Misfolding of Lysosomal α-Galactosidase a in a Fly Model and Its Alleviation by the Pharmacological Chaperone Migalastat

Abstract
:1. Introduction
2. Results
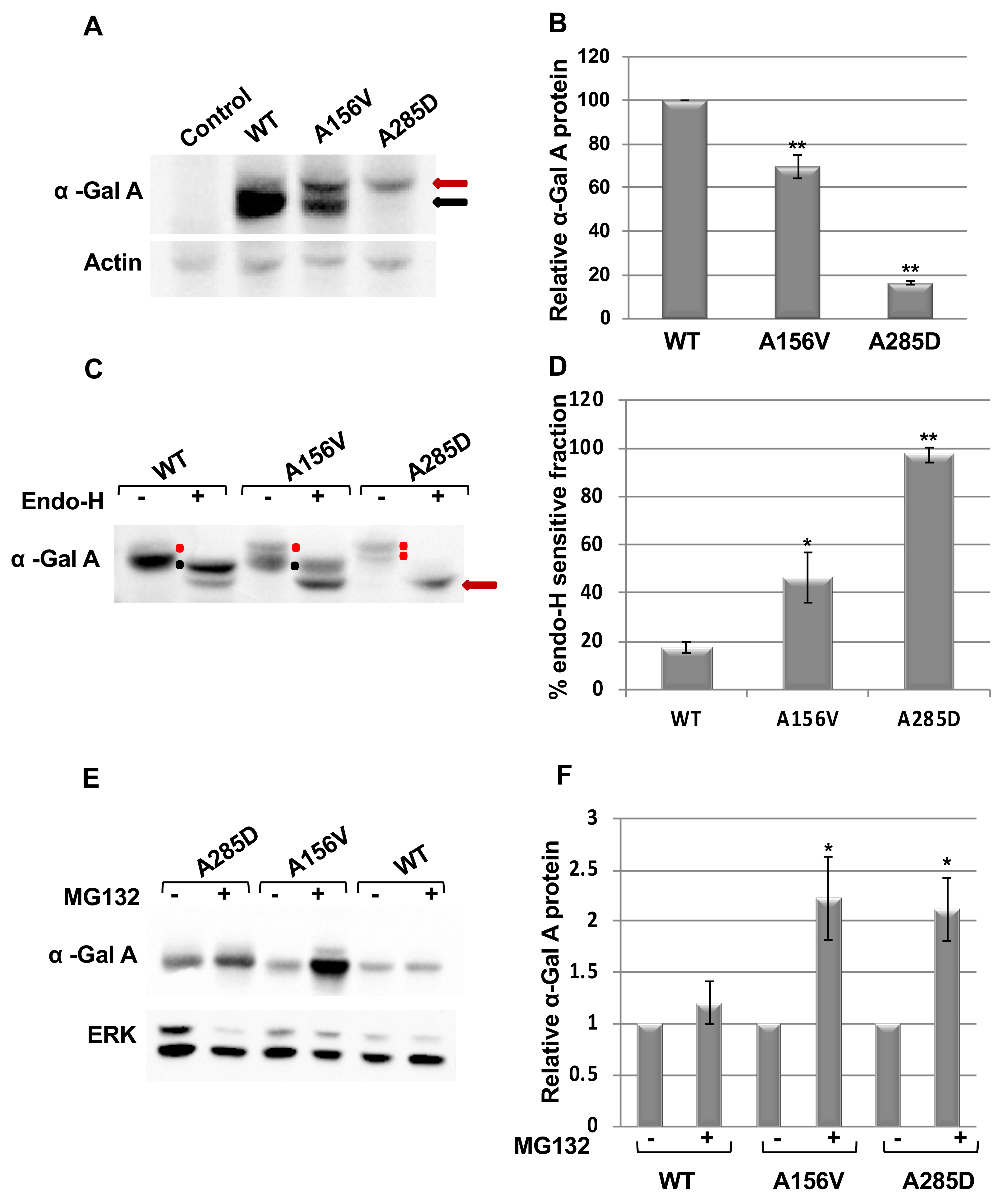
2.1. Alpha-Gal a Is ER Retained and Undergoes ERAD
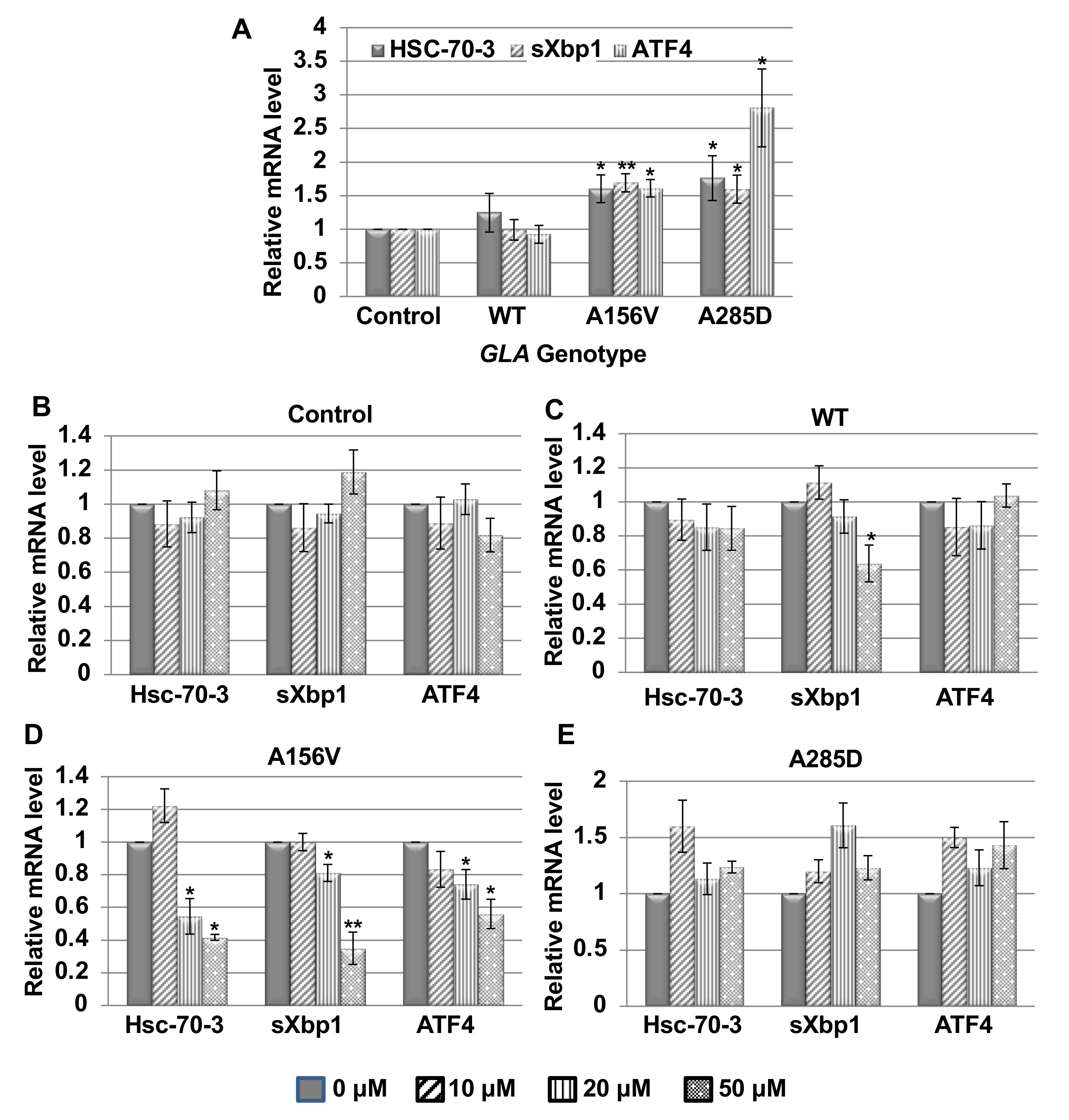
2.2. UPR Activation in Transgenic α-Gal a Expressing Flies
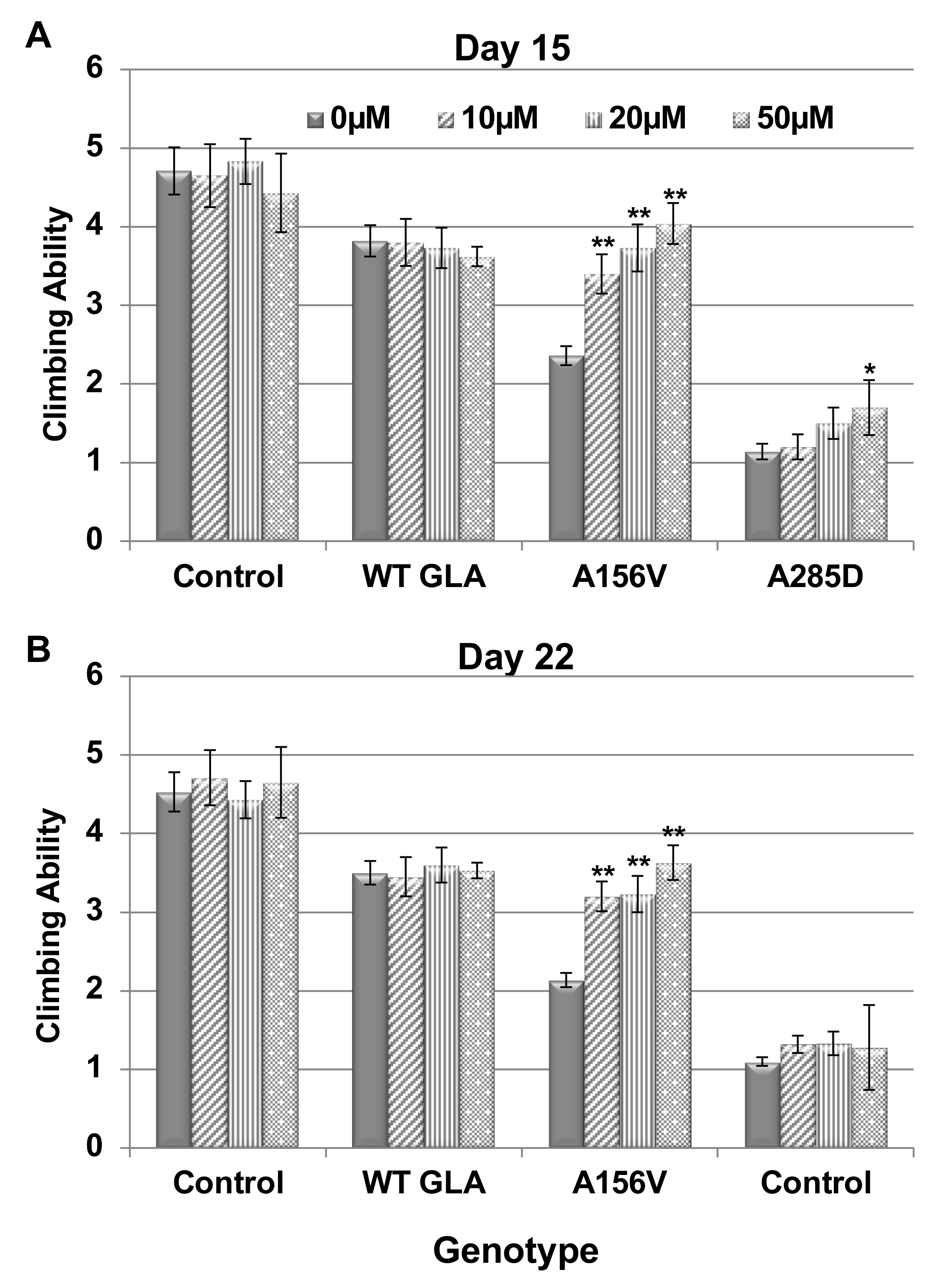
2.3. Climbing Ability of Transgenic α-Gal a Expressing Flies
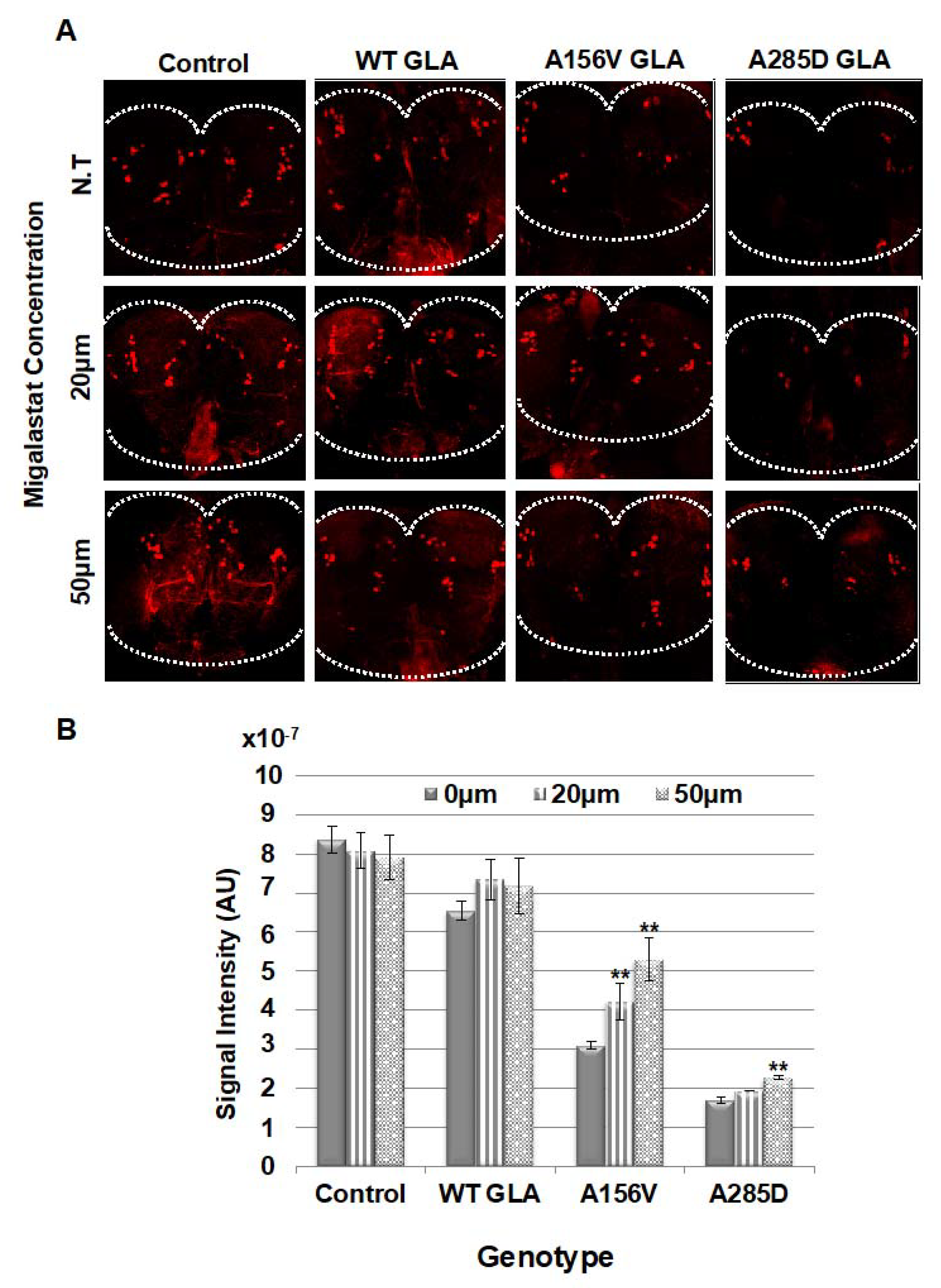
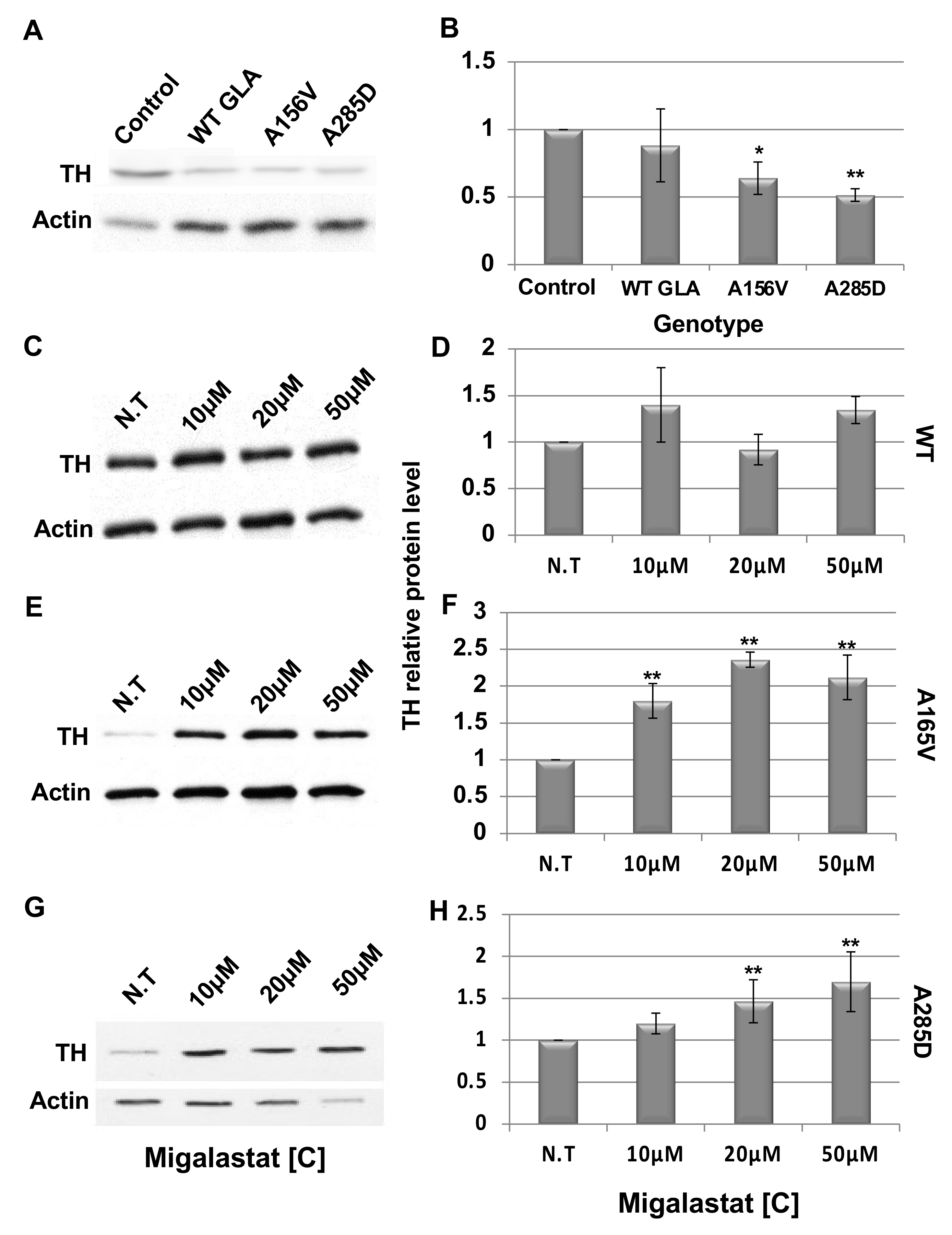
2.4. Death of Dopaminergic Cells in Brains of GLA Flies
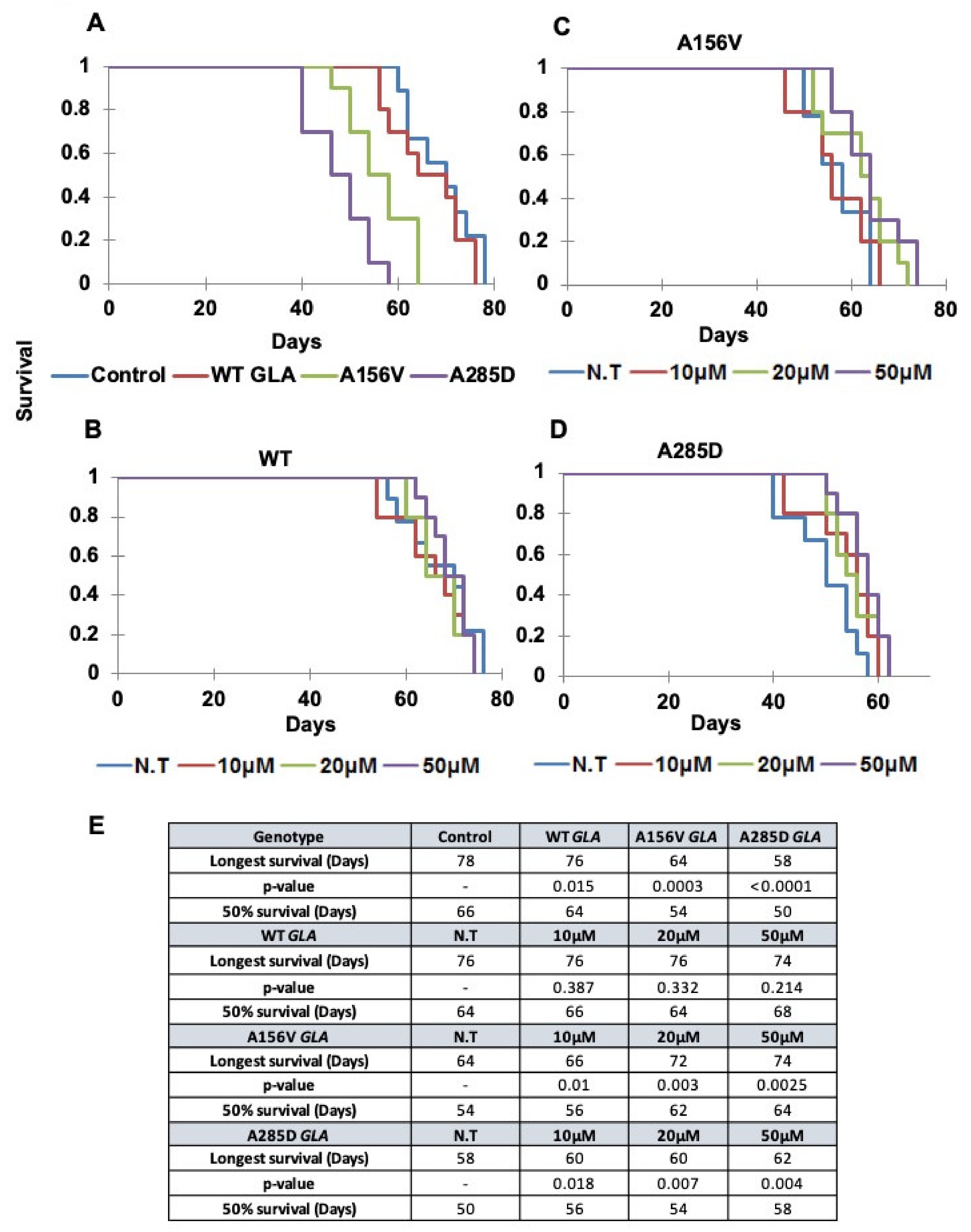
2.5. Survival of Transgenic α-Gal a Expressing Flies
3. Discussion
4. Materials and Methods
4.1. Antibodies
4.2. Fly Strains
4.3. Cell Lines and Transfections
4.4. MG132 (Carbobenzoxy-l-leucyl-l-leucyl-l-leucinal) Treatment
4.5. Plasmid Preparation
4.6. RNA Preparation
4.7. cDNA Preparation and qRT-PCR
4.8. Endonuclease-H Sensitivity
4.9. SDS–PAGE and Western Blotting
4.10. Immunofluorescence and Confocal Microscopy
4.11. Chaperone Treatment
4.12. Climbing Assay
4.13. Survival Assay
4.14. Statistical analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DW | Double distilled water |
| ER | Endoplasmic reticulum |
| ERAD | Endoplasmic reticulum associated degradation |
| M-MLV | Murine Molony leukemia virus |
| PCR | Polymerase chain reaction |
| SDS-PAGE | Sodium dodecyl sulphate polyacrylamise gel electrophoresis |
| SEM | Standard error of mean |
| TH | Tyrosine hydroxylase |
| qRT-PCR | Quantitative real time PCR |
| UPR | Unfolded protein response |
| WT | Wild type |
References
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Kaufman, R.J. The impact of the unfolded protein response on human disease. J. Cell Biol. 2012, 197, 857–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendikov-Bar, I.; Maor, G.; Filocamo, M.; Horowitz, M. Ambroxol as a pharmacological chaperone for mutant glucocerebrosidase. Blood Cells Mol. Dis. 2013, 50, 141–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendikov-Bar, I.; Ron, I.; Filocamo, M.; Horowitz, M. Characterization of the ERAD process of the L444P mutant glucocerebrosidase variant. Blood Cells Mol. Dis. 2011, 46, 4–10. [Google Scholar] [CrossRef]
- Braunstein, H.; Maor, G.; Chicco, G.; Filocamo, M.; Zimran, A.; Horowitz, M. UPR activation and CHOP mediated induction of GBA1 transcription in Gaucher disease. Blood Cells Mol. Dis. 2018, 68, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Maor, G.; Rencus-Lazar, S.; Filocamo, M.; Steller, H.; Segal, D.; Horowitz, M. Unfolded protein response in Gaucher disease: From human to Drosophila. Orphanet J. Rare Dis. 2013, 8, 140. [Google Scholar] [CrossRef] [Green Version]
