Helicobacter pylori Virulence Factor Cytotoxin-Associated Gene A (CagA)-Mediated Gastric Pathogenicity



Abstract
:1. Introduction
2. Cytotoxin-Associated Gene Pathogenicity Island (cagPAI)
3. Cytotoxin-Associated Gene A (CagA)
4. CagA Expression
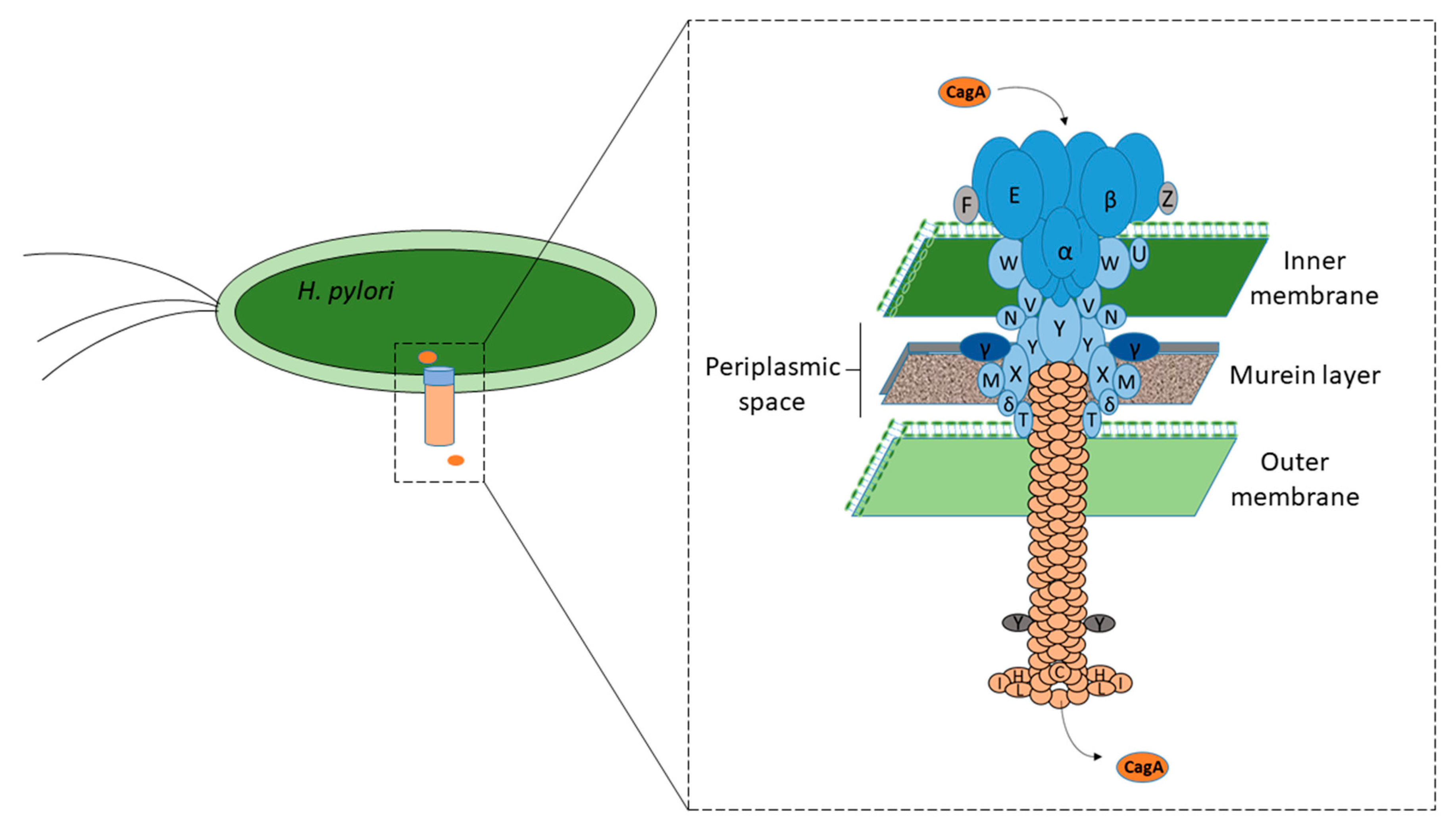
5. CagA Translocation
6. Tyrosine Phosphorylation
7. CagA-Dependent Mechanisms of Pathogenicity
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zamani, M.; Ebrahimtabar, F.; Zamani, V.; Miller, W.H.; Alizadeh-Navaei, R.; Shokri-Shirvani, J.; Derakhshan, M.H. Systematic review with meta-analysis: The worldwide prevalence of Helicobacter pylori infection. Aliment. Pharmacol. Ther. 2018, 47, 868–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooi, J.K.Y.; Lai, W.Y.; Ng, W.K.; Suen, M.M.Y.; Underwood, F.E.; Tanyingoh, D.; Malfertheiner, P.; Graham, D.Y.; Wong, V.W.S.; Wu, J.C.Y.; et al. Global Prevalence of Helicobacter pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology 2017, 153, 420–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peleteiro, B.; Bastos, A.; Ferro, A.; Lunet, N. Prevalence of Helicobacter pylori infection worldwide: A systematic review of studies with national coverage. Dig. Dis. Sci. 2014, 59, 1698–1709. [Google Scholar] [CrossRef] [PubMed]
- Mamishi, S.; Eshaghi, H.; Mahmoudi, S.; Bahador, A.; Hosseinpour Sadeghi, R.; Najafi, M.; Farahmand, F.; Khodadad, A.; Pourakbari, B. Intrafamilial transmission of Helicobacter pylori: Genotyping of faecal samples. Br. J. Biomed. Sci. 2016, 73, 38–43. [Google Scholar] [CrossRef]
- Osaki, T.; Konno, M.; Yonezawa, H.; Hojo, F.; Zaman, C.; Takahashi, M.; Fujiwara, S.; Kamiya, S. Analysis of intra-familial transmission of Helicobacter pylori in Japanese families. J. Med. Microbiol. 2015, 64 Pt 1, 67–73. [Google Scholar] [CrossRef]
- Bui, D.; Brown, H.E.; Harris, R.B.; Oren, E. Serologic Evidence for Fecal-Oral Transmission of Helicobacter pylori. Am. J. Trop. Med. Hyg. 2016, 94, 82–88. [Google Scholar] [CrossRef] [Green Version]
- Yokota, S.; Konno, M.; Fujiwara, S.; Toita, N.; Takahashi, M.; Yamamoto, S.; Ogasawara, N.; Shiraishi, T. Intrafamilial, Preferentially Mother-to-Child and Intraspousal, Helicobacter pylori Infection in Japan Determined by Multilocus Sequence Typing and Random Amplified Polymorphic DNA Fingerprinting. Helicobacter 2015, 20, 334–342. [Google Scholar] [CrossRef]
- Jiang, J.; Chen, Y.; Shi, J.; Song, C.; Zhang, J.; Wang, K. Population attributable burden of Helicobacter pylori-related gastric cancer, coronary heart disease, and ischemic stroke in China. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 199–212. [Google Scholar] [CrossRef]
- Backert, S.; Neddermann, M.; Maubach, G.; Naumann, M. Pathogenesis of Helicobacter pylori infection. Helicobacter 2016, 21, 19–25. [Google Scholar] [CrossRef]
