Immunopathogenesis of ANCA-Associated Vasculitis

 ,
,  , , , , ,
, , , , ,  , , ,
, , ,  add
Show full author list
add
Show full author list
Abstract
:1. Introduction
2. Overview of ANCA-Associated Vasculitis
2.1. Genetic and Epigenetic Changes in ANCA-Associated Vasculitis
2.2. Pathogenesis of ANCA-Associated Vasculitis
3. Classification of ANCA-Associated Vasculitis According to Clinical Phenotypes
3.1. Brief Description of the Different Phenotypes
3.2. Current Classification Criteria
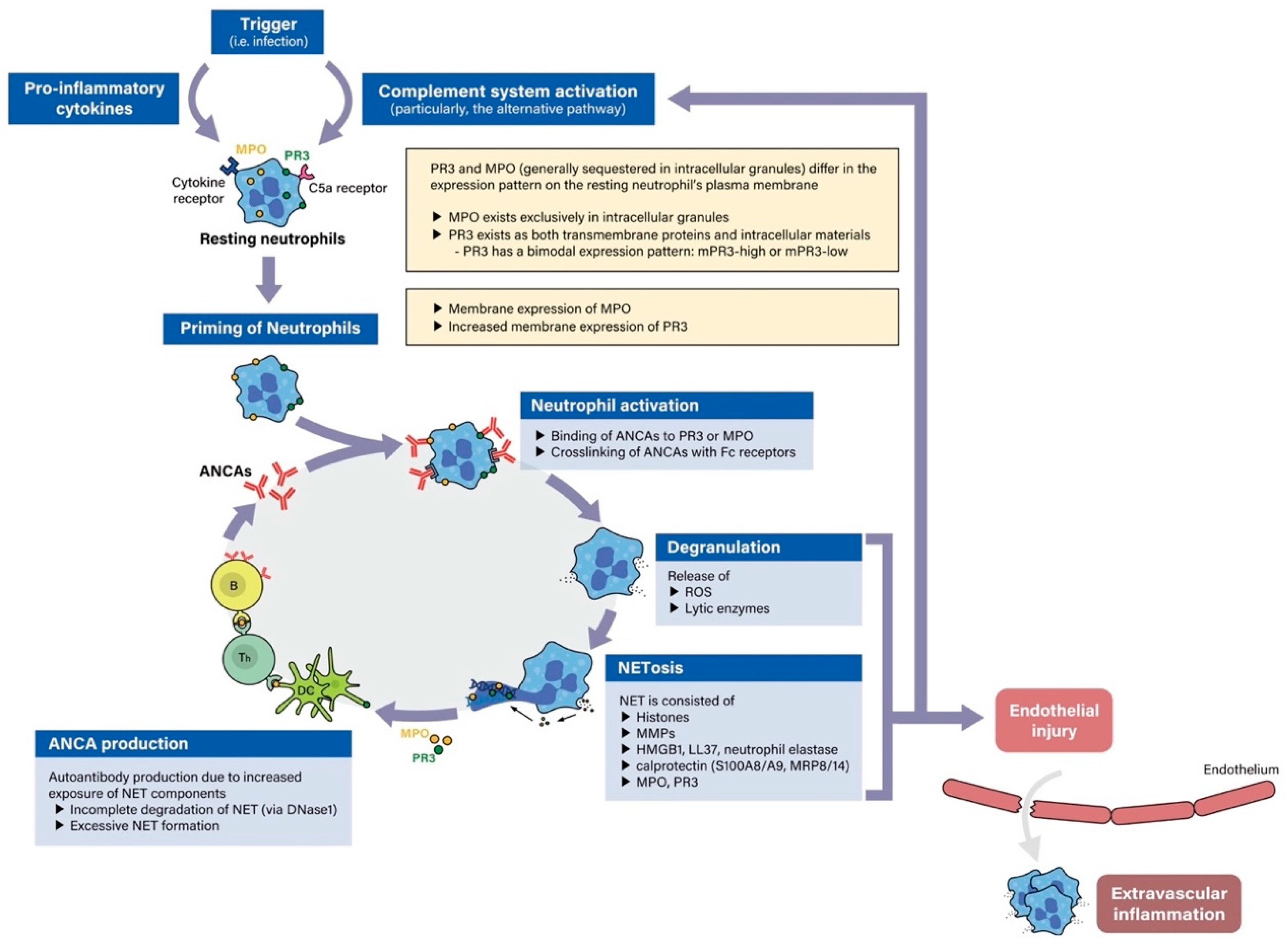
4. Pathogenetic Steps in ANCA-Associated Vasculitis and ANCA Serotype Specificity
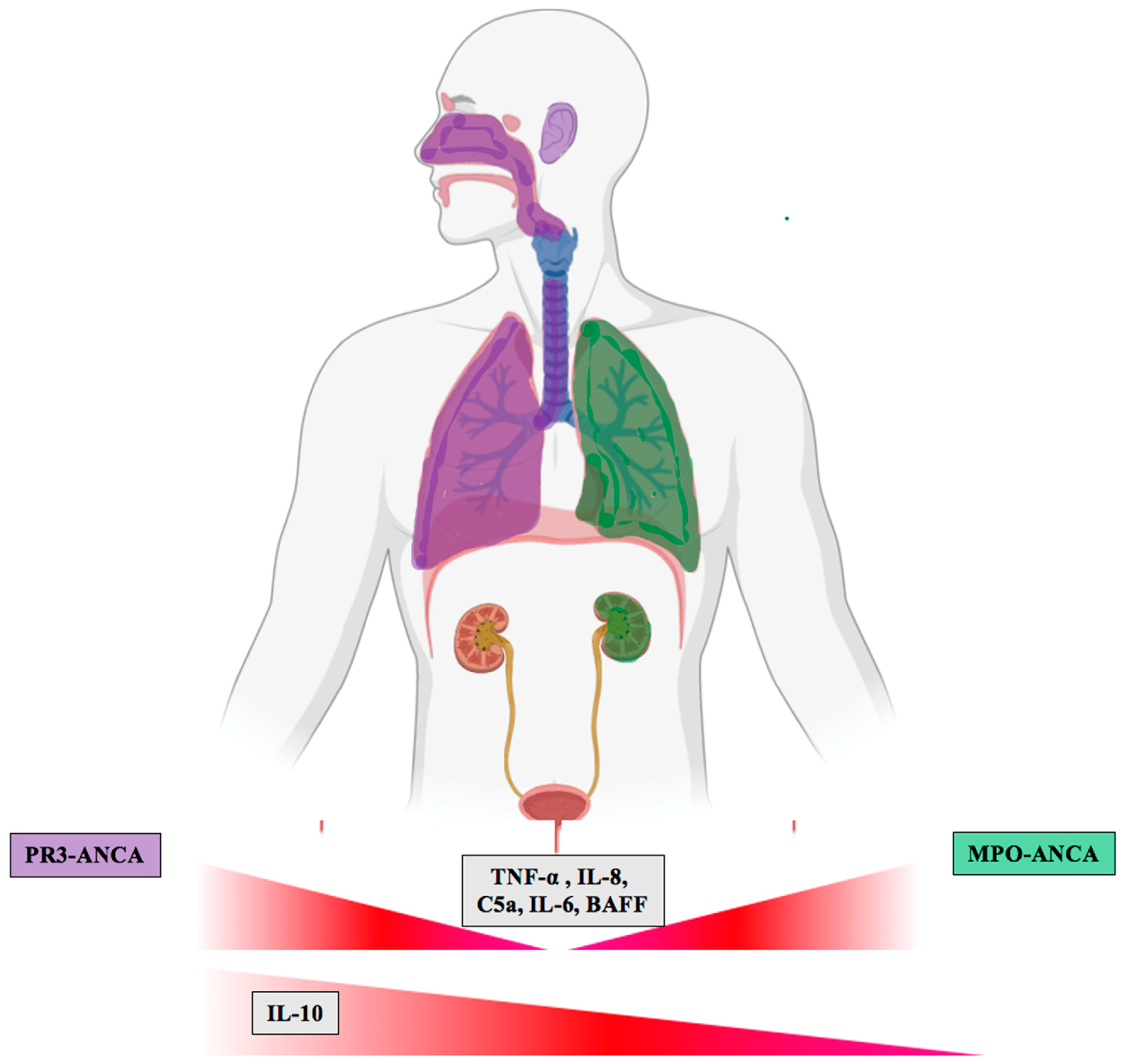
4.1. Regulation of Cytokines and Related Molecules According to ANCA Serotype
4.1.1. Priming of Neutrophils and Monocytes as a Key Step in the Pathogenesis of PR3-and MPO-ANCA Vasculitis
4.1.2. Activation of Neutrophils and Monocytes in PR3-and MPO-ANCA Vasculitis
4.1.3. T Cell Activation in PR3-and MPO-ANCA Vasculitis
4.1.4. B Cell Activation in PR3-and MPO-ANCA Vasculitis
4.1.5. Tissue Damage and Repair in PR3-and MPO-ANCA Vasculitis
4.1.6. Endothelial Injury and Repair in PR3-and MPO-ANCA Vasculitis
4.1.7. Role of Proteinase-3
5. Differences in Biomarker Expression in PR3-ANCA and MPO-ANCA Vasculitis
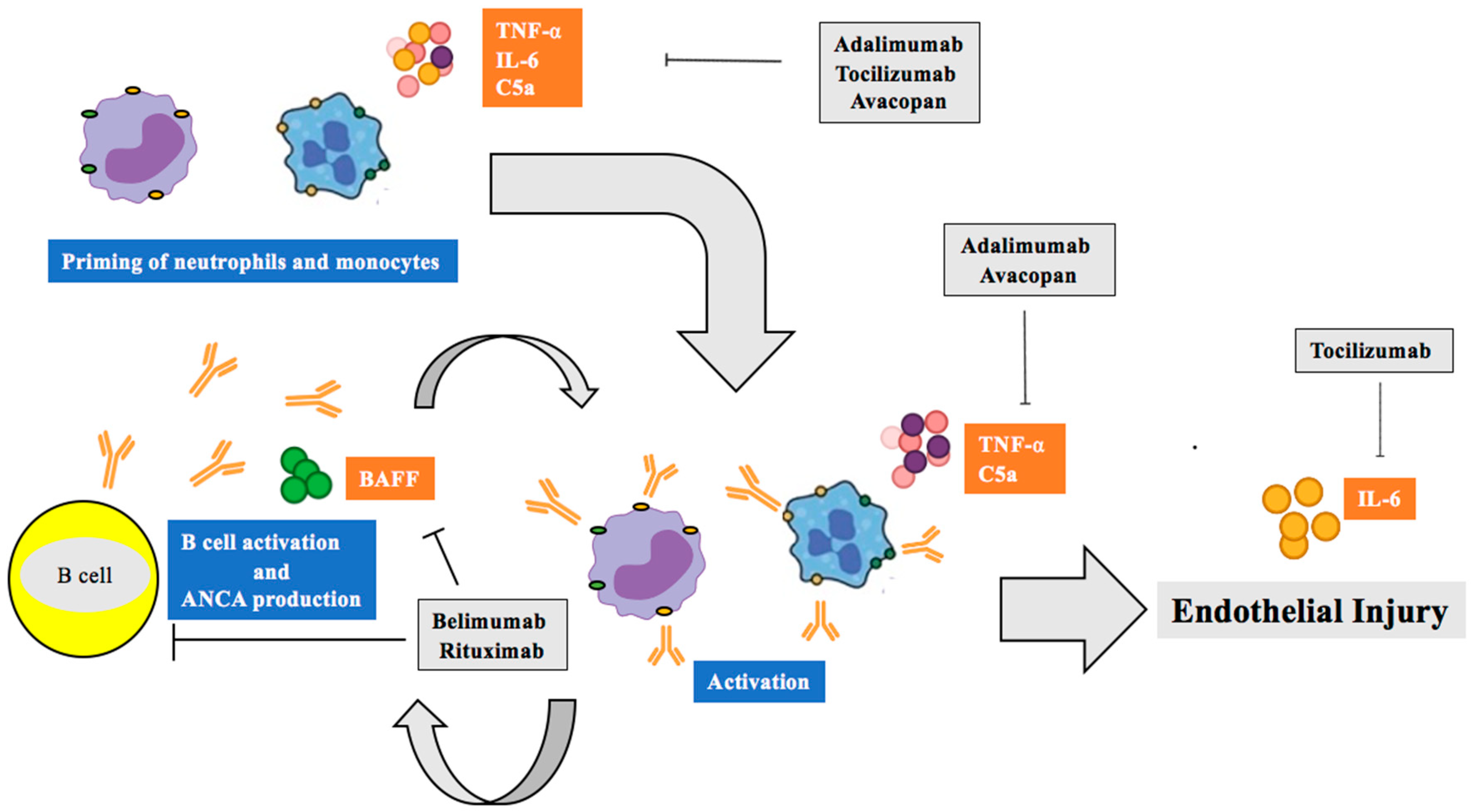
6. Therapeutic Implications of Biomarker Discoveries
6.1. TNF-α Inhibitors
6.2. Interleukin-6 and ANCA-Associated Vasculitis
6.3. B-lymphocyte Stimulator (Blys)/B Cell Activating Factor (BAFF) Inhibition
6.4. Complement C5a/C5ar Inhibition
6.5. Rituximab, the “New Normal”
6.6. Developing Preclinical Targets
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACR | American College of Rheumatology |
| ADAM | ADAM metalloproteinase domain |
| ANCA | Anti-neutrophil cytoplasmic antibody |
| BAFF | B cell activating factor |
| BCA | B cell-activating chemokine |
| BLyS | B-lymphocyte stimulator |
| BREVAS | Belimumab in Remission of VASculitis |
| CALIBRATE | Rituximab and Belimumab for Lupus Nephritis |
| CCR | C-C motif chemokine receptor |
| CD | Cluster of differentiation |
| CHCC | Chapel Hill Consensus Conference |
| COMBIVAS | A study of Rituximab and Belimumab Combination therapy in PR3 |
| COVID-19 | Coronavirus Disease 2019 |
| EGPA | Eosinophilic granulomatosis with polyangiitis |
| EMA | European Medicines Agency |
| ENT | Ear, nose and throat |
| G-CSF | Granulocyte colony-stimulating factor |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| GPA | Granulomatosis with polyangiitis |
| GWAS | Genome-wide association study |
| HLA | Human leukocyte antigen |
| HMGB1 | High-mobility-group-protein B1 |
| ICAM | Intercellular adhesion molecule |
| IFN | Interferon |
| IL | Interleukin |
| KIM | Kidney injury molecule |
| MAINRITSAN | Maintenance of Remission using Rituximab in Systemic ANCA-associated Vasculitis |
| MCP | Monocyte chemoattractant protein-1 |
| MIF | Migration inhibitory factor |
| MMP | Matrix metalloproteinase |
| MPA | Microscopic polyangiitis |
| MPO | Myeloperoxidase |
| NET | Neutrophil extracellular trap |
| NGAL | Neutrophil gelatinase-associated lipocalin |
| NGF | Nerve growth factor |
| PAI | Plasminogen activator inhibitor |
| PDGF | Platelet derived growth factor |
| PR3 | Proteinase 3 |
| RAS | Renin angiotensin-system |
| RAVE | Rituximab in ANCA-associated vasculitis |
| RITAZAREM | Rituximab Vasculitis Maintenance Study |
| RITUXVAS | Rituximab versus cyclophosphamide in ANCA-associated vasculitis |
| ROS | Reactive oxygen species |
| SEMA | Semaphorin |
| sFlt | Soluble Fms-like tyrosine kinase |
| SOC | Standard of care |
| ST | Suppression of tumorigenesis |
| TARC | Thymus and activation-regulated chemokine |
| Th | T helper |
| TKT | Transketolase |
| TNC | Tenascin C |
| TNF | Tumor necrosis factor |
| TXA2 | Thromboxane A2 |
| WGET | Wegener’s Granulomatosis Etanercept Trial |
References
- Jennette, J.C.; Falk, R.J.; Bacon, P.A.; Basu, N.; Cid, M.C.; Ferrario, F.; Flores-Suarez, L.F.; Gross, W.L.; Guillevin, L.; Hagen, E.C.; et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013, 65, 1–11. [Google Scholar] [CrossRef] [PubMed]
- van der Woude, F.J.; Rasmussen, N.; Lobatto, S.; Wiik, A.; Permin, H.; van Es, L.A.; van der Giessen, M.; van der Hem, G.K.; The, T.H. Autoantibodies against neutrophils and monocytes: Tool for diagnosis and marker of disease activity in Wegener’s granulomatosis. Lancet 1985, 1, 425–429. [Google Scholar] [CrossRef]
- Cohen Tervaert, J.W.; Damoiseaux, J. Antineutrophil cytoplasmic autoantibodies: How are they detected and what is their use for diagnosis, classification and follow-up? Clin. Rev. Allergy Immunol. 2012, 43, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Radice, A.; Sinico, R.A. Antineutrophil cytoplasmic antibodies (ANCA). Autoimmunity 2005, 38, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Bossuyt, X.; Cohen Tervaert, J.W.; Arimura, Y.; Blockmans, D.; Flores-Suarez, L.F.; Guillevin, L.; Hellmich, B.; Jayne, D.; Jennette, J.C.; Kallenberg, C.G.M.; et al. Position paper: Revised 2017 international consensus on testing of ANCAs in granulomatosis with polyangiitis and microscopic polyangiitis. Nat. Rev. Rheumatol. 2017, 13, 683–692. [Google Scholar] [CrossRef]
- Miloslavsky, E.M.; Lu, N.; Unizony, S.; Choi, H.K.; Merkel, P.A.; Seo, P.; Spiera, R.; Langford, C.A.; Hoffman, G.S.; Kallenberg, C.G.; et al. Myeloperoxidase-Antineutrophil Cytoplasmic Antibody (ANCA)-Positive and ANCA-Negative Patients With Granulomatosis With Polyangiitis (Wegener’s): Distinct Patient Subsets. Arthritis Rheumatol. 2016, 68, 2945–2952. [Google Scholar] [CrossRef]
- Schirmer, J.H.; Wright, M.N.; Herrmann, K.; Laudien, M.; Nolle, B.; Reinhold-Keller, E.; Bremer, J.P.; Moosig, F.; Holle, J.U. Myeloperoxidase-Antineutrophil Cytoplasmic Antibody (ANCA)-Positive Granulomatosis With Polyangiitis (Wegener’s) Is a Clinically Distinct Subset of ANCA-Associated Vasculitis: A Retrospective Analysis of 315 Patients From a German Vasculitis Referral Center. Arthritis Rheumatol. 2016, 68, 2953–2963. [Google Scholar] [CrossRef] [Green Version]
- Lotscher, F.; Krusche, M.; Ruffer, N.; Kubacki, T.; Person, F.; Kotter, I. Cocaine-induced ANCA-associated renal disease: A case-based review. Rheumatol. Int. 2019, 39, 2005–2014. [Google Scholar] [CrossRef]
- Nakazawa, D.; Masuda, S.; Tomaru, U.; Ishizu, A. Pathogenesis and therapeutic interventions for ANCA-associated vasculitis. Nat. Rev. Rheumatol. 2019, 15, 91–101. [Google Scholar] [CrossRef]
- Lyons, P.A.; Rayner, T.F.; Trivedi, S.; Holle, J.U.; Watts, R.A.; Jayne, D.R.; Baslund, B.; Brenchley, P.; Bruchfeld, A.; Chaudhry, A.N.; et al. Genetically distinct subsets within ANCA-associated vasculitis. N. Engl. J. Med. 2012, 367, 214–223. [Google Scholar] [CrossRef] [Green Version]
- Unizony, S.; Villarreal, M.; Miloslavsky, E.M.; Lu, N.; Merkel, P.A.; Spiera, R.; Seo, P.; Langford, C.A.; Hoffman, G.S.; Kallenberg, C.M.; et al. Clinical outcomes of treatment of anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis based on ANCA type. Ann. Rheum. Dis. 2016, 75, 1166–1169. [Google Scholar] [CrossRef] [PubMed]