- Ron, I.; Horowitz, M. ER retention and degradation as the molecular basis underlying Gaucher disease heterogeneity. Hum. Mol. Genet. 2005, 14, 2387–2398. [Google Scholar] [CrossRef] [Green Version]
- Maor, G.; Cabasso, O.; Krivoruk, O.; Rodriguez, J.; Steller, H.; Segal, D.; Horowitz, M. The contribution of mutant GBA to the development of Parkinson disease in Drosophila. Hum. Mol. Genet. 2016, 25, 2712–2727. [Google Scholar] [CrossRef] [Green Version]
- Aerts, J.M.F.G.; Groener, J.E.; Kuiper, S.; Donker-Koopman, W.E.; Strijland, A.; Ottenhoff, R.; Van Roomen, C.; Mirzaian, M.; Wijburg, F.A.; Linthorst, G.E.; et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc. Natl. Acad. Sci. USA 2008, 105, 2812–2817. [Google Scholar] [CrossRef] [Green Version]
- Brady, R.O.; Gal, A.E.; Bradley, R.M.; Martensson, E.; Warshaw, A.L.; Laster, L. Enzymatic defect in Fabry’s disease. N. Engl. J. Med. 1967, 276, 1163–1167. [Google Scholar] [CrossRef]
- Desnick, R.J.; Wasserstein, M.P. Fabry disease: Clinical features and recent advances in enzyme replacement therapy. Adv. Nephrol. Necker Hosp. 2001, 31, 317–339. [Google Scholar] [PubMed]
- Kint, J.A. The enzyme defect in Fabry’s disease. Nat. Cell Biol. 1970, 227, 1173. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; O’Brien, J.S.; Chase, L.R.; Aurbach, G.D. Generalized gangliosidosis: Beta-galactosidase deficiency. Science 1968, 160, 1002–1004. [Google Scholar] [CrossRef] [PubMed]
- Brady, R.O. Enzymatic abnormalities in diseases of sphingolipid metabolism. Clin. Chem. 1967, 13, 565–577. [Google Scholar] [CrossRef]
- Saifudeen, Z.; Desnick, R.J.; Ehrlich, M. A mutation in the 5′ untranslated region of the human α-galactosidase A gene in high-activity variants inhibits specific protein binding. FEBS Lett. 1995, 371, 181–184. [Google Scholar] [CrossRef] [Green Version]
- Germain, D.P. Fabry disease. Orphanet J. Rare Dis. 2010, 5, 30. [Google Scholar] [CrossRef] [Green Version]
- McCafferty, E.H.; Scott, L.J. Migalastat: A review in Fabry disease. Drugs 2019, 79, 543–554. [Google Scholar] [CrossRef] [Green Version]
- Doheny, D.; Srinivasan, R.; Pagant, S.; Chen, B.; Yasuda, M.; Desnick, R.J. Fabry disease: Prevalence of affected males and heterozygotes with pathogenic GLA mutations identified by screening renal, cardiac and stroke clinics, 1995–2017. J. Med Genet. 2018, 55, 261–268. [Google Scholar] [CrossRef]
