce PRKN Exon 4 Skipping
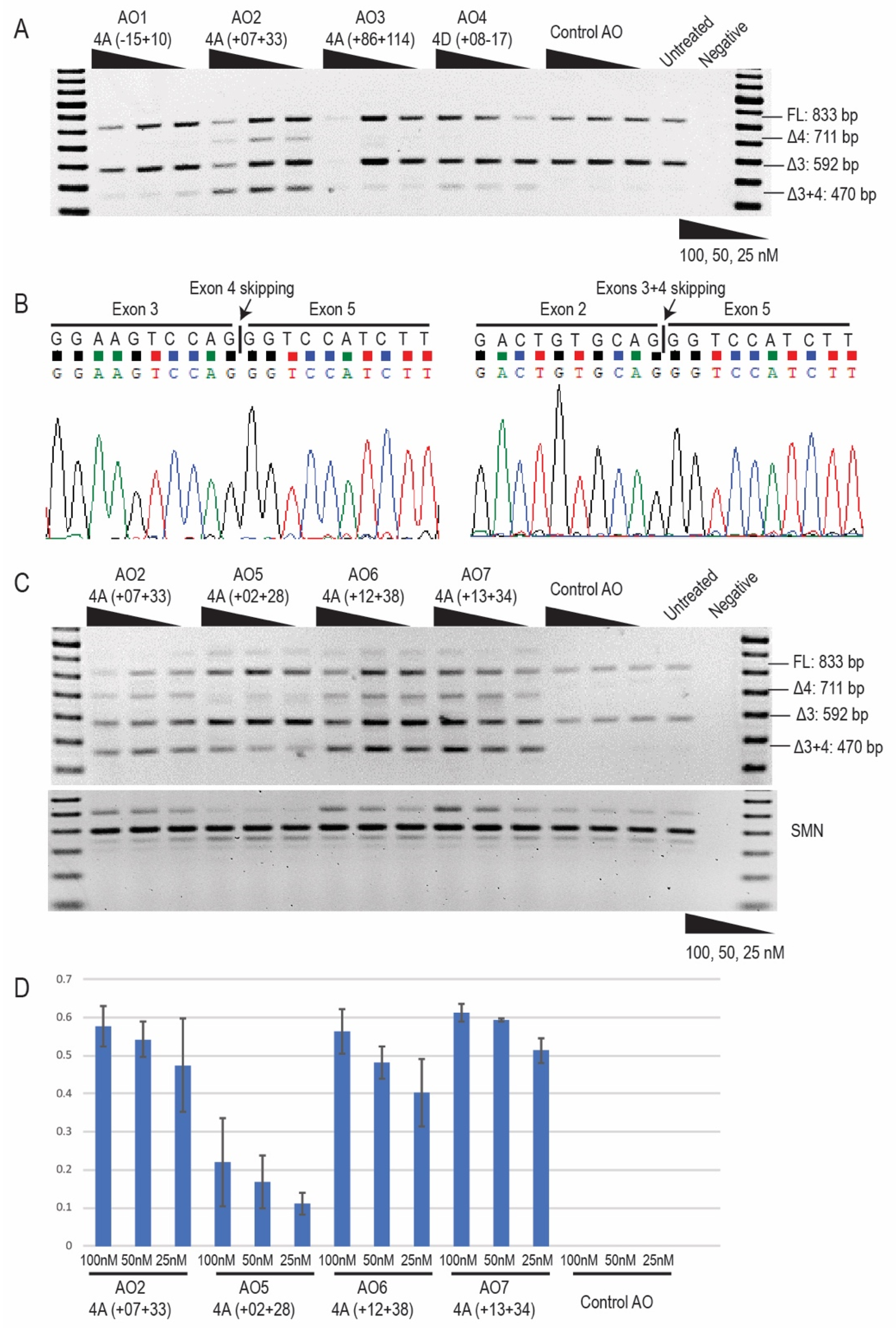
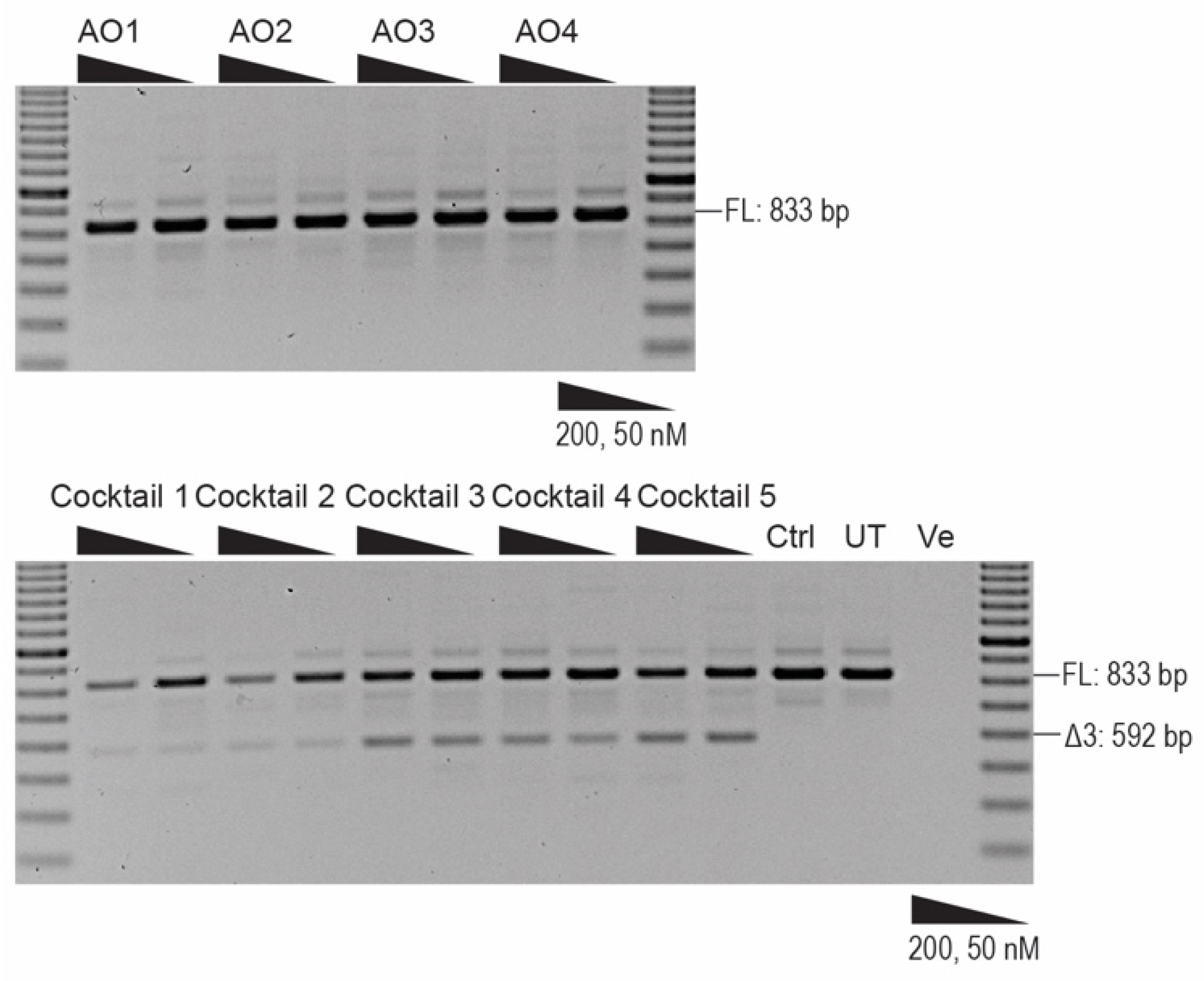
2.2. Antisense Oligonucleotide-Induced Exon 4 Skipping to Restore the PRKN Reading Frame in Patient-Derived Fibroblasts
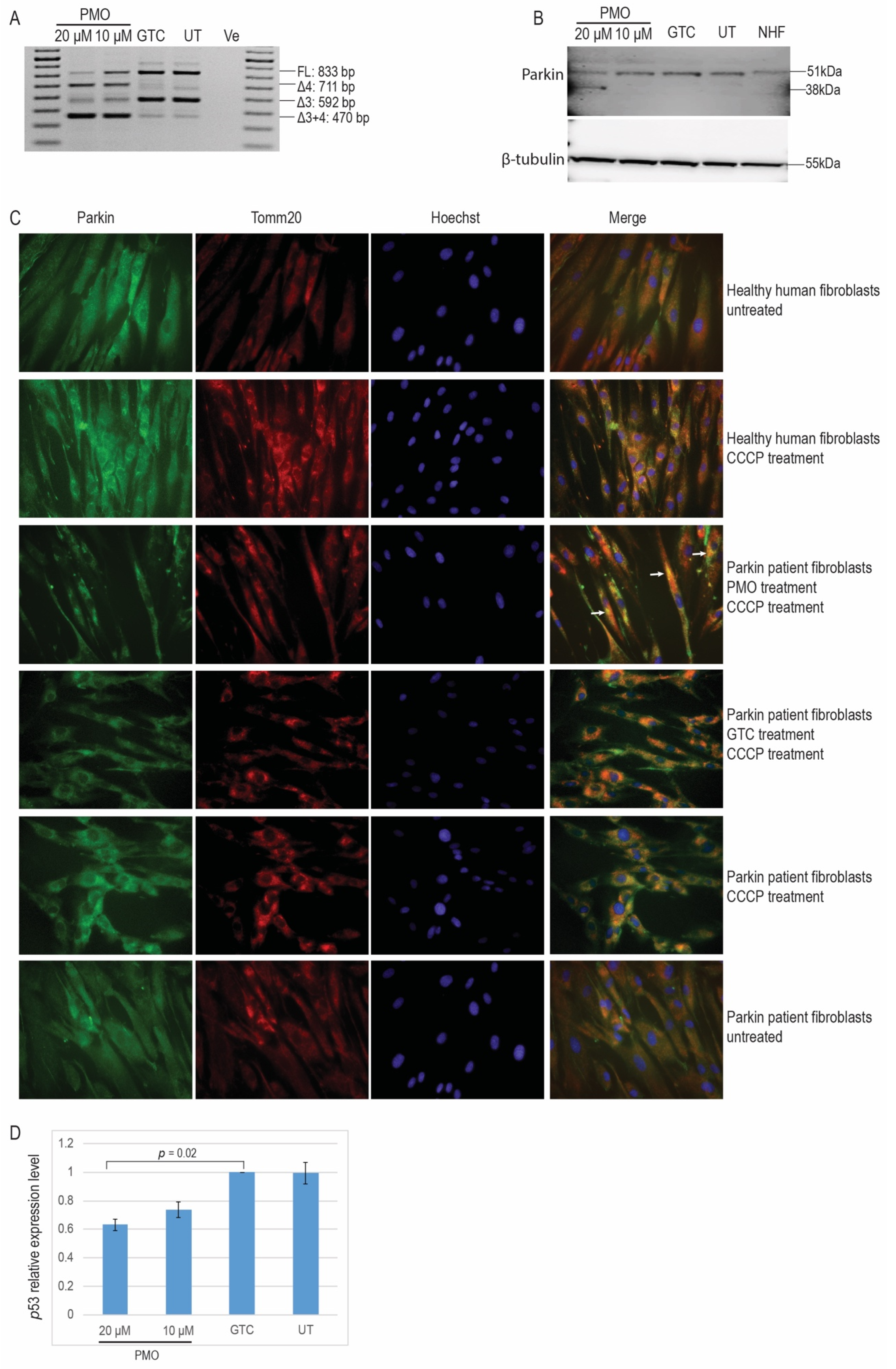
2.3. Morpholino Oligomer-Induced PRKN Exon 4 Skipping and Production of a Shorter Functional Parkin Isoform
2.4. 2′ OMe PS AO Cocktail-Mediated PRKN Exon 3 Skipping in Healthy Human Fibroblasts
3. Discussion
4. Materials and Methods
4.1. Antisense Oligomer Design and Synthesis
4.2. Cell Propagation
4.3. Transfection
4.4. RNA Extraction and PCR
4.5. cDNA Synthesis and Real-Time PCR
4.6. Western Blot
4.7. Immunofluorescence
5. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PD | Parkinson’s disease |
| SNpc | Substantia nigra pars compacta |
| ARJP | Autosomal recessive juvenile-onset Parkinson’s disease |
| AO | Antisense oligonucleotide |
| DMD | Duchenne muscular dystrophy |
| PS | Phosphorothioate |
| PMO | Phosphorodiamidate morpholino oligomer |
| GTC | Gene Tools control |
| CCCP | Carbonyl cyanide m-chlorophenyl hydrazone |
References
- De Lau, L.M.; Breteler, M.M. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006, 5, 525–535. [Google Scholar] [CrossRef]
- Forno, L.S. Neuropathology of Parkinson’s disease. J. Neuropathol. Exp. Neurol. 1996, 55, 259–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lücking, C.B.; Dürr, A.; Bonifati, V.; Vaughan, J.; De Michele, G.; Gasser, T.; Harhangi, B.S.; Meco, G.; Denèfle, P.; Wood, N.W.; et al. Association between early-onset Parkinson’s disease and mutations in the Parkin gene. N. Eng. J. Med. 2000, 342, 1560–1567. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Lohmann, K. Parkinson disease (s): Is “Parkin disease” a distinct clinical entity? Neurology 2009, 72, 106–107. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L. α-Synuclein in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [Green Version]