- Parsonnet, J.; Friedman, G.D.; Vandersteen, D.P.; Chang, Y.; Vogelman, J.H.; Orentreich, N.; Sibley, R.K. Helicobacter pylori infection and the risk of gastric carcinoma. N. Engl. J. Med. 1991, 325, 1127–1131. [Google Scholar] [CrossRef]
- Amieva, M.; Peek, R.M., Jr. Pathobiology of Helicobacter pylori-induced gastric cancer. Gastroenterology 2016, 150, 64–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansari, S.; Yamaoka, Y. Role of vacuolating cytotoxin A in Helicobacter pylori infection and its impact on gastric pathogenesis. Expert Rev. Anti-Infect. Ther. 2020. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, Y. Mechanisms of disease: Helicobacter pylori virulence factors. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 629–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olbermann, P.; Josenhans, C.; Moodley, Y.; Uhr, M.; Stamer, C.; Vauterin, M.; Suerbaum, S.; Achtman, M.; Linz, B. A global overview of the genetic and functional diversity in the Helicobacter pylori cag pathogenicity island. PLoS Genet. 2010, 6, e1001069. [Google Scholar] [CrossRef] [Green Version]
- Blomstergren, A.; Lundin, A.; Nilsson, C.; Engstrand, L.; Lundeberg, J. Comparative analysis of the complete cag pathogenicity island sequence in four Helicobacter pylori isolates. Gene 2004, 328, 85–93. [Google Scholar] [CrossRef]
- Backert, S.; Tegtmeyer, N.; Fischer, W. Composition, structure and function of the Helicobacter pylori cag pathogenicity island encoded type IV secretion system. Future Microbiol. 2015, 10, 955–965. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.M.; Sheedlo, M.J.; Campbell, A.M.; Sawhney, N.; Frick-Cheng, A.E.; Lacy, D.B.; Cover, T.L.; Ohi, M.D. Structure of the Helicobacter pylori Cag type IV secretion system. eLife 2019, 8, e47644. [Google Scholar] [CrossRef]
- Backert, S.; Haas, R.; Gerhard, M.; Naumann, M. The Helicobacter pylori type IV secretion system encoded by the cag pathogenicity island: Architecture, function, and signaling. Curr. Top. Microbiol. Immunol. 2017, 413, 187–220. [Google Scholar]
- Akopyants, N.S.; Clifton, S.W.; Kersulyte, D.; Crabtree, J.E.; Youree, B.E.; Reece, C.A.; Bukanov, N.O.; Drazek, E.S.; Roe, B.A.; Berg, D.E. Analyses of the cag pathogenicity island of Helicobacter pylori. Mol. Microbiol. 1998, 28, 37–53. [Google Scholar] [CrossRef]
- Censini, S.; Lange, C.; Xiang, Z.; Crabtree, J.E.; Ghiara, P.; Borodovsky, M.; Rappuoli, R.; Covacci, A. Cag, a pathogenicity island of Helicobacter pylori, encodes type I- specific and disease- associated virulence factors. Proc. Natl. Acad. Sci. USA 1996, 93, 14648–14653. [Google Scholar] [CrossRef] [Green Version]
- Waskito, L.A.; Miftahussurur, M.; Lusida, M.I.; Syam, A.F.; Suzuki, R.; Subsomwong, P.; Uchida, T.; Hamdan, M.; Nasronudin; Yamaoka, Y. Distribution and clinical associations of integrating conjugative elements and cag pathogenicity islands of Helicobacter pylori in Indonesia. Sci. Rep. 2018, 8, 6073. [Google Scholar] [CrossRef] [PubMed]