- Hogan, S.L.; Falk, R.J.; Chin, H.; Cai, J.; Jennette, C.E.; Jennette, J.C.; Nachman, P.H. Predictors of relapse and treatment resistance in antineutrophil cytoplasmic antibody-associated small-vessel vasculitis. Ann. Intern. Med. 2005, 143, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Tanna, A.; Guarino, L.; Tam, F.W.; Rodriquez-Cubillo, B.; Levy, J.B.; Cairns, T.D.; Griffith, M.; Tarzi, R.M.; Caplin, B.; Salama, A.D.; et al. Long-term outcome of anti-neutrophil cytoplasm antibody-associated glomerulonephritis: Evaluation of the international histological classification and other prognostic factors. Nephrol. Dial. Transplant. 2015, 30, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Mahr, A.; Katsahian, S.; Varet, H.; Guillevin, L.; Hagen, E.C.; Hoglund, P.; Merkel, P.A.; Pagnoux, C.; Rasmussen, N.; Westman, K.; et al. Revisiting the classification of clinical phenotypes of anti-neutrophil cytoplasmic antibody-associated vasculitis: A cluster analysis. Ann. Rheum. Dis. 2013, 72, 1003–1010. [Google Scholar] [CrossRef]
- Kronbichler, A.; Leierer, J.; Shin, J.I.; Merkel, P.A.; Spiera, R.; Seo, P.; Langford, C.A.; Hoffman, G.S.; Kallenberg, C.G.M.; St Clair, E.W.; et al. Association of Pulmonary Hemorrhage, Positive Proteinase 3, and Urinary Red Blood Cell Casts With Venous Thromboembolism in Antineutrophil Cytoplasmic Antibody-Associated Vasculitis. Arthritis Rheumatol. 2019, 71, 1888–1893. [Google Scholar] [CrossRef] [Green Version]
- Wallace, Z.S.; Fu, X.; Harkness, T.; Stone, J.H.; Zhang, Y.; Choi, H. All-cause and cause-specific mortality in ANCA-associated vasculitis: Overall and according to ANCA type. Rheumatology 2019. [Google Scholar] [CrossRef]
- Berti, A.; Warner, R.; Johnson, K.; Cornec, D.; Schroeder, D.; Kabat, B.; Langford, C.A.; Hoffman, G.S.; Fervenza, F.C.; Kallenberg, C.G.M.; et al. Brief Report: Circulating Cytokine Profiles and Antineutrophil Cytoplasmic Antibody Specificity in Patients With Antineutrophil Cytoplasmic Antibody-Associated Vasculitis. Arthritis Rheumatol. 2018, 70, 1114–1121. [Google Scholar] [CrossRef]
- Cui, Z.; Zhao, M.H.; Segelmark, M.; Hellmark, T. Natural autoantibodies to myeloperoxidase, proteinase 3, and the glomerular basement membrane are present in normal individuals. Kidney Int. 2010, 78, 590–597. [Google Scholar] [CrossRef] [Green Version]
- Furuta, S.; Jayne, D.R. Antineutrophil cytoplasm antibody-associated vasculitis: Recent developments. Kidney Int. 2013, 84, 244–249. [Google Scholar] [CrossRef] [Green Version]
- Rahmattulla, C.; Mooyaart, A.L.; van Hooven, D.; Schoones, J.W.; Bruijn, J.A.; Dekkers, O.M.; Bajema, I.M. Genetic variants in ANCA-associated vasculitis: A meta-analysis. Ann. Rheum. Dis. 2016, 75, 1687–1692. [Google Scholar] [CrossRef]
- Lee, K.S.; Kronbichler, A.; Pereira Vasconcelos, D.F.; Pereira da Silva, F.R.; Ko, Y.; Oh, Y.S.; Eisenhut, M.; Merkel, P.A.; Jayne, D.; Amos, C.I.; et al. Genetic Variants in Antineutrophil Cytoplasmic Antibody-Associated Vasculitis: A Bayesian Approach and Systematic Review. J. Clin. Med. 2019, 8, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, B.E.; Yang, J.; Muthigi, A.; Hogan, S.L.; Hu, Y.; Starmer, J.; Henderson, C.D.; Poulton, C.J.; Brant, E.J.; Pendergraft, W.F., 3rd; et al. Gene-Specific DNA Methylation Changes Predict Remission in Patients with ANCA-Associated Vasculitis. J. Am. Soc. Nephrol. 2017, 28, 1175–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessenbrock, K.; Krumbholz, M.; Schonermarck, U.; Back, W.; Gross, W.L.; Werb, Z.; Grone, H.J.; Brinkmann, V.; Jenne, D.E. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 2009, 15, 623–625. [Google Scholar] [CrossRef] [PubMed]
- Sangaletti, S.; Tripodo, C.; Chiodoni, C.; Guarnotta, C.; Cappetti, B.; Casalini, P.; Piconese, S.; Parenza, M.; Guiducci, C.; Vitali, C.; et al. Neutrophil extracellular traps mediate transfer of cytoplasmic neutrophil antigens to myeloid dendritic cells toward ANCA induction and associated autoimmunity. Blood 2012, 120, 3007–3018. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.H.; Kronbichler, A.; Park, D.D.; Park, Y.; Moon, H.; Kim, H.; Choi, J.H.; Choi, Y.; Shim, S.; Lyu, I.S.; et al. Neutrophil extracellular traps (NETs) in autoimmune diseases: A comprehensive review. Autoimmun. Rev. 2017, 16, 1160–1173. [Google Scholar] [CrossRef]
- Savage, C.O.; Gaskin, G.; Pusey, C.D.; Pearson, J.D. Myeloperoxidase binds to vascular endothelial cells, is recognized by ANCA and can enhance complement dependent cytotoxicity. Adv. Exp. Med. Biol. 1993, 336, 121–123. [Google Scholar] [CrossRef]
- Huugen, D.; Xiao, H.; van Esch, A.; Falk, R.J.; Peutz-Kootstra, C.J.; Buurman, W.A.; Tervaert, J.W.; Jennette, J.C.; Heeringa, P. Aggravation of anti-myeloperoxidase antibody-induced glomerulonephritis by bacterial lipopolysaccharide: Role of tumor necrosis factor-alpha. Am. J. Pathol. 2005, 167, 47–58. [Google Scholar] [CrossRef]
- Kronbichler, A.; Kerschbaum, J.; Mayer, G. The Influence and Role of Microbial Factors in Autoimmune Kidney Diseases: A Systematic Review. J. Immunol. Res. 2015, 2015, 858027. [Google Scholar] [CrossRef] [Green Version]
- Flint, S.M.; McKinney, E.F.; Smith, K.G. Emerging concepts in the pathogenesis of antineutrophil cytoplasmic antibody-associated vasculitis. Curr. Opin. Rheumatol. 2015, 27, 197–203. [Google Scholar] [CrossRef]
- Abdulahad, W.H.; Lamprecht, P.; Kallenberg, C.G. T-helper cells as new players in ANCA-associated vasculitides. Arthritis Res. 2011, 13, 236. [Google Scholar] [CrossRef] [Green Version]
- Moiseev, S.; Lee, J.M.; Zykova, A.; Bulanov, N.; Novikov, P.; Gitel, E.; Bulanova, M.; Safonova, E.; Shin, J.I.; Kronbichler, A.; et al. THE alternative complement pathway in ANCA-associated vasculitis: Further evidence and a meta-analysis. Clin. Exp. Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, A.; Xiao, H.; Jennette, J.C.; Schneider, W.; Luft, F.C.; Kettritz, R. C5a receptor mediates neutrophil activation and ANCA-induced glomerulonephritis. J. Am. Soc. Nephrol. 2009, 20, 289–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stegeman, C.A.; Tervaert, J.W.; de Jong, P.E.; Kallenberg, C.G. Trimethoprim-sulfamethoxazole (co-trimoxazole) for the prevention of relapses of Wegener’s granulomatosis. Dutch Co-Trimoxazole Wegener Study Group. N. Engl. J. Med. 1996, 335, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Lepse, N.; Land, J.; Rutgers, A.; Kallenberg, C.G.; Stegeman, C.A.; Abdulahad, W.H.; Heeringa, P. Toll-like receptor 9 activation enhances B cell activating factor and interleukin-21 induced anti-proteinase 3 autoantibody production in vitro. Rheumatology 2016, 55, 162–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, A.; Rousselle, A.; Becker, J.U.; von Mässenhausen, A.; Linkermann, A.; Kettritz, R. Necroptosis controls NET generation and mediates complement activation, endothelial damage, and autoimmune vasculitis. Proc. Natl. Acad. Sci. USA 2017, 114, E9618–E9625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewert, B.H.; Jennette, J.C.; Falk, R.J. Anti-myeloperoxidase antibodies stimulate neutrophils to damage human endothelial cells. Kidney Int. 1992, 41, 375–383. [Google Scholar] [CrossRef] [Green Version]