- Rocha, E.M.; De Miranda, B.; Sanders, L.H. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 2018, 109, 249–257. [Google Scholar] [CrossRef]
- Dagda, R.K.; Chu, C.T. Mitochondrial quality control: Insights on how Parkinson’s disease related genes PINK1, Parkin, and Omi/HtrA2 interact to maintain mitochondrial homeostasis. J. Bioenerg. Biomembr. 2009, 41, 473–479. [Google Scholar] [CrossRef] [Green Version]
- Darios, F.; Corti, O.; Lucking, C.B.; Hampe, C.; Muriel, M.P.; Abbas, N.; Gu, W.J.; Hirsch, E.C.; Rooney, T.; Ruberg, M.; et al. Parkin prevents mitochondrial swelling and cytochrome c release in mitochondria-dependent cell death. Hum. Mol. Genet. 2003, 12, 517–526. [Google Scholar] [CrossRef]
- Sunico, C.R.; Nakamura, T.; Rockenstein, E.; Mante, M.; Adame, A.; Chan, S.F.; Newmeyer, T.F.; Masliah, E.; Nakanishi, N.; Lipton, S.A. S-Nitrosylation of parkin as a novel regulator of p53-mediated neuronal cell death in sporadic Parkinson’s disease. Mol. Neurodegener. 2013, 8, 29. [Google Scholar] [CrossRef] [Green Version]
- Da Costa, C.A.; Sunyach, C.; Giaime, E.; West, A.; Corti, O.; Brice, A.; Safe, S.; Abou-Sleiman, P.M.; Wood, N.W.; Takahashi, H. Transcriptional repression of p53 by parkin and impairment by mutations associated with autosomal recessive juvenile Parkinson’s disease. Nat. Cell Biol. 2009, 11, 1370–1375. [Google Scholar] [CrossRef] [Green Version]