- Wroblewski, L.E.; Peek, R.M., Jr. Helicobacter pylori: Pathogenic enablers-toxic relationships in the stomach. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 317–318. [Google Scholar] [CrossRef] [PubMed]
- Cover, T.L. Helicobacter pylori diversity and gastric cancer risk. mBio 2016, 7, e01869-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miernyk, K.M.; Bruden, D.; Rudolph, K.M.; Hurlburt, D.A.; Sacco, F.; McMahon, B.J.; Bruce, M.G. Presence of cagPAI genes and characterization of vacA s, i and m regions in Helicobacter pylori isolated from Alaskans and their association with clinical pathologies. J. Med. Microbiol. 2020, 69, 218–227. [Google Scholar] [CrossRef]
- Markovska, R.; Boyanova, L.; Yordanov, D.; Stankova, P.; Gergova, G.; Mito, I. Status of Helicobacter pylori cag pathogenicity island (cagPAI) integrity and significance of its individual genes. Infect. Genet. Evol. 2018, 59, 167–171. [Google Scholar] [CrossRef]
- Jiménez-Soto, L.F.; Haas, R. The CagA toxin of Helicobacter pylori: Abundant production but relatively low amount translocated. Sci. Rep. 2016, 6, 23227. [Google Scholar] [CrossRef] [Green Version]
- Higashi, H.; Tsutsumi, R.; Fujita, A.; Yamazaki, S.; Asaka, M.; Azuma, T.; Hatakeyama, M. Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proc. Natl. Acad. Sci. USA 2002, 99, 14428–14433. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Liu, J.; Gong, Y.; Yuan, Y. Association of CagA EPIYA-D or EPIYA-C phosphorylation sites with peptic ulcer and gastric cancer risks: A meta-analysis. Medicine 2017, 96, e6620. [Google Scholar] [CrossRef]
- Zhang, X.S.; Tegtmeyer, N.; Traube, L.; Jindal, S.; Perez-Perez, G.; Sticht, H.; Backert, S.; Blaser, M.J. A specific A/T polymorphism in Western tyrosine phosphorylation B-motifs regulates Helicobacter pylori CagA epithelial cell interactions. PLoS Pathog. 2015, 11, e1004621. [Google Scholar] [CrossRef] [Green Version]
- Canzian, F.; Rizzato, C.; Obazee, O.; Stein, A.; Flores-Luna, L.; Camorlinga-Ponce, M.; Mendez-Tenorio, A.; Vivas, J.; Trujillo, E.; Jang, H.; et al. Genetic polymorphisms in the cag pathogenicity island of Helicobacter pylori and risk of stomach cancer and high-grade premalignant gastric lesions. Int. J. Cancer. 2020, 1–9. [Google Scholar] [CrossRef]
- Rizzato, C.; Torres, J.; Obazee, O.; Camorlinga-Ponce, M.; Trujillo, E.; Stein, A.; Mendez-Tenorio, A.; Bravo, M.M.; Canzian, F.; Kato, I. Variations in cag pathogenicity island genes of Helicobacter pylori from Latin American groups may influence neoplastic progression to gastric cancer. Sci. Rep. 2020, 10, 6570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polk, D.B.; Peek, R.M., Jr. Helicobacter pylori: Gastric cancer and beyond. Nat. Rev. Cancer. 2010, 10, 403–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Meng, W.; Wang, B.; Qiao, L. Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer Lett. 2014, 345, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.C.; Tsai, H.F.; Kuo, S.H.; Wu, M.S.; Lin, C.W.; Hsu, P.I.; Cheng, A.L.; Hsu, P.N. Translocation of Helicobacter pylori CagA into Human B lymphocytes, the origin of mucosa-associated lymphoid tissue lymphoma. Cancer Res. 2010, 70, 5740–5748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, S.H.; Chen, L.T.; Lin, C.W.; Wu, M.S.; Hsu, P.N.; Tsai, H.J.; Chu, C.Y.; Tzeng, Y.S.; Wang, H.P.; Yeh, K.H.; et al. Detection of the Helicobacter pylori CagA protein in gastric mucosa-associated lymphoid tissue lymphoma cells: Clinical and biological significance. Blood Cancer J. 2013, 3, e125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashinaga, M.; Suzuki, R.; Akada, J.; Matsumoto, T.; Kido, Y.; Okimoto, T.; Kodama, M.; Murakami, K.; Yamaoka, Y. Differences in amino acid frequency in CagA and VacA sequences of Helicobacter pylori distinguish gastric cancer from gastric MALT lymphoma. Gut Pathog. 2016, 8, 54. [Google Scholar] [CrossRef] [Green Version]
- Tegtmeyer, N.; Neddermann, M.; Asche, C.I.; Backert, S. Subversion of host kinases: A key network in cellular signaling hijacked by Helicobacter pylori CagA. Mol. Microbiol. 2017, 105, 358–372. [Google Scholar] [CrossRef] [Green Version]
- Vaziri, F.; Peerayeh, S.N.; Alebouyeh, M.; Maghsoudi, N.; Azimzadeh, P.; Siadat, S.D.; Zali, M.R. Novel effects of Helicobacter pylori CagA on key genes of gastric cancer signal transduction: A comparative transfection study. Pathog. Dis. 2015, 73, ftu021. [Google Scholar] [CrossRef] [Green Version]
- Loh, J.T.; Shaffer, C.L.; Piazuelo, M.B.; Bravo, L.E.; McClain, M.S.; Correa, P.; Cover, T.L. Analysis of cagA in Helicobacter pylori strains from Colombian populations with contrasting gastric cancer risk reveals a biomarker for disease severity. Cancer Epidemiol. Biomark. Prev. 2011, 20, 2237–2249. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, R.M.; Pinto-Ribeiro, I.; Wen, X.; Marcos-Pinto, R.; Dinis-Ribeiro, M.; Carneiro, F.; Figueiredo, C. Helicobacter pylori cagA promoter region sequences influence CagA expression and interleukin 8 secretion. J. Infect. Dis. 2016, 213, 669–673. [Google Scholar] [CrossRef]
- Loh, J.T.; Lin, A.S.; Beckett, A.C.; McClain, M.S.; Cover, T.L. Role of a stem-loop structure in Helicobacter pylori cagA transcript stability. Infect. Immun. 2019, 87, e00692-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Necchi, V.; Ricci, V.; Sommi, P.; Solcia, E. CagA Effector Protein in Helicobacter pylori-Infected Human Gastric Epithelium in Vivo: From Bacterial Core and Adhesion/Injection Clusters to Host Cell Proteasome-Rich Cytosol. Toxins 2019, 11, 618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tegtmeyer, N.; Wessler, S.; Necchi, V.; Rohde, M.; Harrer, A.; Rau, T.T. Helicobacter pylori employs a unique basolateral type IV secretion mechanism for CagA delivery. Cell Host Microbe 2017, 22, 552–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata-Kamiya, N.; Kikuchi, K.; Hayashi, T.; Higashi, H.; Hatakeyama, M. Helicobacter