- Whybra, C.; Wendrich, K.; Ries, M.; Gal, A.; Beck, M. Clinical manifestation in female Fabry disease patients. Contrib. Nephrol. 2001, 245–250. [Google Scholar] [CrossRef]
- Wilcox, W.R.; Oliveira, J.P.; Hopkin, R.J.; Ortiz, A.; Banikazemi, M.; Feldt-Rasmussen, U.; Sims, K.; Waldek, S.; Pastores, G.M.; Lee, P.; et al. Females with Fabry disease frequently have major organ involvement: Lessons from the Fabry registry. Mol. Genet. Metab. 2008, 93, 112–128. [Google Scholar] [CrossRef]
- Lukas, J.; Giese, A.-K.; Markoff, A.; Grittner, U.; Kolodny, E.; Mascher, H.; Lackner, K.J.; Meyer, W.; Wree, P.; Saviouk, V.; et al. Functional characterisation of alpha-galactosidase a mutations as a basis for a new classification system in Fabry disease. PLoS Genet. 2013, 9, e1003632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamin, E.R.; Della Valle, M.C.; Wu, X.; Katz, E.; Pruthi, F.; Bond, S.; Bronfin, B.; Williams, H.; Yu, J.; Bichet, D.-G.; et al. The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat. Genet. Med. 2016, 19, 430–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brady, R.O. Enzyme replacement for lysosomal diseases. Annu. Rev. Med. 2006, 57, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Lidove, O.; West, M.L.; Pintos-Morell, G.; Reisin, R.; Nicholls, K.; Figuera, L.E.; Parini, R.; Carvalho, L.R.; Kampmann, C.; Pastores, G.M.; et al. Effects of enzyme replacement therapy in Fabry disease-A comprehensive review of the medical literature. Genet. Med. 2010, 12, 668–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiffmann, R.; Brady, R.O. Development of enzyme replacement therapy for Fabry disease. In Fabry Disease: Perspectives from 5 Years of FOS; Oxford PharmaGenesis: Oxford, UK, 2006. [Google Scholar]
- Barbey, F.; Lidove, O.; Schwarting, A. Fabry nephropathy: 5 years of enzyme replacement therapy-A short review. Clin. Kidney J. 2007, 1, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Schiffmann, R.; Hughes, D.; Linthorst, G.E.; Ortiz, A.; Svarstad, E.; Warnock, D.G.; West, M.L.; Wanner, C.; Bichet, D.-G.; Christensen, E.I.; et al. Screening, diagnosis, and management of patients with Fabry disease: Conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int. 2017, 91, 284–293. [Google Scholar] [CrossRef]
- Araki, K.; Nagata, K. Protein folding and quality control in the ER. Cold Spring Harb. Perspect. Biol. 2011, 3, a007526. [Google Scholar] [CrossRef] [Green Version]