- Haylett, W.; Swart, C.; Van Der Westhuizen, F.; Van Dyk, H.; Van der Merwe, L.; Van Der Merwe, C.; Loos, B.; Carr, J.; Kinnear, C.; Bardien, S. Altered mitochondrial respiration and other features of mitochondrial function in Parkin-mutant fibroblasts from Parkinson’s disease patients. Parkinson’s Dis. 2016, 2016, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asai, H.; Hirano, M.; Kiriyama, T.; Ikeda, M.; Ueno, S. Naturally-and experimentally-designed restorations of the Parkin gene deficit in autosomal recessive juvenile parkinsonism. Biochem. Biophys. Res. Commun. 2010, 391, 800–805. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Corey, D.R. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2017, 46, 1584–1600. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.A.; Castanotto, D. FDA-approved oligonucleotide therapies in 2017. Mol. Ther. 2017, 25, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Piva, F.; Giulietti, M.; Nocchi, L.; Principato, G. SpliceAid: A database of experimental RNA target motifs bound by splicing proteins in humans. Bioinformatics 2009, 25, 1211–1213. [Google Scholar] [CrossRef]
- Carroll, R.G.; Hollville, E.; Martin, S.J. Parkin sensitizes toward apoptosis induced by mitochondrial depolarization through promoting degradation of Mcl-1. Cell Rep. 2014, 9, 1538–1553. [Google Scholar] [CrossRef] [Green Version]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Heffernan, S.M.; Kilduff, L.P.; Day, S.H.; Pitsiladis, Y.P.; Williams, A.G. Genomics in rugby union: A review and future prospects. Eur. J. Sport Sci. 2015, 15, 460–468. [Google Scholar] [CrossRef]
- Nilsen, T.W.; Graveley, B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature 2010, 463, 457. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Schwartz, M.; Dunø, M.; Palle, A.L.; Krag, T.; Vissing, J. Deletion of exon 16 of the dystrophin gene is not associated with disease. Hum. Mutat. 2007, 28, 205. [Google Scholar] [CrossRef] [PubMed]
