STAT3 Mediated miR-30a-5p Inhibition Enhances Proliferation and Inhibits Apoptosis in Colorectal Cancer Cells

 ,
,  ,
,
Abstract
:1. Introduction
2. Results
2.1. Colorectal HCT116 and HT29 Tumor Cells Highly Expressed CD133
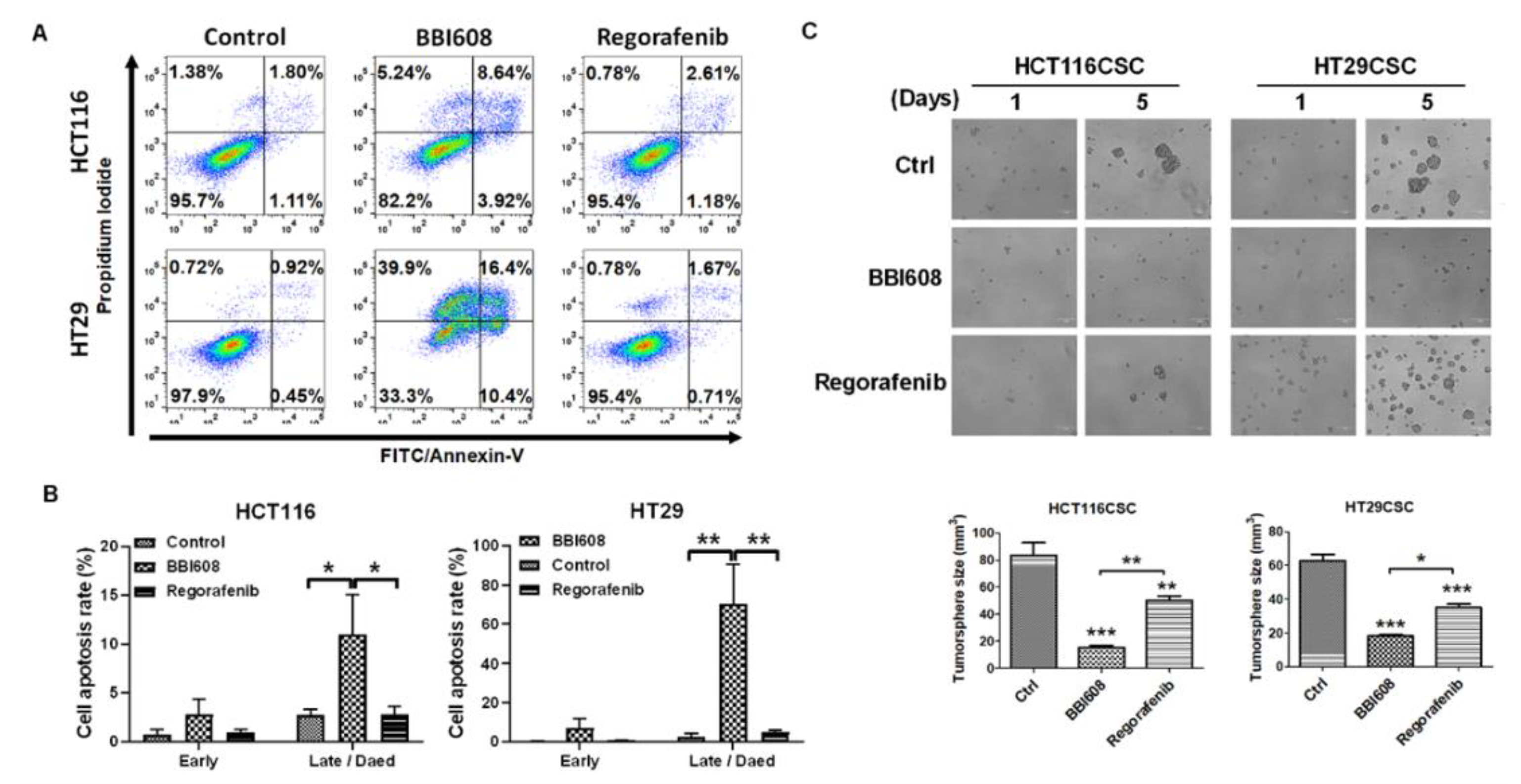
2.2. HCT116- and HT29-Derived Tumorspheres Were More Sensitized to BBI608 Than Regorafenib
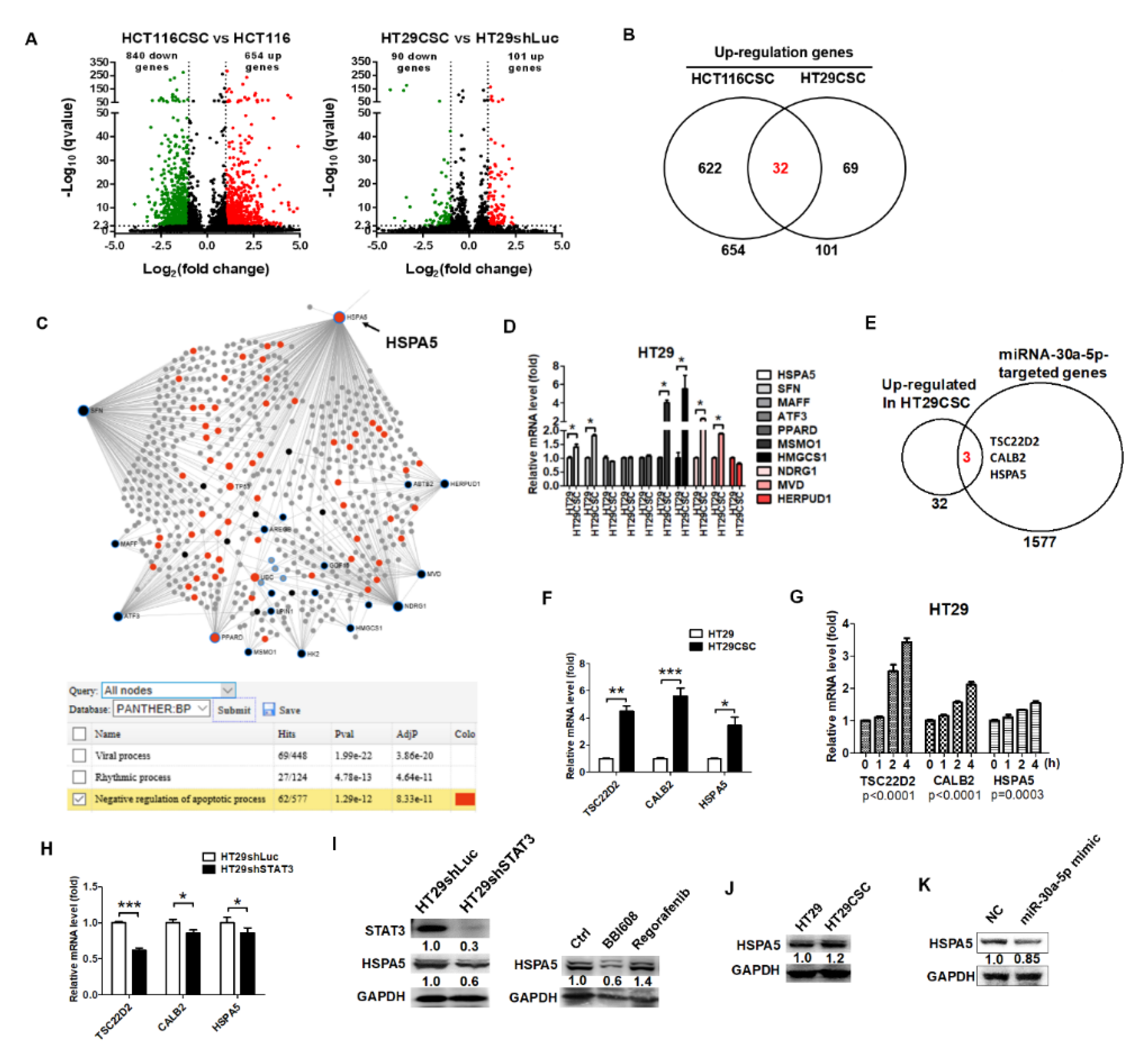
2.3. Identification of STAT3-Mediated miRNAs in HT29-Derived Tumorspheres
2.4. STAT3 Suppressed miR-30a-5p to Inhibit Apoptosis in Colorectal HT29 Cancer Cells
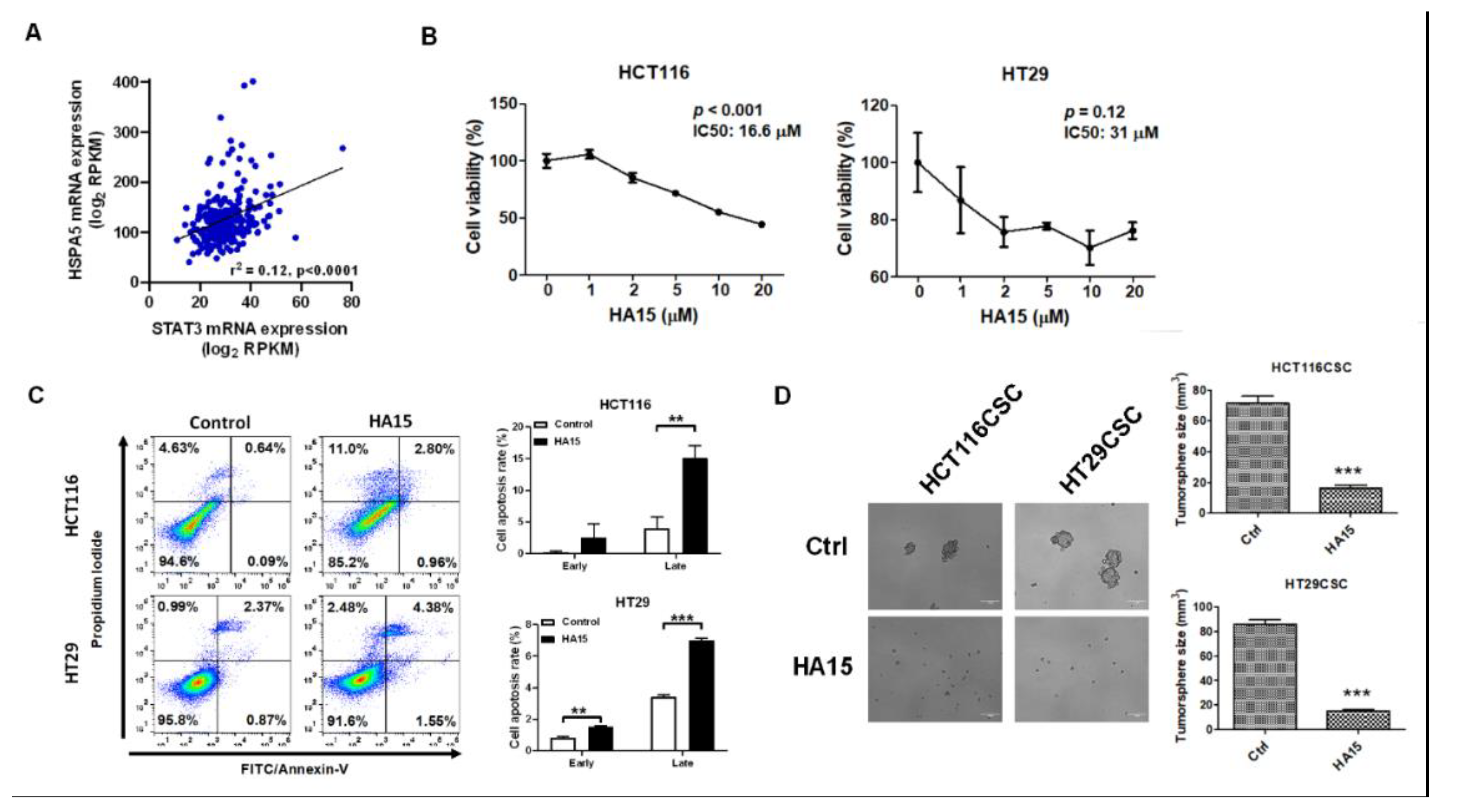
2.5. HSPA5 was the Downstream Target of miR-30a-5p and Was Involved in Anti-Apoptosis
2.6. Inhibition of HSPA5 Reduced Cell Viability and Increased Apoptosis in Colorectal Cancer-Derived Tumorspheres
3. Discussion
4. Materials and Methods
4.1. Cell culture and Tumorsphere Formation
4.2. Small RNAseq Profiling and Bioinformatics Analysis
4.3. RNAseq Profiling and Bioinformatics Analysis
4.4. Quantitative PCR
4.5. Gene Knockdown
4.6. Western Blotting
4.7. Cell Viability
4.8. miR-30a-5p Detection
4.9. Transfection of miR-30a-5p Mimic
4.10. Flow Cytometry
4.11. CIP
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Prz. Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Ducreux, M.; Petersen, L.N.; Ohler, L.; Bergamo, F.; Metges, J.P.; de Groot, J.W.; Wang, J.Y.; Garcia Paredes, B.; Dochy, E.; Fiala-Buskies, S.; et al. Safety and effectiveness of regorafenib in patients with metastatic colorectal cancer in routine clinical practice in the prospective, observational CORRELATE study. Eur. J. Cancer 2019, 123, 146–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, S.Y.; Chen, Y.H.; Lin, P.R.; Chao, T.C.; Su, J.C.; Shiau, C.W.; Su, Y. Two novel SHP-1 agonists, SC-43 and SC-78, are more potent than regorafenib in suppressing the in vitro stemness of human colorectal cancer cells. Cell Death Discov. 2018, 4, 25. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.T.; Chu, P.Y.; Shiau, C.W.; Chen, Y.L.; Li, Y.S.; Hung, M.H.; Chen, L.J.; Chen, P.L.; Su, J.C.; Lin, P.Y.; et al. STAT3 mediates regorafenib-induced apoptosis in hepatocellular carcinoma. Clin. Cancer Res. 2014, 20, 5768–5776. [Google Scholar] [CrossRef] [Green Version]