- Ewert, B.H.; Becker, M.E.; Jennette, J.C.; Falk, R.J. Antimyeloperoxidase antibodies induce neutrophil adherence to cultured human endothelial cells. Ren. Fail. 1995, 17, 125–133. [Google Scholar] [CrossRef]
- Frangou, E.; Vassilopoulos, D.; Boletis, J.; Boumpas, D.T. An emerging role of neutrophils and NETosis in chronic inflammation and fibrosis in systemic lupus erythematosus (SLE) and ANCA-associated vasculitides (AAV): Implications for the pathogenesis and treatment. Autoimmun Rev. 2019, 18, 751–760. [Google Scholar] [CrossRef]
- Cleary, S.J.; Kwaan, N.; Tian, J.J.; Calabrese, D.R.; Mallavia, B.; Magnen, M.; Greenland, J.R.; Urisman, A.; Singer, J.P.; Hays, S.R.; et al. Complement activation on endothelium initiates antibody-mediated acute lung injury. J. Clin. Investig. 2020. [Google Scholar] [CrossRef]
- Jennette, J.C.; Falk, R.J. Small-vessel vasculitis. N. Engl. J. Med. 1997, 337, 1512–1523. [Google Scholar] [CrossRef]
- de Groot, K.; Harper, L.; Jayne, D.R.; Flores Suarez, L.F.; Gregorini, G.; Gross, W.L.; Luqmani, R.; Pusey, C.D.; Rasmussen, N.; Sinico, R.A.; et al. Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: A randomized trial. Ann. Intern. Med. 2009, 150, 670–680. [Google Scholar] [CrossRef]
- De Groot, K.; Rasmussen, N.; Bacon, P.A.; Tervaert, J.W.; Feighery, C.; Gregorini, G.; Gross, W.L.; Luqmani, R.; Jayne, D.R. Randomized trial of cyclophosphamide versus methotrexate for induction of remission in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum. 2005, 52, 2461–2469. [Google Scholar] [CrossRef]
- Jayne, D.; Rasmussen, N.; Andrassy, K.; Bacon, P.; Tervaert, J.W.; Dadoniene, J.; Ekstrand, A.; Gaskin, G.; Gregorini, G.; de Groot, K.; et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N. Engl. J. Med. 2003, 349, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Jayne, D.R.; Gaskin, G.; Rasmussen, N.; Abramowicz, D.; Ferrario, F.; Guillevin, L.; Mirapeix, E.; Savage, C.O.; Sinico, R.A.; Stegeman, C.A.; et al. Randomized trial of plasma exchange or high-dosage methylprednisolone as adjunctive therapy for severe renal vasculitis. J. Am. Soc. Nephrol. 2007, 18, 2180–2188. [Google Scholar] [CrossRef] [PubMed]
- Berti, A.; Kronbichler, A. Orbital masses in ANCA-associated vasculitis: An unsolved challenge? Rheumatology 2019, 58, 1520–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kronbichler, A.; Leierer, J.; Leierer, G.; Mayer, G.; Casian, A.; Hoglund, P.; Westman, K.; Jayne, D. Clinical associations with venous thromboembolism in anti-neutrophil cytoplasm antibody-associated vasculitides. Rheumatology 2017, 56, 704–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millet, A.; Pederzoli-Ribeil, M.; Guillevin, L.; Witko-Sarsat, V.; Mouthon, L. Antineutrophil cytoplasmic antibody-associated vasculitides: Is it time to split up the group? Ann. Rheum. Dis. 2013, 72, 1273–1279. [Google Scholar] [CrossRef] [Green Version]
- Kemna, M.J.; Damoiseaux, J.; Austen, J.; Winkens, B.; Peters, J.; van Paassen, P.; Cohen Tervaert, J.W. ANCA as a predictor of relapse: Useful in patients with renal involvement but not in patients with nonrenal disease. J. Am. Soc. Nephrol. 2015, 26, 537–542. [Google Scholar] [CrossRef]
- McClure, M.E.; Wason, J.; Gopaluni, S.; Tieu, J.; Smith, R.M.; Jayne, D.R.; Jones, R.B. Evaluation of PR3-ANCA Status After Rituximab for ANCA-Associated Vasculitis. J. Clin. Rheumatol. 2019, 25, 217–223. [Google Scholar] [CrossRef]
- Kronbichler, A.; Jayne, D.R.W. ANCA Renal Risk Score: Is prediction of end-stage renal disease at baseline possible? Kidney Int. 2018, 94, 1045–1047. [Google Scholar] [CrossRef]
- Kronbichler, A.; Jayne, D.R.W. Estimating the epidemiology of anti-neutrophil cytoplasm antibody-associated renal vasculitis and the role of histologic chronicity in predicting renal outcomes. Nephrol. Dial. Transplant. 2019, 34, 1429–1432. [Google Scholar] [CrossRef] [PubMed]
- Hilhorst, M.; van Paassen, P.; Tervaert, J.W.; Registry, L.R. Proteinase 3-ANCA Vasculitis versus Myeloperoxidase-ANCA Vasculitis. J. Am. Soc. Nephrol. 2015, 26, 2314–2327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franssen, C.F.; Stegeman, C.A.; Kallenberg, C.G.; Gans, R.O.; De Jong, P.E.; Hoorntje, S.J.; Tervaert, J.W. Antiproteinase 3- and antimyeloperoxidase-associated vasculitis. Kidney Int. 2000, 57, 2195–2206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leavitt, R.Y.; Fauci, A.F.; Bloch, D.A.; Michel, B.A.; Hunder, G.G.; Arend, W.P.; Calabrese, L.H.; Fries, J.F.; Lie, J.T.; Lightfoot, R.W., Jr.; et al. The American College of Rheumatology 1990 criteria for the classification of Wegener’s granulomatosis. Arthritis Rheum. 1990, 33, 1101–1107. [Google Scholar]
- Seeliger, B.; Sznajd, J.; Robson, J.C.; Judge, A.; Craven, A.; Grayson, P.C.; Suppiah, R.S.; Watts, R.A.; Merkel, P.A.; Luqmani, R.A. Are the 1990 American College of Rheumatology vasculitis classification criteria still valid? Rheumatology 2017, 56, 1154–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennette, J.C.; Falk, R.J.; Andrassy, K.; Bacon, P.A.; Churg, J.; Gross, W.L.; Hagen, E.C.; Hoffman, G.S.; Hunder, G.G.; Kallenberg, C.G.; et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum. 1994, 37, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Watts, R.; Lane, S.; Hanslik, T.; Hauser, T.; Hellmich, B.; Koldingsnes, W.; Mahr, A.; Segelmark, M.; Cohen-Tervaert, J.W.; Scott, D. Development and validation of a consensus methodology for the classification of the ANCA-associated vasculitides and polyarteritis nodosa for epidemiological studies. Ann. Rheum. Dis. 2007, 66, 222–227. [Google Scholar] [CrossRef]
- Abdulkader, R.; Lane, S.E.; Scott, D.G.; Watts, R.A. Classification of vasculitis: EMA classification using CHCC 2012 definitions. Ann. Rheum. Dis. 2013, 72, 1888. [Google Scholar] [CrossRef]
- Wilde, B.; Thewissen, M.; Damoiseaux, J.; van Paassen, P.; Witzke, O.; Tervaert, J.W. T cells in ANCA-associated vasculitis: What can we learn from lesional versus circulating T cells? Arthritis Res. 2010, 12, 204. [Google Scholar] [CrossRef] [Green Version]
- Popa, E.R.; Stegeman, C.A.; Bos, N.A.; Kallenberg, C.G.; Tervaert, J.W. Differential B- and T-cell activation in Wegener’s granulomatosis. J. Allergy Clin. Immunol. 1999, 103, 885–894. [Google Scholar] [CrossRef]
- Dumoitier, N.; Terrier, B.; London, J.; Lofek, S.; Mouthon, L. Implication of B lymphocytes in the pathogenesis of ANCA-associated vasculitides. Autoimmun. Rev. 2015, 14, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Jennette, J.C.; Falk, R.J. B cell-mediated pathogenesis of ANCA-mediated vasculitis. Semin. Immunopathol. 2014, 36, 327–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Specks, U.; Merkel, P.A.; Seo, P.; Spiera, R.; Langford, C.A.; Hoffman, G.S.; Kallenberg, C.G.; St Clair, E.W.; Fessler, B.J.; Ding, L.; et al. Efficacy of remission-induction regimens for ANCA-associated vasculitis. N. Engl. J. Med. 2013, 369, 417–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, J.H.; Merkel, P.A.; Spiera, R.; Seo, P.; Langford, C.A.; Hoffman, G.S.; Kallenberg, C.G.; St Clair, E.W.; Turkiewicz, A.; Tchao, N.K.; et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N. Engl. J. Med. 2010, 363, 221–232. [Google Scholar] [CrossRef]