- Dawson, T.M.; Dawson, V.L. The role of parkin in familial and sporadic Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2010, 25, S32–S39. [Google Scholar] [CrossRef] [PubMed]
- Seirafi, M.; Kozlov, G.; Gehring, K. Parkin structure and function. FEBS J. 2015, 282, 2076–2088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gladkova, C.; Maslen, S.L.; Skehel, J.M.; Komander, D. Mechanism of parkin activation by PINK1. Nature 2018, 559, 410–414. [Google Scholar] [CrossRef]
- Trempe, J.-F.; Sauvé, V.; Grenier, K.; Seirafi, M.; Tang, M.Y.; Ménade, M.; Al-Abdul-Wahid, S.; Krett, J.; Wong, K.; Kozlov, G. Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 2013, 340, 1451–1455. [Google Scholar] [CrossRef]
- Xiao, B.; Goh, J.-Y.; Xiao, L.; Xian, H.; Lim, K.-L.; Liou, Y.-C. Reactive oxygen species trigger Parkin/PINK1 pathway–Dependent mitophagy by inducing mitochondrial recruitment of Parkin. J. Biol. Chem. 2017, 292, 16697–16708. [Google Scholar] [CrossRef] [Green Version]
- Cesari, R.; Martin, E.S.; Calin, G.A.; Pentimalli, F.; Bichi, R.; McAdams, H.; Trapasso, F.; Drusco, A.; Shimizu, M.; Masciullo, V.; et al. Parkin, a gene implicated in autosomal recessive juvenile parkinsonism, is a candidate tumor suppressor gene on chromosome 6q25–q27. Proc. Natl. Acad. Sci. USA 2003, 100, 5956–5961. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, Y.; Hattori, N.; Mori, H.; Suzuki, T.; Tanaka, K. Parkin and Parkinson’s disease. Curr. Opin. Neurol. 2001, 14, 477–482. [Google Scholar] [CrossRef]
- Hedrich, K.; Eskelson, C.; Wilmot, B.; Marder, K.; Harris, J.; Garrels, J.; Meija-Santana, H.; Vieregge, P.; Jacobs, H.; Bressman, S.B. Distribution, type, and origin of Parkin mutations: Review and case studies. Mov. Disord. 2004, 19, 1146–1157. [Google Scholar] [CrossRef]