- Min, S.J.; Lim, J.Y.; Kim, H.R.; Kim, S.J.; Kim, Y. Sasa quelpaertensis leaf extract inhibits colon cancer by regulating cancer cell stemness in vitro and in vivo. Int. J. Mol. Sci. 2015, 16, 9976–9997. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Guo, X.; Huang, X.; Wu, M.; Li, X.; Wu, S.; Luo, X. Metformin reverses stem cell like HepG2 sphere formation and resistance to sorafenib by attenuating epithelial mesenchymal transformation. Mol. Med. Rep. 2018, 18, 3866–3872. [Google Scholar] [CrossRef] [Green Version]
- Goel, G. Evolution of regorafenib from bench to bedside in colorectal cancer: Is it an attractive option or merely a “me too” drug? Cancer Manag. Res. 2018, 10, 425–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Xie, J.; Guo, J.; Manning, H.C.; Gore, J.C.; Guo, N. Evaluation of CD44 and CD133 as cancer stem cell markers for colorectal cancer. Oncol. Rep. 2012, 28, 1301–1308. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.C.; Liao, P.N.; Ho, A.S.; Lim, K.H.; Chang, J.; Su, Y.W.; Chen, C.G.; Chiang, Y.W.; Yang, B.L.; Lin, H.C.; et al. STAT3 exacerbates survival of cancer stem-like tumorspheres in EGFR-positive colorectal cancers: RNAseq analysis and therapeutic screening. J. Biomed. Sci. 2018, 25, 60. [Google Scholar] [CrossRef]
- Ou, H.; Li, Y.; Kang, M. Activation of miR-21 by STAT3 induces proliferation and suppresses apoptosis in nasopharyngeal carcinoma by targeting PTEN gene. PLoS ONE 2014, 9, e109929. [Google Scholar] [CrossRef]