- Cortez, L.; Sim, V. The therapeutic potential of chemical chaperones in protein folding diseases. Prion 2014, 8, 197–202. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.-Q. A contradictory treatment for lysosomal storage disorders: Inhibitors enhance mutant enzyme activity. Trends Pharmacol. Sci. 2003, 24, 355–360. [Google Scholar] [CrossRef]
- Fan, J.-Q.; Ishii, S.; Asano, N.; Suzuki, Y. Accelerated transport and maturation of lysosomal α-galactosidase A in Fabry lymphoblasts by an enzyme inhibitor. Nat. Med. 1999, 5, 112–115. [Google Scholar] [CrossRef]
- Asano, N.; Ishii, S.; Ikeda, K.; Kato, A.; Fan, J.-Q.; Kizu, H.; Yasuda, K.; Martin, O.R. In vitro inhibition and intracellular enhancement of lysosomal α-galactosidase A activity in Fabry lymphoblasts by 1-deoxygalactonojirimycin and its derivatives. JBIC J. Biol. Inorg. Chem. 2000, 267, 4179–4186. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.R.; Flanagan, J.J.; Schilling, A.; Chang, H.H.; Agarwal, L.; Katz, E.; Wu, X.; Pine, C.; Wustman, B.; Desnick, R.J.; et al. The pharmacological chaperone 1-deoxygalactonojirimycin increases α-galactosidase A levels in Fabry patient cell lines. J. Inherit. Metab. Dis. 2009, 32, 424–440. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P.; Fan, J.-Q. Pharmacological chaperone therapy by active-site-specific chaperones in Fabry disease: In vitro and preclinical studies. Int. J. Clin. Pharmacol. Ther. 2009, 47, S111–S117. [Google Scholar] [PubMed]
- Hamanaka, R.; Shinohara, T.; Yano, S.; Nakamura, M.; Yasuda, A.; Yokoyama, S.; Fan, J.-Q.; Kawasaki, K.; Watanabe, M.; Ishii, S. Rescue of mutant α-galactosidase A in the endoplasmic reticulum by 1-deoxygalactonojirimycin leads to trafficking to lysosomes. Biochim. et Biophys. Acta (BBA)-Mol. Basis Dis. 2008, 1782, 408–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukas, J.; Cimmaruta, C.; Liguori, L.; Pantoom, S.; Iwanov, K.; Petters, J.; Hund, C.; Bunschkowski, M.; Hermann, A.; Cubellis, M.V.; et al. Assessment of gene variant amenability for pharmacological chaperone therapy with 1-deoxygalactonojirimycin in Fabry disease. Int. J. Mol. Sci. 2020, 21, 956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seemann, S.; Ernst, M.; Cimmaruta, C.; Struckmann, S.; Cozma, C.; Koczan, D.; Knospe, A.-M.; Haake, L.R.; Citro, V.; Bräuer, A.U.; et al. Proteostasis regulators modulate proteasomal activity and gene expression to attenuate multiple phenotypes in Fabry disease. Biochem. J. 2020, 477, 359–380. [Google Scholar] [CrossRef] [PubMed]