- Mitrpant, C.; Adams, A.M.; Meloni, P.L.; Muntoni, F.; Fletcher, S.; Wilton, S.D. Rational design of antisense oligomers to induce dystrophin exon skipping. Mol. Ther. 2009, 17, 1418–1426. [Google Scholar] [CrossRef]
- Adams, A.M.; Harding, P.L.; Iversen, P.L.; Coleman, C.; Fletcher, S.; Wilton, S.D. Antisense oligonucleotide induced exon skipping and the dystrophin gene transcript: Cocktails and chemistries. BMC Mol. Biol. 2007, 8, 57. [Google Scholar] [CrossRef] [Green Version]
- Wilton, S.D.; Fall, A.M.; Harding, P.L.; McClorey, G.; Coleman, C.; Fletcher, S. Antisense oligonucleotide-induced exon skipping across the human dystrophin gene transcript. Mol. Ther. 2007, 15, 1288–1296. [Google Scholar] [CrossRef]
- Auburger, G.; Klinkenberg, M.; Drost, J.; Marcus, K.; Morales-Gordo, B.; Kunz, W.S.; Brandt, U.; Broccoli, V.; Reichmann, H.; Gispert, S.; et al. Primary skin fibroblasts as a model of Parkinson’s disease. Mol. Neurobiol. 2012, 46, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Soldner, F.; Hockemeyer, D.; Beard, C.; Gao, Q.; Bell, G.W.; Cook, E.G.; Hargus, G.; Blak, A.; Cooper, O.; Mitalipova, M.; et al. Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 2009, 136, 964–977. [Google Scholar] [CrossRef] [Green Version]
- Mann, C.J.; Honeyman, K.; McClorey, G.; Fletcher, S.; Wilton, S.D. Improved antisense oligonucleotide induced exon skipping in the mdx mouse model of muscular dystrophy. J. Gene Med. 2002, 4, 644–654. [Google Scholar] [CrossRef]
- Aung-Htut, M.T.; McIntosh, C.S.; Ham, K.A.; Pitout, I.L.; Flynn, L.L.; Greer, K.; Fletcher, S.; Wilton, S.D. Systematic approach to developing splice modulating antisense oligonucleotides. Int. J. Mol. Sci. 2019, 20, 5030. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [Green Version]