pylori Exploits Host Membrane Phosphatidylserine for Delivery, Localization, and Pathophysiological Action of the CagA Oncoprotein. Cell Host Microbe 2010, 7, 399–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, N.; Shariq, M.; Kumar, A.; Kumari, R.; Subbarao, N.; Tyagi, R.K.; Mukhopadhyay, G. Analyzing the role of CagV, a VirB8 homolog of the type IV secretion system of Helicobacter pylori. FEBS Open Bio 2017, 7, 915–993. [Google Scholar] [CrossRef]
- Tegtmeyer, N.; Wessler, S.; Backert, S. Role of the cag-pathogenicity island encoded type IV secretion system in Helicobacter pylori pathogenesis. FEBS J. 2011, 278, 1190–1202. [Google Scholar] [CrossRef]
- Conradi, J.; Tegtmeyer, N.; Wozna, M.; Wissbrock, M.; Michalek, C.; Gagell, C.; Cover, T.L.; Frank, R.; Sewald, N.; Backert, S. An RGD helper sequence in CagL of Helicobacter pylori assists in interactions with integrins and injection of CagA. Front. Cell. Infect. Microbiol. 2012, 2, 70. [Google Scholar] [CrossRef] [Green Version]
- Gorrell, R.J.; Guan, J.; Xin, Y.; Tafreshi, M.A.; Hutton, M.L.; McGuckin, M.A.; Ferrero, R.L.; Kwok, T. A novel NOD1- and CagA-independent pathway of interleukin-8 induction mediated by the Helicobacter pylori type IV secretion system. Cell. Microbiol. 2013, 15, 554–570. [Google Scholar] [CrossRef]
- Saha, A.; Backert, S.; Hammond, C.E.; Gooz, M.; Smolka, A.J. Helicobacter pylori CagL activates ADAM17 to induce repression of the gastric H, K-ATPase alpha subunit. Gastroenterology 2010, 139, 239–248. [Google Scholar] [CrossRef] [Green Version]
- Tegtmeyer, N.; Hartig, R.; Delahay, R.M.; Rohde, M.; Brandt, S.; Conradi, J.; Takahashi, S.; Smolka, A.J.; Sewald, N.; Backert, S. A small fibronectin-mimicking protein from bacteria induces cell spreading and focal adhesion formation. J. Biol. Chem. 2010, 285, 23515–23526. [Google Scholar] [CrossRef] [Green Version]
- Tafreshi, M.; Zwickel, N.; Gorrell, R.J.; Kwok, T. Preservation of Helicobacter pylori CagA translocation and host cell pro-inflammatory responses in the face of CagL hyper-variability at amino acid residues 58/59. PLoS ONE 2015, 10, e0133531. [Google Scholar] [CrossRef] [PubMed]
- Gorrell, R.J.; Zwickel, N.; Reynolds, J.; Bulach, D.; Kwok, T. Helicobacter pylori CagL hypervariable motif: A global analysis of geographical diversity and association with gastric cancer. J. Infect. Dis. 2016, 213, 1927–1931. [Google Scholar] [CrossRef] [PubMed]
- Román Roman, A.; Martínez Santos, V.I.; Castañón Sánchez, C.A.; Albañil Muñoz, A.J.; Mendoza, P.G.; Soto Flores, D.G.; Martínez Carrillo, D.N.; Tilapa, G.F. CagL polymorphisms D58/K59 are predominant in Helicobacter pylori strains isolated from Mexican patients with chronic gastritis. Gut Pathog. 2019, 11, 5. [Google Scholar] [CrossRef] [PubMed]