- Duffy, J.B. GAL4 system indrosophila: A fly geneticist’s swiss army knife. Genesis 2002, 34, 1–15. [Google Scholar] [CrossRef]
- Brand, A.H.; Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993, 118, 401–415. [Google Scholar]
- Smith, J.E.; Cronmiller, C. Faculty of 1000 evaluation for The Drosophila daughterless gene autoregulates and is controlled by both positive and negative cis regulation. Development 2001, 128, 4705–4714. [Google Scholar] [CrossRef]
- Maley, F.; Trimble, R.B.; Tarentino, A.L.; Plummer, T.H. Characterization of glycoproteins and their associated oligosaccharides through the use of endoglycosidases. Anal. Biochem. 1989, 180, 195–204. [Google Scholar] [CrossRef]
- Trimble, R.B.; Tarentino, A.L. Identification of distinct endoglycosidase (endo) activities in Flavobacterium meningosepticum: Endo F1, endo F2, and endo F3. Endo F1 and endo H hydrolyze only high mannose and hybrid glycans. J. Boil. Chem. 1991, 266, 1646–1651. [Google Scholar]
- Narasimhan, S.; Stanley, P.; Schachter, H. Control of glycoprotein synthesis. Lectin-resistant mutant containing only one of two distinct N-acetylglucosaminyltransferase activities present in wild type Chinese hamster ovary cells. J. Biol. Chem. 1977, 252, 3926–3933. [Google Scholar] [PubMed]
- Tai, T.; Yamashita, K.; Ito, S.; Kobata, A. Structures of the carbohydrate moiety of ovalbumin glycopeptide III and the difference in specificity of endo-beta-N-acetylglucosaminidases CII and H. J. Boil. Chem. 1977, 252, 6687–6694. [Google Scholar]
- Goldberg, A.L. Selective inhibitors of the proteasome-dependent and vacuolar pathways of protein degradation in Saccharomyces cerevisiae. J. Biol. Chem. 1996, 271, 27280–27284. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.H.; Goldberg, A.L. Proteasome inhibitors: Valuable new tools for cell biologists. Trends Cell Boil. 1998, 8, 397–403. [Google Scholar] [CrossRef]
- Rock, K.L.; Gramm, C.; Rothstein, L.; Clark, K.; Stein, R.; Dick, L.; Hwang, D.; Goldberg, A.L. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 1994, 78, 761–771. [Google Scholar] [CrossRef]
- Feany, M.B.; Bender, W.W. A Drosophila model of Parkinson’s disease. Nat. Cell Biol. 2000, 404, 394–398. [Google Scholar] [CrossRef]
- Mao, Z.; Davis, R.L. Eight different types of dopaminergic neurons innervate the Drosophila mushroom body neuropil: Anatomical and physiological heterogeneity. Front. Neural Circuits 2009, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Tuttolomondo, A.; Simonetta, I.; Duro, G.; Pecoraro, R.; Miceli, S.; Colomba, P.; Zizzo, C.; Nucera, A.; Daidone, M.; Di Chiara, T.; et al. Inter-familial and intra-familial phenotypic variability in three Sicilian families with Anderson-Fabry disease. Oncotarget 2017, 8, 61415–61424. [Google Scholar] [CrossRef] [Green Version]