- Wilton, S.D.; Lim, L.; Dye, D.; Laing, N. Bandstab: A PCR-based alternative to cloning PCR products. BioTechniques 1997, 22, 642–645. [Google Scholar] [CrossRef]
- Cooper, S.T.; Lo, H.P.; North, K.N. Single section Western blot: Improving the molecular diagnosis of the muscular dystrophies. Neurology 2003, 61, 93–97. [Google Scholar] [CrossRef]
- Nicholson, L.V.; Davison, K.; Falkous, G.; Harwood, C.; O’Donnell, E.; Slater, C.R.; Harris, J.B. Dystrophin in skeletal muscle I. Western blot analysis using a monoclonal antibody. J. Neurol. Sci. 1989, 94, 125–136. [Google Scholar] [CrossRef]
- Nicholson, L.; Johnson, M.; Davison, K.; O’Donnell, E.; Falkous, G.; Barron, M.; Harris, J. Dystrophin or a “related protein” in Duchenne muscular dystrophy? Acta Neurol. Scand. 1992, 86, 8–14. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AO Number | AO Nomenclature | Sequence 5′→3′ | Length (bp) |
|---|---|---|---|
| 1 | PRKN_H4A (−15 + 10) | GAUCUACCUGCUGGAGAAGAAAAAG | 25 |
| 2 | PRKN_H4A (+07 + 33) | ACACAUAAAAGCUGUUGUAGAUUGAUC | 27 |
| 3 | PRKN_H4A (+86 + 114) | GGUGAGCGUUGCCUGCCUGCAGGUGCUGC | 29 |
| 4 | PRKN_H4D (+08 − 17) | ACACUGCAUUUCCUUACCUGGGUCA | 25 |
| 5 | PRKN_H4A (+02 + 28) | UAAAAGCUGUUGUAGAUUGAUCUACCU | 27 |
| 6 | PRKN_H4A (+12 + 38) | GCAAUACACAUAAAAGCUGUUGUAGAU | 27 |
| 7 | PRKN_H4A (+13 + 34) | UACACAUAAAAGCUGUUGUAGA | 22 |
| AO Number | AO Nomenclature | Sequence 5′→3′ | Length (bp) |
|---|---|---|---|
| 1 | PRKN_H3A (+09 + 35) | AUGUGAACAAUGCUCUGCUGAUCCAGG | 27 |
| 2 | PRKN_H3A (+205 + 230) | CUGGUGGUGAGUCCUUCCUGCUGUCA | 26 |
| 3 | PRKN_H3A (−05 + 22) | UCUGCUGAUCCAGGUCACAAUUCUGUU | 27 |
| 4 | PRKN_H3A (−05 + 21) | AUUACCUGGACUUCCAGCUGGUGGUG | 26 |
| 5 | PRKN_H3A (+137 + 161) | CUGAGCUGCUGAGGUCCACCCGAGU | 25 |