- Yadegar, A.; Mohabati Mobarez, A.; Zali, M.R. Genetic diversity and amino acid sequence polymorphism in Helicobacter pylori CagL hypervariable motif and its association with virulence markers and gastroduodenal diseases. Cancer Med. 2019, 8, 1619–1632. [Google Scholar] [CrossRef]
- McClain, M.S.; Duncan, S.S.; Gaddy, J.A.; Cover, T.L. Control of gene expression in Helicobacter pylori using the Tet repressor. J. Microbiol. Methods 2013, 95, 336–341. [Google Scholar] [CrossRef] [Green Version]
- Johnson, E.M.; Gaddy, J.A.; Voss, B.J.; Hennig, E.E.; Cover, T.L. Genes required for assembly of pili associated with the Helicobacter pylori cag type IV secretion system. Infect. Immun. 2014, 82, 3457–3470. [Google Scholar] [CrossRef] [Green Version]
- Fischer, W.; Puls, J.; Buhrdorf, R.; Gebert, B.; Odenbreit, S.; Haas, R. Systematic mutagenesis of the Helicobacter pylori cag pathogenicity island: Essential genes for CagA translocation in host cells and induction of interleukin-8. Mol. Microbiol. 2001, 42, 1337–1348. [Google Scholar] [CrossRef]
- Frick-Cheng, A.E.; Pyburn, T.M.; Voss, B.J.; McDonald, W.H.; Ohi, M.D.; Cover, T.L. Molecular and Structural Analysis of the Helicobacter pylori cag Type IV Secretion System Core Complex. mBio 2016, 7, e02001–e02015. [Google Scholar] [CrossRef] [Green Version]
- McClain, M.S.; Voss, B.J.; Cover, T.L. Lipoprotein processing and sorting in Helicobacter pylori. mBio 2020, 11, e00911–00920. [Google Scholar] [CrossRef]
- Barrozo, R.M.; Cooke, C.L.; Hansen, L.M.; Lam, A.M.; Gaddy, J.A.; Johnson, E.M.; Cariaga, T.A.; Suarez, G.; Peek, R.M., Jr.; Cover, T.L.; et al. Functional plasticity in the type IV secretion system of Helicobacter pylori. PLoS Pathog. 2013, 9, e1003189. [Google Scholar]
- Suarez, G.; Romero-Gallo, J.; Sierra, J.C.; Piazuelo, M.B.; Krishna, U.S.; Gomez, M.A.; Wilson, K.T.; Peek, R.M., Jr. Genetic manipulation of Helicobacter pylori virulence function by host carcinogenic phenotypes. Cancer Res. 2017, 77, 2401–2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skoog, E.C.; Morikis, V.A.; Martin, M.E.; Foster, G.A.; Cai, L.P.; Hansen, L.M.; Li, B.; Gaddy, J.A.; Simon, S.I.; Solnick, J.V. CagY-dependent regulation of type IV secretion in Helicobacter pylori is associated with alterations in integrin binding. mBio 2018, 9, e00717–e00718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sierra, J.C.; Suarez, G.; Piazuelo, M.B.; Luis, P.B.; Baker, D.R.; Romero-Gallo, J.; Barry, D.P.; Schneider, C.; Morgan, D.R.; Peek, R.M., Jr.; et al. α-Difluoromethylornithine reduces gastric carcinogenesis by causing mutations in Helicobacter pylori cagY. Proc. Natl. Acad. Sci. USA 2019, 116, 5077–5085. [Google Scholar] [CrossRef] [Green Version]