- Eng, C.M.; Desnick, R.J. Molecular basis of fabry disease: Mutations and polymorphisms in the human α-galactosidase A gene. Hum. Mutat. 1994, 3, 103–111. [Google Scholar] [CrossRef]
- Okumiya, T.; Ishii, S.; Kase, R.; Kamei, S.; Sakuraba, H.; Suzuki, Y. α-Galactosidase gene mutations in Fabry disease: Heterogeneous expressions of mutant enzyme proteins. Qual. Life Res. 1995, 95, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Romeo, G.; D’Urso, M.; Pisacane, A.; Blum, E.; De Falco, A.; Ruffilli, A. Residual activity of α-galactosidase A in Fabry’s disease. Biochem. Genet. 1975, 13, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Shabbeer, J.; Yasuda, M.; Benson, S.D.; Desnick, R.J. Fabry disease: Identification of 50 novel α-galactosidase A mutations causing the classic phenotype and three-dimensional structural analysis of 29 missense mutations. Hum. Genom. 2006, 2, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ishii, S.; Kase, R.; Okumiya, T.; Sakuraba, H.; Suzuki, Y. Aggregation of the inactive form of human α -galactosidase in the endoplasmic reticulum. Biochem. Biophys. Res. Commun. 1996, 220, 812–815. [Google Scholar] [CrossRef]
- Yam, G.H.-F.; Bosshard, N.; Zuber, C.; Steinmann, B.; Roth, J. Pharmacological chaperone corrects lysosomal storage in Fabry disease caused by trafficking-incompetent variants. Am. J. Physiol. Physiol. 2006, 290, C1076–C1082. [Google Scholar] [CrossRef] [Green Version]
- Yam, G.H.-F.; Zuber, C.; Roth, J. A synthetic chaperone corrects the trafficking defect and disease phenotype in a protein misfolding disorder. FASEB J. 2005, 19, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Movsichoff, F.; Castro, O.A.; Parodi, A.J. Characterization of Schizosaccharomyces pombe ER α-Mannosidase: A reevaluation of the role of the enzyme on ER-associated degradation. Mol. Biol. Cell 2005, 16, 4714–4724. [Google Scholar] [CrossRef] [Green Version]
- Fenteany, G.; Standaert, R.; Lane, W.; Choi, S.; Corey, E.; Schreiber, S. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science 1995, 268, 726–731. [Google Scholar] [CrossRef]
- Ishii, S.; Chang, H.-H.; Kawasaki, K.; Yasuda, K.; Wu, H.-L.; Garman, S.C.; Fan, J.-Q. Mutant α-galactosidase A enzymes identified in Fabry disease patients with residual enzyme activity: Biochemical characterization and restoration of normal intracellular processing by 1-deoxygalactonojirimycin. Biochem. J. 2007, 406, 285–295. [Google Scholar] [CrossRef]