- Jones, R.B.; Tervaert, J.W.; Hauser, T.; Luqmani, R.; Morgan, M.D.; Peh, C.A.; Savage, C.O.; Segelmark, M.; Tesar, V.; van Paassen, P.; et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N. Engl. J. Med. 2010, 363, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Falk, R.J.; Terrell, R.S.; Charles, L.A.; Jennette, J.C. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc. Natl. Acad. Sci. USA 1990, 87, 4115–4119. [Google Scholar] [CrossRef] [Green Version]
- Charles, L.A.; Caldas, M.L.; Falk, R.J.; Terrell, R.S.; Jennette, J.C. Antibodies against granule proteins activate neutrophils in vitro. J. Leukoc. Biol. 1991, 50, 539–546. [Google Scholar] [CrossRef]
- Little, M.A.; Smyth, C.L.; Yadav, R.; Ambrose, L.; Cook, H.T.; Nourshargh, S.; Pusey, C.D. Antineutrophil cytoplasm antibodies directed against myeloperoxidase augment leukocyte-microvascular interactions in vivo. Blood 2005, 106, 2050–2058. [Google Scholar] [CrossRef]
- Csernok, E.; Ernst, M.; Schmitt, W.; Bainton, D.F.; Gross, W.L. Activated neutrophils express proteinase 3 on their plasma membrane in vitro and in vivo. Clin. Exp. Immunol. 1994, 95, 244–250. [Google Scholar] [CrossRef]
- Uehara, A.; Sato, T.; Iwashiro, A.; Yokota, S. PR3-ANCA in Wegener’s granulomatosis prime human mononuclear cells for enhanced activation via TLRs and NOD1/2. Diagn. Pathol. 2009, 4, 23. [Google Scholar] [CrossRef] [Green Version]
- Berti, A.; Cavalli, G.; Campochiaro, C.; Guglielmi, B.; Baldissera, E.; Cappio, S.; Sabbadini, M.G.; Doglioni, C.; Dagna, L. Interleukin-6 in ANCA-associated vasculitis: Rationale for successful treatment with tocilizumab. Semin. Arthritis. Rheum. 2015, 45, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Hewins, P.; Morgan, M.D.; Holden, N.; Neil, D.; Williams, J.M.; Savage, C.O.; Harper, L. IL-18 is upregulated in the kidney and primes neutrophil responsiveness in ANCA-associated vasculitis. Kidney Int. 2006, 69, 605–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monach, P.A.; Warner, R.L.; Tomasson, G.; Specks, U.; Stone, J.H.; Ding, L.; Fervenza, F.C.; Fessler, B.J.; Hoffman, G.S.; Ikle, D.; et al. Serum proteins reflecting inflammation, injury and repair as biomarkers of disease activity in ANCA-associated vasculitis. Ann. Rheum. Dis. 2013, 72, 1342–1350. [Google Scholar] [CrossRef] [PubMed]
- Wilde, B.; Hoerning, A.; Kribben, A.; Witzke, O.; Dolff, S. Abnormal expression pattern of the IL-2 receptor beta-chain on CD4+ T cells in ANCA-associated vasculitis. Dis. Markers 2014, 2014, 249846. [Google Scholar] [CrossRef] [Green Version]
- Kronbichler, A.; Kerschbaum, J.; Grundlinger, G.; Leierer, J.; Mayer, G.; Rudnicki, M. Evaluation and validation of biomarkers in granulomatosis with polyangiitis and microscopic polyangiitis. Nephrol. Dial. Transplant. 2016, 31, 930–936. [Google Scholar] [CrossRef]
- Hellmich, B.; Csernok, E.; Trabandt, A.; Gross, W.L.; Ernst, M. Granulocyte-macrophage colony-stimulating factor (GM-CSF) but not granulocyte colony-stimulating factor (G-CSF) induces plasma membrane expression of proteinase 3 (PR3) on neutrophils in vitro. Clin. Exp. Immunol. 2000, 120, 392–398. [Google Scholar] [CrossRef]
- Wang, C.; Wang, H.; Chang, D.Y.; Hao, J.; Zhao, M.H.; Chen, M. High mobility group box 1 contributes to anti-neutrophil cytoplasmic antibody-induced neutrophils activation through receptor for advanced glycation end products (RAGE) and Toll-like receptor 4. Arthritis Res. 2015, 17, 64. [Google Scholar] [CrossRef] [Green Version]
- Hao, J.; Lv, T.G.; Wang, C.; Xu, L.P.; Zhao, J.R. Macrophage migration inhibitory factor contributes to anti-neutrophil cytoplasmic antibody-induced neutrophils activation. Hum. Immunol. 2016, 77, 1209–1214. [Google Scholar] [CrossRef]
- Bertram, A.; Lovric, S.; Engel, A.; Beese, M.; Wyss, K.; Hertel, B.; Park, J.K.; Becker, J.U.; Kegel, J.; Haller, H.; et al. Circulating ADAM17 Level Reflects Disease Activity in Proteinase-3 ANCA-Associated Vasculitis. J. Am. Soc. Nephrol. 2015, 26, 2860–2870. [Google Scholar] [CrossRef] [Green Version]
- Morris, H.; Morgan, M.D.; Wood, A.M.; Smith, S.W.; Ekeowa, U.I.; Herrmann, K.; Holle, J.U.; Guillevin, L.; Lomas, D.A.; Perez, J.; et al. ANCA-associated vasculitis is linked to carriage of the Z allele of alpha(1) antitrypsin and its polymers. Ann. Rheum. Dis. 2011, 70, 1851–1856. [Google Scholar] [CrossRef]
- Freeley, S.J.; Coughlan, A.M.; Popat, R.J.; Dunn-Walters, D.K.; Robson, M.G. Granulocyte colony stimulating factor exacerbates antineutrophil cytoplasmic antibody vasculitis. Ann. Rheum. Dis. 2013, 72, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Popa, E.R.; Franssen, C.F.; Limburg, P.C.; Huitema, M.G.; Kallenberg, C.G.; Tervaert, J.W. In vitro cytokine production and proliferation of T cells from patients with anti-proteinase 3- and antimyeloperoxidase-associated vasculitis, in response to proteinase 3 and myeloperoxidase. Arthritis Rheum. 2002, 46, 1894–1904. [Google Scholar] [CrossRef] [PubMed]
- Salama, A.D. Relapse in Anti-Neutrophil Cytoplasm Antibody (ANCA)-Associated Vasculitis. Kidney Int. Rep. 2020, 5, 7–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csernok, E.; Holle, J.U.; Gross, W.L. Proteinase 3, protease-activated receptor-2 and interleukin-32: Linking innate and autoimmunity in Wegener’s granulomatosis. Clin. Exp. Rheumatol. 2008, 26, S112–S117. [Google Scholar]
- Ohlsson, S.; Holm, L.; Hansson, C.; Ohlsson, S.M.; Gunnarsson, L.; Pettersson, A.; Skattum, L. Neutrophils from ANCA-associated vasculitis patients show an increased capacity to activate the complement system via the alternative pathway after ANCA stimulation. PLoS ONE 2019, 14, e0218272. [Google Scholar] [CrossRef]
- Hu, N.; Westra, J.; Huitema, M.G.; Bijl, M.; Brouwer, E.; Stegeman, C.A.; Heeringa, P.; Limburg, P.C.; Kallenberg, C.G. Coexpression of CD177 and membrane proteinase 3 on neutrophils in antineutrophil cytoplasmic autoantibody-associated systemic vasculitis: Anti-proteinase 3-mediated neutrophil activation is independent of the role of CD177-expressing neutrophils. Arthritis Rheum. 2009, 60, 1548–1557. [Google Scholar] [CrossRef]
- Abdgawad, M.; Gunnarsson, L.; Bengtsson, A.A.; Geborek, P.; Nilsson, L.; Segelmark, M.; Hellmark, T. Elevated neutrophil membrane expression of proteinase 3 is dependent upon CD177 expression. Clin. Exp. Immunol. 2010, 161, 89–97. [Google Scholar] [CrossRef]
- Choi, M.; Eulenberg, C.; Rolle, S.; von Kries, J.P.; Luft, F.C.; Kettritz, R. The use of small molecule high-throughput screening to identify inhibitors of the proteinase 3-NB1 interaction. Clin. Exp. Immunol. 2010, 161, 389–396. [Google Scholar] [CrossRef]
- Nishide, M.; Nojima, S.; Ito, D.; Takamatsu, H.; Koyama, S.; Kang, S.; Kimura, T.; Morimoto, K.; Hosokawa, T.; Hayama, Y.; et al. Semaphorin 4D inhibits neutrophil activation and is involved in the pathogenesis of neutrophil-mediated autoimmune vasculitis. Ann. Rheum. Dis. 2017, 76, 1440–1448. [Google Scholar] [CrossRef]
- Nishide, M.; Kumanogoh, A. The role of semaphorins in immune responses and autoimmune rheumatic diseases. Nat. Rev. Rheumatol. 2018, 14, 19–31. [Google Scholar] [CrossRef]