- Barrozo, R.M.; Hansen, L.M.; Lam, A.M.; Skoog, E.C.; Martin, M.E.; Cai, L.P.; Lin, Y.; Latoscha, A.; Suerbaum, S.; Canfield, D.R. CagY is an Immune-Sensitive Regulator of the Helicobacter pylori Type IV Secretion System. Gastroenterology 2016, 151, 1164–1175. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Ling, F.; Wang, H.; Yu, M.; Zhu, H.; Chen, C.; Qian, J.; Liu, C.; Zhang, Y.; Shao, S. The Helicobacter pylori Cag pathogenicity island protein Cag1 is associated with the function of T4SS. Curr. Microbiol. 2016, 73, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Shen, Y.; Zhu, L.; Ni, Y.; Wang, H.; Shao, S. Preliminary study and bioinformatics analysis on the potential role of CagQ in type IV secretion system of H. pylori. Microb. Pathog. 2018, 116, 1–7. [Google Scholar] [CrossRef]
- Javaheri, A.; Kruse, T.; Moonens, K.; Mejias-Luque, R.; Debraekeleer, A.; Asche, C.I.; Tegtmeyer, N.; Kalali, B.; Bach, N.C.; Sieber, S.A.; et al. Helicobacter pylori adhesin HopQ engages in a virulence-enhancing interaction with human CEACAMs. Nat. Microbiol. 2016, 2, 16189. [Google Scholar] [CrossRef] [Green Version]
- Koniger, V.; Holsten, L.; Harrison, U.; Busch, B.; Loell, E.; Zhao, Q.; Bonsor, D.A.; Roth, A.; Kengmo-Tchoupa, A.; Smith, S.I.; et al. Helicobacter pylori exploits human CEACAMs via HopQ for adherence and translocation of CagA. Nat. Microbiol. 2016, 2, 16188. [Google Scholar] [CrossRef]
- Zhao, Q.; Busch, B.; Jimenez-Soto, L.F.; Ishikawa-Ankerhold, H.; Massberg, S.; Terradot, L.; Fischer, W.; Haas, R. Integrin but not CEACAM receptors are dispensable for Helicobacter pylori CagA translocation. PLoS Pathog. 2018, 14, e1007359. [Google Scholar] [CrossRef] [Green Version]
- Tegtmeyer, N.; Harrer, A.; Schmitt, V.; Singer, B.B.; Backert, S. Expression of CEACAM1 or CEACAM5 in AZ-521 cells restores the type IV secretion deficiency for translocation of CagA by Helicobacter pylori. Cell Microbiol. 2019, 21, e12965. [Google Scholar] [CrossRef] [Green Version]
- Tegtmeyer, N.; Ghete, T.D.; Schmitt, V.; Remmerbach, T.; Cortes, M.C.C.; Bondoc, E.M.; Graf, H.L.; Singer, B.B.; Hirsch, C.; Backert, S. Type IV secretion of Helicobacter pylori CagA into oral epithelial cells is prevented by the absence of CEACAM receptor expression. Gut Pathog. 2020, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Mueller, D.; Tegtmeyer, N.; Brandt, S.; Yamaoka, Y.; Poire, E.D.; Sgouras, D.; Wessler, S.; Torres, J.; Smolka, A.; Backert, S. c-Src and c-Abl kinases control hierarchic phosphorylation and function of the CagA effector protein in Western and East Asian Helicobacter pylori strains. J. Clin. Investig. 2012, 122, 1553–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanotti, G.; Cendron, L. Structural and functional aspects of the Helicobacter pylori secretome. World J. Gastroenterol. 2014, 20, 1402–1423. [Google Scholar] [CrossRef] [PubMed]
- Khatri, A.; Wang, J.; Pendergast, A.M. Multifunctional Abl kinases in health and disease. J. Cell Sci. 2016, 129, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashi, H.; Tsutsumi, R.; Muto, S.; Sugiyama, T.; Azuma, T.; Asaka, M.; Hatakeyama, M. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science 2002, 295, 683–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mimuro, H.; Suzuki, T.; Tanaka, J.; Asahi, M.; Haas, R.; Sasakawa, C. Grb2 is a key mediator of Helicobacter pylori CagA protein activities. Mol. Cell 2002, 10, 745–755. [Google Scholar] [CrossRef]