- Singh, M.; Garg, N.; Venugopal, C.; Hallett, R.; Tokar, T.; McFarlane, N.; Mahendram, S.; Bakhshinyan, D.; Manoranjan, B.; Vora, P.; et al. STAT3 pathway regulates lung-derived brain metastasis initiating cell capacity through miR-21 activation. Oncotarget 2015, 6, 27461–27477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.C.; Wu, M.J.; Yang, J.Y.; Camarillo, I.G.; Chang, C.J. Leptin-STAT3-G9a signaling promotes obesity-mediated breast cancer progression. Cancer Res. 2015, 75, 2375–2386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.F.; Lim, K.H.; Chiang, Y.W.; Sie, Z.L.; Chang, J.; Ho, A.S.; Cheng, C.C. STAT3 induces G9a to exacerbate HER3 expression for the survival of epidermal growth factor receptor-tyrosine kinase inhibitors in lung cancers. BMC Cancer 2019, 19, 959. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Lee, M.R.; Kim, T.; Kim, Y.W.; Cho, M.Y. Activated STAT3 may participate in tumor progression through increasing CD133/surviving expression in early stage of colon cancer. Biochem. Biophys. Res. Commun. 2018, 497, 354–361. [Google Scholar] [CrossRef]
- Lin, L.; Fuchs, J.; Li, C.; Olson, V.; Bekaii-Saab, T.; Lin, J. STAT3 signaling pathway is necessary for cell survival and tumorsphere forming capacity in ALDH(+)/CD133(+) stem cell-like human colon cancer cells. Biochem. Biophys. Res. Commun. 2011, 416, 246–251. [Google Scholar] [CrossRef]
- Galoczova, M.; Coates, P.; Vojtesek, B. STAT3, stem cells, cancer stem cells and p63. Cell. Mol. Biol. Lett. 2018, 23, 12. [Google Scholar] [CrossRef] [Green Version]
- Obata, T.; Brown, G.E.; Yaffe, M.B. MAP kinase pathways activated by stress: The p38 MAPK pathway. Crit. Care Med. 2000, 28, N67–N77. [Google Scholar] [CrossRef]
- Coulthard, L.R.; White, D.E.; Jones, D.L.; McDermott, M.F.; Burchill, S.A. p38(MAPK): Stress responses from molecular mechanisms to therapeutics. Trends Mol. Med. 2009, 15, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, R.; Kumar, B.; Chen, Z.; Chen, X.; Muller, D.; Lele, S.M.; Washington, M.K.; Batra, S.K.; Dhawan, P.; Singh, A.B. Loss of claudin-3 expression induces IL6/gp130/Stat3 signaling to promote colon cancer malignancy by hyperactivating Wnt/beta-catenin signaling. Oncogene 2017, 36, 6592–6604. [Google Scholar] [CrossRef]