- Ishizaki, J.; Takemori, A.; Suemori, K.; Matsumoto, T.; Akita, Y.; Sada, K.E.; Yuzawa, Y.; Amano, K.; Takasaki, Y.; Harigai, M.; et al. Targeted proteomics reveals promising biomarkers of disease activity and organ involvement in antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Res. 2017, 19, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdgawad, M.; Pettersson, A.; Gunnarsson, L.; Bengtsson, A.A.; Geborek, P.; Nilsson, L.; Segelmark, M.; Hellmark, T. Decreased neutrophil apoptosis in quiescent ANCA-associated systemic vasculitis. PLoS ONE 2012, 7, e32439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, A.; Nagao, T.; Nagi-Miura, N.; Ohno, N.; Yasuhara, M.; Yamamoto, K.; Nakayama, T.; Suzuki, K. MPO-ANCA induces IL-17 production by activated neutrophils in vitro via classical complement pathway-dependent manner. J. Autoimmun. 2008, 31, 79–89. [Google Scholar] [CrossRef]
- Gan, P.Y.; Steinmetz, O.M.; Tan, D.S.; O’Sullivan, K.M.; Ooi, J.D.; Iwakura, Y.; Kitching, A.R.; Holdsworth, S.R. Th17 cells promote autoimmune anti-myeloperoxidase glomerulonephritis. J. Am. Soc. Nephrol. 2010, 21, 925–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casselman, B.L.; Kilgore, K.S.; Miller, B.F.; Warren, J.S. Antibodies to neutrophil cytoplasmic antigens induce monocyte chemoattractant protein-1 secretion from human monocytes. J. Lab. Clin. Med. 1995, 126, 495–502. [Google Scholar]
- Moran, S.M.; Monach, P.A.; Zgaga, L.; Cuthbertson, D.; Carette, S.; Khalidi, N.A.; Koening, C.L.; Langford, C.A.; McAlear, C.A.; Moreland, L.; et al. Urinary soluble CD163 and monocyte chemoattractant protein-1 in the identification of subtle renal flare in anti-neutrophil cytoplasmic antibody-associated vasculitis. Nephrol. Dial. Transplant. 2020, 35, 283–291. [Google Scholar] [CrossRef]
- Hattar, K.; Bickenbach, A.; Csernok, E.; Rosseau, S.; Grandel, U.; Seeger, W.; Grimminger, F.; Sibelius, U. Wegener’s granulomatosis: Antiproteinase 3 antibodies induce monocyte cytokine and prostanoid release-role of autocrine cell activation. J. Leukoc. Biol. 2002, 71, 996–1004. [Google Scholar]
- O’Brien, E.C.; Abdulahad, W.H.; Rutgers, A.; Huitema, M.G.; O’Reilly, V.P.; Coughlan, A.M.; Harrington, M.; Heeringa, P.; Little, M.A.; Hickey, F.B. Intermediate monocytes in ANCA vasculitis: Increased surface expression of ANCA autoantigens and IL-1beta secretion in response to anti-MPO antibodies. Sci. Rep. 2015, 5, 11888. [Google Scholar] [CrossRef] [Green Version]
- Sanada, S.; Akiyama, Y.; Sato, M.; Sato, T.; Taguma, Y. Chemokine Receptor 8 Can Distinguish Antineutrophil Cytoplasmic Antibody-Associated Vasculitis From Infectious Complications. Kidney Int. Rep. 2019, 4, 447–454. [Google Scholar] [CrossRef] [Green Version]
- Popat, R.J.; Hakki, S.; Thakker, A.; Coughlan, A.M.; Watson, J.; Little, M.A.; Spickett, C.M.; Lavender, P.; Afzali, B.; Kemper, C.; et al. Anti-myeloperoxidase antibodies attenuate the monocyte response to LPS and shape macrophage development. JCI Insight 2017, 2, e87379. [Google Scholar] [CrossRef] [Green Version]
- Abdulahad, W.H.; Lepse, N.; Stegeman, C.A.; Huitema, M.G.; Doornbos-van der Meer, B.; Tadema, H.; Rutgers, A.; Limburg, P.C.; Kallenberg, C.G.; Heeringa, P. Increased frequency of circulating IL-21 producing Th-cells in patients with granulomatosis with polyangiitis (GPA). Arthritis Res. 2013, 15, R70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogueira, E.; Hamour, S.; Sawant, D.; Henderson, S.; Mansfield, N.; Chavele, K.M.; Pusey, C.D.; Salama, A.D. Serum IL-17 and IL-23 levels and autoantigen-specific Th17 cells are elevated in patients with ANCA-associated vasculitis. Nephrol. Dial. Transplant. 2010, 25, 2209–2217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzen, J.; Lovric, S.; Kramer, R.; Haller, H.; Haubitz, M. Osteopontin in antineutrophil cytoplasmic autoantibody-associated vasculitis: Relation to disease activity, organ manifestation and immunosuppressive therapy. Ann. Rheum. Dis. 2010, 69, 1169–1171. [Google Scholar] [CrossRef]
- Sanders, J.S.; Huitma, M.G.; Kallenberg, C.G.; Stegeman, C.A. Plasma levels of soluble interleukin 2 receptor, soluble CD30, interleukin 10 and B cell activator of the tumour necrosis factor family during follow-up in vasculitis associated with proteinase 3-antineutrophil cytoplasmic antibodies: Associations with disease activity and relapse. Ann. Rheum. Dis. 2006, 65, 1484–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lúdvíksson, B.R.; Sneller, M.C.; Chua, K.S.; Talar-Williams, C.; Langford, C.A.; Ehrhardt, R.O.; Fauci, A.S.; Strober, W. Active Wegener’s granulomatosis is associated with HLA-DR+ CD4+ T cells exhibiting an unbalanced Th1-type T cell cytokine pattern: Reversal with IL-10. J. Immunol. 1998, 160, 3602–3609. [Google Scholar] [PubMed]
- Csernok, E.; Trabandt, A.; Müller, A.; Wang, G.C.; Moosig, F.; Paulsen, J.; Schnabel, A.; Gross, W.L. Cytokine profiles in Wegener’s granulomatosis: Predominance of type 1 (Th1) in the granulomatous inflammation. Arthritis. Rheum. 1999, 42, 742–750. [Google Scholar] [CrossRef]
- Xin, G.; Chen, M.; Su, Y.; Xu, L.X.; Zhao, M.H.; Li, K.S. Serum B-cell activating factor in myeloperoxiase-antineutrophil cytoplasmic antibodies-associated vasculitis. Am. J. Med. Sci. 2014, 348, 25–29. [Google Scholar] [CrossRef]
- Nagai, M.; Hirayama, K.; Ebihara, I.; Shimohata, H.; Kobayashi, M.; Koyama, A. Serum levels of BAFF and APRIL in myeloperoxidase anti-neutrophil cytoplasmic autoantibody-associated renal vasculitis: Association with disease activity. Nephron. Clin. Pr. 2011, 118, c339–c345. [Google Scholar] [CrossRef]
- Muller Kobold, A.C.; van Wijk, R.T.; Franssen, C.F.; Molema, G.; Kallenberg, C.G.; Tervaert, J.W. In vitro up-regulation of E-selectin and induction of interleukin-6 in endothelial cells by autoantibodies in Wegener’s granulomatosis and microscopic polyangiitis. Clin. Exp. Rheumatol. 1999, 17, 433–440. [Google Scholar]
- Le Roux, S.; Pepper, R.J.; Dufay, A.; Neel, M.; Meffray, E.; Lamande, N.; Rimbert, M.; Josien, R.; Hamidou, M.; Hourmant, M.; et al. Elevated soluble Flt1 inhibits endothelial repair in PR3-ANCA-associated vasculitis. J. Am. Soc. Nephrol. 2012, 23, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Hladinova, Z.; Hruskova, Z.; Svobodova, B.; Malickova, K.; Lanska, V.; Konopasek, P.; Jancova, E.; Rysava, R.; Edelstein, C.L.; Tesar, V. Increased levels of soluble ST2 in patients with active newly diagnosed ANCA-associated vasculitis. Mediat. Inflamm. 2015, 2015, 603750. [Google Scholar] [CrossRef] [PubMed]
- Noronha, I.L.; Kruger, C.; Andrassy, K.; Ritz, E.; Waldherr, R. In situ production of TNF-alpha, IL-1 beta and IL-2R in ANCA-positive glomerulonephritis. Kidney Int. 1993, 43, 682–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunini, F.; Page, T.H.; Gallieni, M.; Pusey, C.D. The role of monocytes in ANCA-associated vasculitides. Autoimmun Rev. 2016, 15, 1046–1053. [Google Scholar] [CrossRef]