- Andreotti, G.; Citro, V.; De Crescenzo, A.; Orlando, P.; Cammisa, M.; Correra, A.; Cubellis, M.V. Therapy of Fabry disease with pharmacological chaperones: From in silico predictions to in vitro tests. Orphanet J. Rare Dis. 2011, 6, 66. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.-H.; Murray, G.J.; Kluepfel-Stahl, S.; Cooney, A.M.; Quirk, J.M.; Schiffmann, R.; Brady, R.O.; Kaneski, C.R. Screening for pharmacological chaperones in Fabry disease. Biochem. Biophys. Res. Commun. 2007, 359, 168–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukas, J.; Scalia, S.; Eichler, S.; Pockrandt, A.-M.; Dehn, N.; Cozma, C.; Giese, A.-K.; Rolfs, A. Functional and clinical consequences of novel α-galactosidase a mutations in Fabry disease. Hum. Mutat. 2015, 37, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Katz, E.; Della Valle, M.C.; Mascioli, K.; Flanagan, J.J.; Castelli, J.P.; Schiffmann, R.; Boudes, P.; Lockhart, D.J.; Valenzano, K.J.; et al. A pharmacogenetic approach to identify mutant forms of α-galactosidase a that respond to a pharmacological chaperone for Fabry disease. Hum. Mutat. 2011, 32, 965–977. [Google Scholar] [CrossRef] [PubMed]
- Lukas, J.; Pockrandt, A.-M.; Seemann, S.; Sharif, M.; Runge, F.; Pohlers, S.; Zheng, C.; Glaser, A.; Beller, M.; Rolfs, A.; et al. Enzyme enhancers for the treatment of Fabry and Pompe disease. Mol. Ther. 2015, 23, 456–464. [Google Scholar] [CrossRef] [Green Version]
- Maegawa, G.; Tropak, M.B.; Buttner, J.D.; Rigat, B.A.; Fuller, M.; Pandit, D.; Tang, L.; Kornhaber, G.J.; Hamuro, Y.; Clarke, J.T.R.; et al. Identification and characterization of ambroxol as an enzyme enhancement agent for Gaucher disease. J. Biol. Chem. 2009, 284, 23502–23516. [Google Scholar] [CrossRef] [Green Version]
- Khanna, R.; Soska, R.; Lun, Y.; Feng, J.; Frascella, M.; Young, B.; Brignol, N.; Pellegrino, L.; Sitaraman, S.A.; Desnick, R.J.; et al. The pharmacological chaperone 1-deoxygalactonojirimycin reduces tissue globotriaosylceramide levels in a mouse model of Fabry disease. Mol. Ther. 2010, 18, 23–33. [Google Scholar] [CrossRef]
- Young-Gqamana, B.; Brignol, N.; Chang, H.-H.; Khanna, R.; Soska, R.; Fuller, M.; Sitaraman, S.A.; Germain, D.P.; Giugliani, R.; Hughes, D.A.; et al. Migalastat HCl reduces globotriaosylsphingosine (Lyso-Gb3) in Fabry transgenic mice and in the plasma of Fabry patients. PLoS ONE 2013, 8, e57631. [Google Scholar] [CrossRef] [Green Version]