- Lu, L.; Dong, J.; Wang, L.; Xia, Q.; Zhang, D.; Kim, H.; Yin, T.; Fan, S.; Shen, Q. Activation of STAT3 and Bcl-2 and reduction of reactive oxygen species (ROS) promote radioresistance in breast cancer and overcome of radioresistance with niclosamide. Oncogene 2018, 37, 5292–5304. [Google Scholar] [CrossRef]
- Tscherner, A.; Brown, A.C.; Stalker, L.; Kao, J.; Dufort, I.; Sirard, M.A.; LaMarre, J. STAT3 signaling stimulates miR-21 expression in bovine cumulus cells during in vitro oocyte maturation. Sci. Rep. 2018, 8, 11527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Q.; Li, Y.Y.; He, W.F.; Zhang, Z.Z.; Zhou, Q.; Liu, X.; Shen, Y.; Huang, T.T. Interplay between microRNAs and the STAT3 signaling pathway in human cancers. Physiol. Genom. 2013, 45, 1206–1214. [Google Scholar] [CrossRef] [Green Version]
- Haghikia, A.; Hoch, M.; Stapel, B.; Hilfiker-Kleiner, D. STAT3 regulation of and by microRNAs in development and disease. JAKSTAT 2012, 1, 143–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Li, J.; Wang, Q.; Meng, G.; Lv, X.; Zhou, H.; Li, W.; Zhang, J. The relationship between microRNAs and the STAT3-related signaling pathway in cancer. Tumour Biol. 2017, 39, 1010428317719869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.M.; Wang, C.G.; Zhu, M.; Xing, R.; Cui, J.T.; Li, W.M.; Yu, D.D.; Wang, S.B.; Zhu, W.; Ye, Y.J.; et al. STAT3 signaling drives EZH2 transcriptional activation and mediates poor prognosis in gastric cancer. Mol. Cancer 2016, 15, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Guo, W.; Li, Z.; Wu, Y.; Jing, C.; Ren, Y.; Zhao, M.; Kong, L.; Zhang, C.; Dong, J.; et al. Role of the EZH2/miR-200 axis in STAT3-mediated OSCC invasion. Int. J. Oncol. 2018, 52, 1149–1164. [Google Scholar] [CrossRef]