- Jayne, D.R.W.; Bruchfeld, A.N.; Harper, L.; Schaier, M.; Venning, M.C.; Hamilton, P.; Burst, V.; Grundmann, F.; Jadoul, M.; Szombati, I.; et al. Randomized Trial of C5a Receptor Inhibitor Avacopan in ANCA-Associated Vasculitis. J. Am. Soc. Nephrol. 2017, 28, 2756–2767. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, V.P.; Wong, L.; Kennedy, C.; Elliot, L.A.; O’Meachair, S.; Coughlan, A.M.; O’Brien, E.C.; Ryan, M.M.; Sandoval, D.; Connolly, E.; et al. Urinary Soluble CD163 in Active Renal Vasculitis. J. Am. Soc. Nephrol. 2016, 27, 2906–2916. [Google Scholar] [CrossRef] [PubMed]
- Seino, K.; Iwabuchi, K.; Kayagaki, N.; Miyata, R.; Nagaoka, I.; Matsuzawa, A.; Fukao, K.; Yagita, H.; Okumura, K. Chemotactic activity of soluble Fas ligand against phagocytes. J. Immunol. 1998, 161, 4484–4488. [Google Scholar]
- Tarzi, R.M.; Liu, J.; Schneiter, S.; Hill, N.R.; Page, T.H.; Cook, H.T.; Pusey, C.D.; Woollard, K.J. CD14 expression is increased on monocytes in patients with anti-neutrophil cytoplasm antibody (ANCA)-associated vasculitis and correlates with the expression of ANCA autoantigens. Clin. Exp. Immunol. 2015, 181, 65–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogt, W. Complement activation by myeloperoxidase products released from stimulated human polymorphonuclear leukocytes. Immunobiology 1996, 195, 334–346. [Google Scholar] [CrossRef]
- Brilland, B.; Garnier, A.S.; Chevailler, A.; Jeannin, P.; Subra, J.F.; Augusto, J.F. Complement alternative pathway in ANCA-associated vasculitis: Two decades from bench to bedside. Autoimmun Rev. 2020, 19, 102424. [Google Scholar] [CrossRef]
- Xiao, H.; Schreiber, A.; Heeringa, P.; Falk, R.J.; Jennette, J.C. Alternative complement pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmic autoantibodies. Am. J. Pathol. 2007, 170, 52–64. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, R.; Maeda, T.; Zhang, H.; Berry, G.J.; Zeisbrich, M.; Brockett, R.; Greenstein, A.E.; Tian, L.; Goronzy, J.J.; Weyand, C.M. MMP (Matrix Metalloprotease)-9-Producing Monocytes Enable T Cells to Invade the Vessel Wall and Cause Vasculitis. Circ. Res. 2018, 123, 700–715. [Google Scholar] [CrossRef] [PubMed]
- Hattar, K.; van Burck, S.; Bickenbach, A.; Grandel, U.; Maus, U.; Lohmeyer, J.; Csernok, E.; Hartung, T.; Seeger, W.; Grimminger, F.; et al. Anti-proteinase 3 antibodies (c-ANCA) prime CD14-dependent leukocyte activation. J. Leukoc. Biol. 2005, 78, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- von Borstel, A.; Sanders, J.S.; Rutgers, A.; Stegeman, C.A.; Heeringa, P.; Abdulahad, W.H. Cellular immune regulation in the pathogenesis of ANCA-associated vasculitides. Autoimmun Rev. 2018, 17, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Dolff, S.; Witzke, O.; Wilde, B. Th17 cells in renal inflammation and autoimmunity. Autoimmun Rev. 2019, 18, 129–136. [Google Scholar] [CrossRef]
- Tesmer, L.A.; Lundy, S.K.; Sarkar, S.; Fox, D.A. Th17 cells in human disease. Immunol. Rev. 2008, 223, 87–113. [Google Scholar] [CrossRef]
- Dinarello, C.A.; Novick, D.; Kim, S.; Kaplanski, G. Interleukin-18 and IL-18 binding protein. Front. Immunol. 2013, 4, 289. [Google Scholar] [CrossRef] [Green Version]
- Lokau, J.; Agthe, M.; Garbers, C. Generation of Soluble Interleukin-11 and Interleukin-6 Receptors: A Crucial Function for Proteases during Inflammation. Mediat. Inflamm. 2016, 2016, 1785021. [Google Scholar] [CrossRef] [Green Version]
- Berti, A.; Warner, R.; Johnson, K.; Cornec, D.; Schroeder, D.R.; Kabat, B.F.; Langford, C.A.; Kallenberg, C.G.M.; Seo, P.; Spiera, R.F.; et al. The association of serum interleukin-6 levels with clinical outcomes in antineutrophil cytoplasmic antibody-associated vasculitis. J. Autoimmun. 2019, 105, 102302. [Google Scholar] [CrossRef]
- Icer, M.A.; Gezmen-Karadag, M. The multiple functions and mechanisms of osteopontin. Clin. Biochem. 2018, 59, 17–24. [Google Scholar] [CrossRef]
- Masutani, K.; Tokumoto, M.; Nakashima, H.; Tsuruya, K.; Kashiwagi, M.; Kudoh, Y.; Fukuda, K.; Kanai, H.; Akahoshi, M.; Otsuka, T.; et al. Strong polarization toward Th1 immune response in ANCA-associated glomerulonephritis. Clin. Nephrol. 2003, 59, 395–405. [Google Scholar] [CrossRef]
- Martinez Valenzuela, L.; Bordignon Draibe, J.; Fulladosa Oliveras, X.; Bestard Matamoros, O.; Cruzado Garrit, J.M.; Torras Ambrós, J. T-lymphocyte in ANCA-associated vasculitis: What do we know? A pathophysiological and therapeutic approach. Clin. Kidney J. 2019, 12, 503–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczeklik, W.; Jakieła, B.; Wawrzycka-Adamczyk, K.; Sanak, M.; Hubalewska-Mazgaj, M.; Padjas, A.; Surmiak, M.; Szczeklik, K.; Sznajd, J.; Musiał, J. Skewing toward Treg and Th2 responses is a characteristic feature of sustained remission in ANCA-positive granulomatosis with polyangiitis. Eur. J. Immunol. 2017, 47, 724–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holden, N.J.; Williams, J.M.; Morgan, M.D.; Challa, A.; Gordon, J.; Pepper, R.J.; Salama, A.D.; Harper, L.; Savage, C.O. ANCA-stimulated neutrophils release BLyS and promote B cell survival: A clinically relevant cellular process. Ann. Rheum. Dis. 2011, 70, 2229–2233. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, P.; Sandell, C.; Backteman, K.; Ernerudh, J. Expansions of CD4+CD28- and CD8+CD28- T cells in granulomatosis with polyangiitis and microscopic polyangiitis are associated with cytomegalovirus infection but not with disease activity. J. Rheumatol. 2012, 39, 1840–1843. [Google Scholar] [CrossRef]
- Chevrier, S.; Genton, C.; Kallies, A.; Karnowski, A.; Otten, L.A.; Malissen, B.; Malissen, M.; Botto, M.; Corcoran, L.M.; Nutt, S.L.; et al. CD93 is required for maintenance of antibody secretion and persistence of plasma cells in the bone marrow niche. Proc. Natl. Acad. Sci. USA 2009, 106, 3895–3900. [Google Scholar] [CrossRef] [Green Version]
- Jenh, C.H.; Cox, M.A.; Hipkin, W.; Lu, T.; Pugliese-Sivo, C.; Gonsiorek, W.; Chou, C.C.; Narula, S.K.; Zavodny, P.J. Human B cell-attracting chemokine 1 (BCA-1; CXCL13) is an agonist for the human CXCR3 receptor. Cytokine 2001, 15, 113–121. [Google Scholar] [CrossRef]
- Wang, J.C. Importance of plasma matrix metalloproteinases (MMP) and tissue inhibitors of metalloproteinase (TIMP) in development of fibrosis in agnogenic myeloid metaplasia. Leuk. Lymphoma 2005, 46, 1261–1268. [Google Scholar] [CrossRef]
- Midwood, K.S.; Chiquet, M.; Tucker, R.P.; Orend, G. Tenascin-C at a glance. J. Cell Sci. 2016, 129, 4321–4327. [Google Scholar] [CrossRef] [Green Version]
- Alexander-Kaufman, K.; Harper, C. Transketolase: Observations in alcohol-related brain damage research. Int. J. Biochem. Cell Biol. 2009, 41, 717–720. [Google Scholar] [CrossRef]
- van Roeyen, C.R.; Ostendorf, T.; Floege, J. The platelet-derived growth factor system in renal disease: An emerging role of endogenous inhibitors. Eur. J. Cell Biol. 2012, 91, 542–551. [Google Scholar] [CrossRef]
- Leung, K. Microbubbles coated with antibody to intracellular adhesion molecule-1. In Molecular Imaging and Contrast Agent Database (MICAD); National Center for Biotechnology Information: Bethesda, MD, USA, 2004. [Google Scholar]