| 6 | PRKN_H3A (+36 + 60) | ACCUUUUCUCCACGGUCUCUGCACA | 25 |
| 7 | PRKN_H3A (+62 + 86) | UCGCCUCCAGUUGCAUUCAUUUCUU | 25 |
| 8 | PRKN_H3A (+50 + 69) | CAUUUCUUGACCUUUUCUCC | 20 |
| 9 | PRKN_H3A (+55 + 81) | CCAGUUGCAUUCAUUUCUUGACCUUU | 26 |
| 10 | PRKN_H3A (+195 + 219) | GUCCUUCCUGCUGUCAGUGUGCAGA | 25 |
| 11 | PRKN_H3D (+05 − 20) | UCUUAGAGCAUUCCAAUUACCUGGA | 25 |
| 12 | PRKN_H3A (−17 + 09) | GUCACAAUUCUGUUUGGGAGCAAGGU | 26 |
| 13 | PRKN_H3A (+86 + 110) | GACGACCCCAGAAACGCGGCGGGAG | 25 |
| 14 | PRKN_H3A (+111 + 135) | GCUGUGAGCGGGAGCCCCAGAGCUU | 25 |
| 15 | PRKN_H3A (+162 + 186) | UCCUCCCAGGAGACUCUGUGGGGCU | 25 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, D.; Aung-Htut, M.T.; Ham, K.A.; Fletcher, S.; Wilton, S.D. A Splice Intervention Therapy for Autosomal Recessive Juvenile Parkinson’s Disease Arising from Parkin Mutations. Int. J. Mol. Sci. 2020, 21, 7282. https://doi.org/10.3390/ijms21197282
Li D, Aung-Htut MT, Ham KA, Fletcher S, Wilton SD. A Splice Intervention Therapy for Autosomal Recessive Juvenile Parkinson’s Disease Arising from Parkin Mutations. International Journal of Molecular Sciences. 2020; 21(19):7282. https://doi.org/10.3390/ijms21197282
Chicago/Turabian StyleLi, Dunhui, May T. Aung-Htut, Kristin A. Ham, Sue Fletcher, and Steve D. Wilton. 2020. "A Splice Intervention Therapy for Autosomal Recessive Juvenile Parkinson’s Disease Arising from Parkin Mutations" International Journal of Molecular Sciences 21, no. 19: 7282. https://doi.org/10.3390/ijms21197282
APA StyleLi, D., Aung-Htut, M. T., Ham, K. A., Fletcher, S., & Wilton, S. D. (2020). A Splice Intervention Therapy for Autosomal Recessive Juvenile Parkinson’s Disease Arising from Parkin Mutations. International Journal of Molecular Sciences, 21(19), 7282. https://doi.org/10.3390/ijms21197282