- Zhang, P.; Garnett, J.; Creighton, C.J.; Al Sannaa, G.A.; Igram, D.R.; Lazar, A.; Liu, X.; Liu, C.; Pollock, R.E. EZH2-miR-30d-KPNB1 pathway regulates malignant peripheral nerve sheath tumour cell survival and tumourigenesis. J. Pathol. 2014, 232, 308–318. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Yang, X.; Ma, X.; Ingram, D.R.; Lazar, A.J.; Torres, K.E.; Pollock, R.E. Antitumor effects of pharmacological EZH2 inhibition on malignant peripheral nerve sheath tumor through the miR-30a and KPNB1 pathway. Mol. Cancer 2015, 14, 55. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.H.; Zhang, H.D.; Tang, J.H. MiR-30a: A novel biomarker and potential therapeutic target for cancer. J. Oncol. 2018, 2018, 5167829. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Wang, X.; Huo, Q.; Sun, M.; Cai, C.; Liu, Z.; Hu, G.; Yang, Q. MicroRNA-30a suppresses breast tumor growth and metastasis by targeting metadherin. Oncogene 2014, 33, 3119–3128. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Yan, N.; Sun, W.; Zheng, S.; Jiang, S.; Wang, J.; Guo, C.; Hao, L.; Tian, Y.; Liu, S.; et al. miR-4521-FAM129A axial regulation on ccRCC progression through TIMP-1/MMP2/MMP9 and MDM2/p53/Bcl2/Bax pathways. Cell Death Discov. 2019, 5, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, N.; Osaki, M.; Onuma, K.; Yumioka, T.; Iwamoto, H.; Sejima, T.; Kugoh, H.; Takenaka, A.; Okada, F. Identification of microRNAs involved in resistance to sunitinib in renal cell carcinoma cells. Anticancer Res. 2017, 37, 2985–2992. [Google Scholar] [PubMed]
- Shiba-Ishii, A.; Kim, Y.; Shiozawa, T.; Iyama, S.; Satomi, K.; Kano, J.; Sakashita, S.; Morishita, Y.; Noguchi, M. Stratifin accelerates progression of lung adenocarcinoma at an early stage. Mol. Cancer 2015, 14, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Shiba-Ishii, A.; Nakagawa, T.; Iemura, S.I.; Natsume, T.; Nakano, N.; Matsuoka, R.; Sakashita, S.; Lee, S.; Kawaguchi, A.; et al. Stratifin regulates stabilization of receptor tyrosine kinases via interaction with ubiquitin-specific protease 8 in lung adenocarcinoma. Oncogene 2018, 37, 5387–5402. [Google Scholar] [CrossRef]