- De Francesco, P.N.; Mucci, J.M.; Ceci, R.; Fossati, C.A.; Rozenfeld, P.A. Higher apoptotic state in Fabry disease peripheral blood mononuclear cells. Mol. Genet. Metab. 2011, 104, 319–324. [Google Scholar] [CrossRef]
- Inagaki, H.K.; Kamikouchi, A.; Ito, K. Methods for quantifying simple gravity sensing in Drosophila melanogaster. Nat. Protoc. 2009, 5, 20–25. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Sequence |
|---|---|
| FP-GLA(ATG) + pUAST/mycHis-left arm | 5′-CAGATCTGCGGCCGCGGCTCGAGGATGCAGCTGAGGAACCCAGAACTAC-3′ |
| RP-GLA(TAA) + pUAST/mycHis-right arm | 5′-GCCCTCTAGAGGTACCCTCGAGCCAAGTAAGTCTTTTAATGACATCTG-3′ |
| FP-GLA(ATG) + pcDNA4/myc-His-B-left arm | 5′-CAGCACAGTGGCGGCCGGTCGAGTATGCAGCTGAGGAACCCAGAA-3 |
| RP-GLA(TAA) + pcDNA4/myc-His-B-right arm | 5′-CCGCGGGCCCTCTAGACTCGAGCGAAGTAAGTCTTTTAATGACATC-3′ |
| Primer Name | Primer Sequence |
|---|---|
| ATF4 F | 5′-AGACGCTGCTTCGCTTCCTTC-3′ |
| ATF4 R | 5′-GCCCGTAAGTGCGAGTACGCT-3′ |
| Hsc 70-3 F | 5′-GCTGGTGTTATTGCCGGTCTGC-3′ |
| Hsc 70-3 R | 5′-GATGCCTCGGGATGGTTCCTTGC-3′ |
| Xbp1 F | 5′-CCGAACTGAAGCAGCAACAGC-3′ |
| Xbp1 R | 5′-GTATACCCTGCGGCAGATCC-3′ |
| RP49 F | 5′-TAAGAAGCGCACAAAGCACT-3′ |
| RP49 R | 5′-GGGCATCAGATATTGTCCCT-3′ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Braunstein, H.; Papazian, M.; Maor, G.; Lukas, J.; Rolfs, A.; Horowitz, M. Misfolding of Lysosomal α-Galactosidase a in a Fly Model and Its Alleviation by the Pharmacological Chaperone Migalastat. Int. J. Mol. Sci. 2020, 21, 7397. https://doi.org/10.3390/ijms21197397
Braunstein H, Papazian M, Maor G, Lukas J, Rolfs A, Horowitz M. Misfolding of Lysosomal α-Galactosidase a in a Fly Model and Its Alleviation by the Pharmacological Chaperone Migalastat. International Journal of Molecular Sciences. 2020; 21(19):7397. https://doi.org/10.3390/ijms21197397
Chicago/Turabian StyleBraunstein, Hila, Maria Papazian, Gali Maor, Jan Lukas, Arndt Rolfs, and Mia Horowitz. 2020. "Misfolding of Lysosomal α-Galactosidase a in a Fly Model and Its Alleviation by the Pharmacological Chaperone Migalastat" International Journal of Molecular Sciences 21, no. 19: 7397. https://doi.org/10.3390/ijms21197397
APA StyleBraunstein, H., Papazian, M., Maor, G., Lukas, J., Rolfs, A., & Horowitz, M. (2020). Misfolding of Lysosomal α-Galactosidase a in a Fly Model and Its Alleviation by the Pharmacological Chaperone Migalastat. International Journal of Molecular Sciences, 21(19), 7397. https://doi.org/10.3390/ijms21197397