- Moschen, A.R.; Adolph, T.E.; Gerner, R.R.; Wieser, V.; Tilg, H. Lipocalin-2: A Master Mediator of Intestinal and Metabolic Inflammation. Trends Endocrinol. Metab. 2017, 28, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Koller, L.; Richter, B.; Winter, M.P.; Sulzgruber, P.; Potolidis, C.; Liebhart, F.; Mortl, D.; Berger, R.; Goliasch, G.; Lang, I.; et al. Clusterin/apolipoprotein J is independently associated with survival in patients with chronic heart failure. J. Clin. Lipidol. 2017, 11, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Abraham, S.; McKenzie, J.A.G.; Jeffs, N.; Swire, M.; Tripathi, V.B.; Luhmann, U.F.O.; Lange, C.A.K.; Zhai, Z.; Arthur, H.M.; et al. LRG1 promotes angiogenesis by modulating endothelial TGF-beta signalling. Nature 2013, 499, 306–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pepper, R.J.; Draibe, J.B.; Caplin, B.; Fervenza, F.C.; Hoffman, G.S.; Kallenberg, C.G.; Langford, C.A.; Monach, P.A.; Seo, P.; Spiera, R.; et al. Association of Serum Calprotectin (S100A8/A9) Level With Disease Relapse in Proteinase 3-Antineutrophil Cytoplasmic Antibody-Associated Vasculitis. Arthritis Rheumatol. 2017, 69, 185–193. [Google Scholar] [CrossRef]
- Kronbichler, A.; Leierer, J.; Gauckler, P.; Shin, J.I. Comorbidities in ANCA-associated vasculitis. Rheumatology 2020, 59. [Google Scholar] [CrossRef]
- Aimo, A.; Migliorini, P.; Vergaro, G.; Franzini, M.; Passino, C.; Maisel, A.; Emdin, M. The IL-33/ST2 pathway, inflammation and atherosclerosis: Trigger and target? Int J. Cardiol. 2018, 267, 188–192. [Google Scholar] [CrossRef]
- Martin, K.R.; Witko-Sarsat, V. Proteinase 3: The odd one out that became an autoantigen. J. Leukoc. Biol. 2017, 102, 689–698. [Google Scholar] [CrossRef]
- Jerke, U.; Marino, S.F.; Daumke, O.; Kettritz, R. Characterization of the CD177 interaction with the ANCA antigen proteinase 3. Sci. Rep. 2017, 7, 43328. [Google Scholar] [CrossRef] [Green Version]
- Kantari, C.; Pederzoli-Ribeil, M.; Amir-Moazami, O.; Gausson-Dorey, V.; Moura, I.C.; Lecomte, M.C.; Benhamou, M.; Witko-Sarsat, V. Proteinase 3, the Wegener autoantigen, is externalized during neutrophil apoptosis: Evidence for a functional association with phospholipid scramblase 1 and interference with macrophage phagocytosis. Blood 2007, 110, 4086–4095. [Google Scholar] [CrossRef] [Green Version]
- Gabillet, J.; Millet, A.; Pederzoli-Ribeil, M.; Tacnet-Delorme, P.; Guillevin, L.; Mouthon, L.; Frachet, P.; Witko-Sarsat, V. Proteinase 3, the autoantigen in granulomatosis with polyangiitis, associates with calreticulin on apoptotic neutrophils, impairs macrophage phagocytosis, and promotes inflammation. J. Immunol. 2012, 189, 2574–2583. [Google Scholar] [CrossRef] [Green Version]
- Millet, A.; Martin, K.R.; Bonnefoy, F.; Saas, P.; Mocek, J.; Alkan, M.; Terrier, B.; Kerstein, A.; Tamassia, N.; Satyanarayanan, S.K.; et al. Proteinase 3 on apoptotic cells disrupts immune silencing in autoimmune vasculitis. J. Clin. Investig. 2015, 125, 4107–4121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everts-Graber, J.; Martin, K.R.; Thieblemont, N.; Mocek, J.; Roccabianca, A.; Chafey, P.; Le Gall, M.; Tacnet-Delorme, P.; Reutelingsperger, C.P.; Naccache, J.M.; et al. Proteomic analysis of neutrophils in ANCA-associated vasculitis reveals a dysregulation in proteinase 3-associated proteins such as annexin-A1 involved in apoptotic cell clearance. Kidney Int. 2019, 96, 397–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Dam, L.S.; Dirikgil, E.; Bredewold, E.W.; Ray, A.; Bakker, J.A.; van Kooten, C.; Rabelink, T.J.; Teng, Y.K.O. Proteinase-3-anti-neutrophil cytoplasmic antibodies (PR3-ANCAs) predict relapses in ANCA-associated vasculitis patients after rituximab. Nephrol. Dial. Transplant. 2020. [Google Scholar] [CrossRef]
- Lee, A.; Nissen, M.J.; Beroukas, D.; Ahern, M.J.; Barbara, J.A. Detectable anti-proteinase-3 antibodies precede clinical manifestations in a case of anti-neutrophil cytoplasmic antibody-associated vasculitis. Scand. J. Rheumatol. 2020, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Booth, A.; Harper, L.; Hammad, T.; Bacon, P.; Griffith, M.; Levy, J.; Savage, C.; Pusey, C.; Jayne, D. Prospective study of TNFalpha blockade with infliximab in anti-neutrophil cytoplasmic antibody-associated systemic vasculitis. J. Am. Soc. Nephrol. 2004, 15, 717–721. [Google Scholar] [CrossRef]
- Kronbichler, A.; Kerschbaum, J.; Gopaluni, S.; Tieu, J.; Alberici, F.; Jones, R.B.; Smith, R.M.; Jayne, D.R.W. Trimethoprim-sulfamethoxazole prophylaxis prevents severe/life-threatening infections following rituximab in antineutrophil cytoplasm antibody-associated vasculitis. Ann. Rheum. Dis. 2018, 77, 1440–1447. [Google Scholar] [CrossRef] [Green Version]
- Laurino, S.; Chaudhry, A.; Booth, A.; Conte, G.; Jayne, D. Prospective study of TNFalpha blockade with adalimumab in ANCA-associated systemic vasculitis with renal involvement. Nephrol. Dial. Transplant. 2010, 25, 3307–3314. [Google Scholar] [CrossRef]
- The Wegener’s Granulomatosis Etanercept Trial (WGET) Research Group. Etanercept plus standard therapy for Wegener’s granulomatosis. N. Engl. J. Med. 2005, 352, 351–361. [Google Scholar] [CrossRef]
- Sakai, R.; Kondo, T.; Kurasawa, T.; Nishi, E.; Okuyama, A.; Chino, K.; Shibata, A.; Okada, Y.; Takei, H.; Nagasawa, H.; et al. Current clinical evidence of tocilizumab for the treatment of ANCA-associated vasculitis: A prospective case series for microscopic polyangiitis in a combination with corticosteroids and literature review. Clin. Rheumatol. 2017, 36, 2383–2392. [Google Scholar] [CrossRef]
- Kronbichler, A.; Gauckler, P.; Windpessl, M.; Il Shin, J.; Jha, V.; Rovin, B.H.; Oberbauer, R. COVID-19: Implications for immunosuppression in kidney disease and transplantation. Nat. Rev. Nephrol. 2020. [Google Scholar] [CrossRef]
- Jayne, D.; Blockmans, D.; Luqmani, R.; Moiseev, S.; Ji, B.; Green, Y.; Hall, L.; Roth, D.; Henderson, R.B.; Merkel, P.A.; et al. Efficacy and Safety of Belimumab and Azathioprine for Maintenance of Remission in Antineutrophil Cytoplasmic Antibody-Associated Vasculitis: A Randomized Controlled Study. Arthritis Rheumatol. 2019, 71, 952–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kronbichler, A.; Brezina, B.; Gauckler, P.; Quintana, L.F.; Jayne, D.R.W. Refractory lupus nephritis: When, why and how to treat. Autoimmun Rev. 2019, 18, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Atisha-Fregoso, Y.; Malkiel, S.; Harris, K.M.; Byron, M.; Ding, L.; Kanaparthi, S.; Barry, W.T.; Gao, W.; Ryker, K.; Tosta, P.; et al. CALIBRATE: A Phase 2 Randomized Trial of Rituximab Plus Cyclophosphamide Followed by Belimumab for the Treatment of Lupus Nephritis. Arthritis Rheumatol. 2020. [Google Scholar] [CrossRef]
- Quintana, L.F.; Kronbichler, A.; Blasco, M.; Zhao, M.H.; Jayne, D. ANCA associated vasculitis: The journey to complement-targeted therapies. Mol. Immunol. 2019, 112, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Dairaghi, D.J.; Powers, J.P.; Ertl, L.S.; Baumgart, T.; Wang, Y.; Seitz, L.C.; Penfold, M.E.; Gan, L.; Hu, P.; et al. C5a receptor (CD88) blockade protects against MPO-ANCA GN. J. Am. Soc. Nephrol. 2014, 25, 225–231. [Google Scholar] [CrossRef] [Green Version]