- Kondo, A.; Yamamoto, S.; Nakaki, R.; Shimamura, T.; Hamakubo, T.; Sakai, J.; Kodama, T.; Yoshida, T.; Aburatani, H.; Osawa, T. Extracellular acidic pH activates the sterol regulatory element-binding protein 2 to promote tumor progression. Cell Rep. 2017, 18, 2228–2242. [Google Scholar] [CrossRef] [Green Version]
- Ashida, S.; Kawada, C.; Inoue, K. Stromal regulation of prostate cancer cell growth by mevalonate pathway enzymes HMGCS1 and HMGCR. Oncol. Lett. 2017, 14, 6533–6542. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Yu, C.; Yang, X.; Hong, H.; Lu, J.; Hu, W.; Hao, X.; Li, S.; Aikemu, B.; Yang, G.; et al. N-myc downstream-regulated gene 1 inhibits the proliferation of colorectal cancer through emulative antagonizing NEDD4-mediated ubiquitylation of p21. J. Exp. Clin. Cancer Res. 2019, 38, 490. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.C.; Chang, J.; Huang, S.C.; Lin, H.C.; Ho, A.S.; Lim, K.H.; Chang, C.C.; Huang, L.; Chang, Y.C.; Chang, Y.F.; et al. YM155 as an inhibitor of cancer stemness simultaneously inhibits autophosphorylation of epidermal growth factor receptor and G9a-mediated stemness in lung cancer cells. PLoS ONE 2017, 12, e0182149. [Google Scholar] [CrossRef]
- Cheng, C.C.; Hsu, P.J.; Sie, Z.L.; Chen, F.H. Discovery of driver genes in colorectal HT29-derived cancer stem-like tumorspheres. J. Vis. Exp. 2020, 161, e61077. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Primer Sequence | |
|---|---|---|
| HSPA5 | Forward | TAGCGTATGGTGCTGCTGTC |
| Reverse | TTTGTCAGGGGTCTTTCACC | |
| SFN | Forward | AGAGCGAAACCTGCTCTCAG |
| Reverse | CTCCTTGATGAGGTGGCTGT | |
| MAFF | Forward | TCTGTGGATCCCCTATCCAG |
| Reverse | CTTCTGCTTCTGCAGCTCCT | |
| ATF3 | Forward | GTGCCGAAACAAGAAGAAGG |
| Reverse | TGGAGTCCTCCCATTCTGAG | |
| PPARD | Forward | ACTGAGTTCGCCAAGAGCAT |
| Reverse | GCGTTGAACTTGACAGCAAA | |
| MSMO1 | Forward | ATCCAGCTGCCTTTGATTTG |
| Reverse | TTCCAAATGGAGCCTGAAAC | |
| HMGCS1 | Forward | CAAAAAGATCCATGCCCAGT |
| Reverse | AAAGGCTTCCAGGCCACTAT | |
| NDRG1 | Forward | ACAACCCTGAGATGGTGGAG |
| Reverse | TGTGGACCACTTCCACGTTA | |
| MVD | Forward | AGGACAGCAACCAGTTCCAC |
| Reverse | GTGTCGTCCAGGGTGAAGAT | |
| HERPUD1 | Forward | ACTTGCTTCCAAAGCAGGAA |
| Reverse | CCCTTTGCCTTAAACCATCA | |
| TSC22D2 | Forward | GCAGCGTCCTGACTAGATCC |
| Reverse | GAGCTGTCAGTTCCCGACTC | |
| CALB2 | Forward | GCTCCAGGAATACACCCAAA |
| Reverse | CAGCTCATGCTCGTCAATGT | |
| GAPDH | Forward | GAGTCAACGGATTTGGTCGT |
| Reverse | TTGATTTTGGAGGGATCTCG | |
| ChIP-miR-30a-5p | Forward | CACACCCACTTTTCTGTATCAACT |
| Reverse | GAATTCCACTCCCATTCTCTTATG |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, C.-C.; Yang, B.-L.; Chen, W.-C.; Ho, A.-S.; Sie, Z.-L.; Lin, H.-C.; Chang, C.-C. STAT3 Mediated miR-30a-5p Inhibition Enhances Proliferation and Inhibits Apoptosis in Colorectal Cancer Cells. Int. J. Mol. Sci. 2020, 21, 7315. https://doi.org/10.3390/ijms21197315
Cheng C-C, Yang B-L, Chen W-C, Ho A-S, Sie Z-L, Lin H-C, Chang C-C. STAT3 Mediated miR-30a-5p Inhibition Enhances Proliferation and Inhibits Apoptosis in Colorectal Cancer Cells. International Journal of Molecular Sciences. 2020; 21(19):7315. https://doi.org/10.3390/ijms21197315
Chicago/Turabian StyleCheng, Chun-Chia, Bi-Ling Yang, Wen-Chao Chen, Ai-Sheng Ho, Zong-Lin Sie, Hsin-Chi Lin, and Chun-Chao Chang. 2020. "STAT3 Mediated miR-30a-5p Inhibition Enhances Proliferation and Inhibits Apoptosis in Colorectal Cancer Cells" International Journal of Molecular Sciences 21, no. 19: 7315. https://doi.org/10.3390/ijms21197315
APA StyleCheng, C. -C., Yang, B. -L., Chen, W. -C., Ho, A. -S., Sie, Z. -L., Lin, H. -C., & Chang, C. -C. (2020). STAT3 Mediated miR-30a-5p Inhibition Enhances Proliferation and Inhibits Apoptosis in Colorectal Cancer Cells. International Journal of Molecular Sciences, 21(19), 7315. https://doi.org/10.3390/ijms21197315