- Merkel, P.A.; Niles, J.; Jimenez, R.; Spiera, R.F.; Rovin, B.H.; Bomback, A.; Pagnoux, C.; Potarca, A.; Schall, T.J.; Bekker, P. A Randomized Clinical Trial of CCX168, an Orally Administered C5aR Inhibitor for Treatment of Patients with ANCA-Associated Vasculitis; Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Jayne, D.; Merkel, P.; Yue, H.; Schall, T.J.; Kelleher, C.; Bekker, P. A Randomized, Double-Blind, Active Controlled Study Of Avacopan In Anti-Neutrophil Cytoplasmic Antibody-Associated Vasculitis. In Nephrology Dialysis Transplantation; Oxford University Press: Oxford, UK, 2020. [Google Scholar]
- Wallace, Z.S.; Miloslavsky, E.M. Management of ANCA associated vasculitis. BMJ 2020, 368, m421. [Google Scholar] [CrossRef] [Green Version]
- van Daalen, E.E.; Rizzo, R.; Kronbichler, A.; Wolterbeek, R.; Bruijn, J.A.; Jayne, D.R.; Bajema, I.M.; Rahmattulla, C. Effect of rituximab on malignancy risk in patients with ANCA-associated vasculitis. Ann. Rheum. Dis. 2017, 76, 1064–1069. [Google Scholar] [CrossRef]
- Smith, R.M.; Jones, R.B.; Specks, U.; Bond, S.; Nodale, M.; Aljayyousi, R.; Andrews, J.; Bruchfeld, A.; Camilleri, B.; Carette, S.; et al. Rituximab as therapy to induce remission after relapse in ANCA-associated vasculitis. Ann. Rheum. Dis. 2020. [Google Scholar] [CrossRef]
- Guillevin, L.; Pagnoux, C.; Karras, A.; Khouatra, C.; Aumaître, O.; Cohen, P.; Maurier, F.; Decaux, O.; Ninet, J.; Gobert, P.; et al. Rituximab versus azathioprine for maintenance in ANCA-associated vasculitis. N. Engl. J. Med. 2014, 371, 1771–1780. [Google Scholar] [CrossRef] [Green Version]
- Charles, P.; Perrodeau, É.; Samson, M.; Bonnotte, B.; Néel, A.; Agard, C.; Huart, A.; Karras, A.; Lifermann, F.; Godmer, P.; et al. Long-Term Rituximab Use to Maintain Remission of Antineutrophil Cytoplasmic Antibody-Associated Vasculitis: A Randomized Trial. Ann. Intern. Med. 2020, 173, 179–187. [Google Scholar] [CrossRef]
- Kronbichler, A.; Windpessl, M.; Pieringer, H.; Jayne, D.R.W. Rituximab for immunologic renal disease: What the nephrologist needs to know. Autoimmun. Rev. 2017, 16, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Antonelou, M.; Michaëlsson, E.; Evans, R.D.R.; Wang, C.J.; Henderson, S.R.; Walker, L.S.K.; Unwin, R.J.; Salama, A.D.; Investigators, R.-I. Therapeutic Myeloperoxidase Inhibition Attenuates Neutrophil Activation, ANCA-Mediated Endothelial Damage, and Crescentic GN. J. Am. Soc. Nephrol. 2020, 31, 350–364. [Google Scholar] [CrossRef] [PubMed]
- Uozumi, R.; Iguchi, R.; Masuda, S.; Nishibata, Y.; Nakazawa, D.; Tomaru, U.; Ishizu, A. Pharmaceutical immunoglobulins reduce neutrophil extracellular trap formation and ameliorate the development of MPO-ANCA-associated vasculitis. Mod. Rheumatol. 2020, 30, 544–550. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| PR3-AAV (vs. Healthy Control) | MPO-AAV (vs. Healthy Control) | ||||
|---|---|---|---|---|---|
| ↑ | ↓ | ↑ | ↓ | ||
| Priming | Neutrophil | TNFα [66,67,68,69,70] IL-6 [71] IL-18 [72,73] IL-2Rα (CD25) [74] C5a [32,75] G-CSF [73] GM-CSF [73,76] HMGB1 [77] MIF [78] ADAM17 [79] α1AT polymers [80] | IL-2Rβ (CD122) [74] | TNFα [66,67,68] IL-6 [71] IL-18 [72,73] IL-2Rα (CD25) [74] C5a [32,75] G-CSF [73,81] GM-CSF [73] HMGB1 [77] MIF [78] | IL-2Rβ (CD122) [74] |
| Monocyte | TNFα [66,67,68,69,70] ADAM17 [79] | ||||
| Neutrophil/ Monocyte activation | Neutrophil | C3a [75] IL-10 [82] IL-17A, IL23 [82,83] IL-32 [84] C3bBbP [85] CD177 [86,87,88] CD14 [70] Semaphorin 4D [89,90] MIF [78] MMP9 [91] sFAS [92] | C3a [75] IL-17A, IL-23 [83,93,94] Semaphorin 4D [89,90] MIF [78] MMP9 [91] | ||
| Monocyte | MCP-1 [95] Urinary sCD163 [96] TNF-a, IL-1β, IL-6, IL-8, TXA2 [97] CD14 [70] TARC [73] | MCP-1 [95] Urinary sCD163 IL-1β, IL-6, IL-8 [98] TARC [73] CCL18, CCR8 [99] | IL-10 [100] | ||
| T cell activation | IL-21 [101] IL-17A, IL-23 [93,102] IL-18BP [73,75] IL-18, sIL-6R, TARC [73] Osteopontin [73,103] sIL2R, sCD30 [104] IFN-γ [105,106] | IL-17A [93,94,102] IL-23 [93,102] IL-18BP [73,75] IL-18, sIL-6R, TARC [73] Osteopontin [73,103] | |||
| B cell activation | BAFF [104] TARC [73] CD93 [91] BCA-1 [41] | BAFF [107,108] TARC [73] CD93 [91] BCA-1 [41] | |||
| Tissue damage and repair | NGFβ [17,73], KIM-1, NGAL, MMP-3, MMP-9, TIMP-1 [73] TNC, TKT [91] | PDGF-AB [73] | NGFβ [17,73], KIM-1, NGAL, MMP-3, MMP-9, TIMP-1 [73] TNC, TKT [91] | PDGF-AB [73] | |
| Endothelial injury and repair | E-selectin, IL-6 [109] NGAL, ICAM-1 [17,73] Clusterin [73], sFlt1 [110] LRG1, S100A8/A9 [91] sST2, IL-33 [111] | PAI-1 [73] | E-selectin, IL-6 [109] NGAL, ICAM-1 [17,73] Clusterin [73], sFlt1 [110] LRG1, S100A8/A9 [91] sST2, IL-33 [111] | PAI-1 [73] | |
| Biomarker in AAV Pathogenesis | Pathogenesis of AAV (vs. Healthy Controls) | |
|---|---|---|
| ↑ | ↓ | |
| Priming of neutrophils (results in ANCA antigen expression on neutrophils’ cell membranes) | TNF-α [66,67,68] IL-6 [71,73] IL-18 [72,73] IL-2Rα (CD25) [74] C5a [32,75] G-CSF, GM-CSF [73] HMGB1 [77] | IL-2Rβ (CD122) [74] |
| Activation of neutrophils | IL-1β [97,98] C3a [75] Semaphrorin 4D [89,90] MIF [78] | |
| Endothelial injury | E-selectin, IL-6 [109] NGFβ, NGAL, ICAM-1 [17,73] Clusterin [73] sFlt1 [110] sST2, IL-33 [111] | PAI-1 [73] |
| Others | IL-8 [97,98] IL-17, IL-23 [93,94,102] MCP-1 [95] BAFF [101,107] C/EBP-α, C/EBP-β, sFAS [92] | |
| Biomarker physiological function | ||
| Cytokine | G-CSF, GM-CSF, IL-6, IL-15, IL-18 [73] Osteopontin [73,103] | |
| Chemokine | BCA-1, IL-8, IP-10, TARC [73] | |
| Soluble receptor | IL-18BP [73,75] sIL-6R, sTNF- RII [73] | |
| Tissue damage and repair | KIM-1, MMP-3, NGFβ, TIMP-1 [73] TNC, CD93, TKT [91] Urinary MCP-1 [75] | PDGF-AB [73] |
| Inflammation and vascular injury | Clusterin, CRP, ESR, ICAM-1, NGAL [73] LRG1, MMP9, S100A8/A9 [91] | PAI-1 [73] |
| Others | Semaphrorin 4D [89,90] | |
| PR3-ANCA (vs. Healthy Control) | MPO-ANCA (vs. Healthy Control) | |||
|---|---|---|---|---|
| ↑ | ↓ | ↑ | ↓ | |
| Cytokine/cytokine receptors | IL-10 [82] IL-21 [101] IL-32 [84] sIL-2R, sCD30 [17] | IL-10 [100] | ||
| Chemokine/chemokine receptors | CD177 [86,87,88] CD14 [70] | CCR8 [99] | ||
| Complement system | C3bBbP [85] | |||
| Others | ADAM17 [79] TXA2 [97] α1AT polymers [80] | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kronbichler, A.; Lee, K.H.; Denicolo, S.; Choi, D.; Lee, H.; Ahn, D.; Kim, K.H.; Lee, J.H.; Kim, H.; Hwang, M.; et al. Immunopathogenesis of ANCA-Associated Vasculitis. Int. J. Mol. Sci. 2020, 21, 7319. https://doi.org/10.3390/ijms21197319
Kronbichler A, Lee KH, Denicolo S, Choi D, Lee H, Ahn D, Kim KH, Lee JH, Kim H, Hwang M, et al. Immunopathogenesis of ANCA-Associated Vasculitis. International Journal of Molecular Sciences. 2020; 21(19):7319. https://doi.org/10.3390/ijms21197319
Chicago/Turabian StyleKronbichler, Andreas, Keum Hwa Lee, Sara Denicolo, Daeun Choi, Hyojeong Lee, Donghyun Ahn, Kang Hyun Kim, Ji Han Lee, HyungTae Kim, Minha Hwang, and et al. 2020. "Immunopathogenesis of ANCA-Associated Vasculitis" International Journal of Molecular Sciences 21, no. 19: 7319. https://doi.org/10.3390/ijms21197319
APA StyleKronbichler, A., Lee, K. H., Denicolo, S., Choi, D., Lee, H., Ahn, D., Kim, K. H., Lee, J. H., Kim, H., Hwang, M., Jung, S. W., Lee, C., Lee, H., Sung, H., Lee, D., Hwang, J., Kim, S., Hwang, I., Kim, D. Y., ... Shin, J. I. (2020). Immunopathogenesis of ANCA-Associated Vasculitis. International Journal of Molecular Sciences, 21(19), 7319. https://doi.org/10.3390/ijms21197319