- Hatakeyama, M. Anthropological and clinical implications for the structural diversity of the Helicobacter pylori CagA oncoprotein. Cancer Sci. 2011, 102, 36–43. [Google Scholar] [CrossRef]
- Hatakeyama, M. Helicobacter pylori CagA and gastric cancer: A paradigm for hit-and-run carcinogenesis. Cell Host Microbe 2014, 15, 306–316. [Google Scholar] [CrossRef] [Green Version]
- Asati, V.; Mahapatra, D.K.; Bharti, S.K. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: Structural and pharmacological perspectives. Eur. J. Med. Chem. 2016, 15, 314–341. [Google Scholar] [CrossRef]
- Wei, L.; Li, Y.; Suo, Z. TSPAN8 promotes gastric cancer growth and metastasis via ERK MAPK pathway. Int. J. Clin. Exp. Med. 2015, 8, 8599–8607. [Google Scholar]
- Servetas, S.L.; Bridge, D.R.; Merrell, D.S. Molecular mechanisms of gastric cancer initiation and progression by Helicobacter pylori. Curr. Opin. Infect. Dis. 2016, 29, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.M.; Ferreira, R.M.; Pinto-Ribeiro, I.; Sougleri, I.S.; Oliveira, M.J.; Carreto, L.; Santos, M.A.; Sgouras, D.N.; Carneiro, F.; Leite, M.; et al. Helicobacter pylori activates matrix metalloproteinase 10 in gastric epithelial cells via EGFR and ERK-mediated pathways. J. Infect. Dis. 2016, 213, 1767–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Senda, M.; Suzuki, N.; Nishikawa, H.; Ben, C.; Tang, C.; Nagase, L.; Inoue, K.; Senda, T.; Hatakeyama, M. Differential Mechanisms for SHP2 binding and activation are exploited by geographically distinct Helicobacter pylori CagA Oncoproteins. Cell Rep. 2017, 20, 2876–2890. [Google Scholar] [CrossRef] [Green Version]
- Fujii, Y.; Murata-Kamiya, N.; Hatakeyama, M. Helicobacter pylori CagA oncoprotein interacts with SHIP2 to increase its delivery into gastric epithelial cells. Cancer Sci. 2020, 111, 1596–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, Y.; Ren, H.; Zhao, R.; Song, L.; Liu, Z.; Xu, W.; Liu, Y.; Wang, S. Helicobacter pylori CagA promotes the malignant transformation of gastric mucosal epithelial cells through the dysregulation of the miR-155/KLF4 signaling pathway. Mol. Carcinogenes. 2019, 58, 1427–1437. [Google Scholar] [CrossRef]
- Naumann, M.; Sokolova, O.; Tegtmeyer, N.; Backert, S. Helicobacter pylori: A Paradigm Pathogen for Subverting Host Cell Signal Transmission. Trends Microbiol. 2017, 25, 316–328. [Google Scholar] [CrossRef]
- Noto, J.M.; Zackular, J.P.; Varga, M.G.; Delgado, A.; Romero-gallo, J.; Scholz, M.B. Crossm Modification of the Gastric Mucosal Microbiota by a Strain-Specific Helicobacter pylori Oncoprotein and Carcinogenic. mBio 2019, 10, e00955-19. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, M.J.; Costa, A.M.; Costa, A.C.; Ferreira, R.M.; Sampaio, P.; Machado, J.C. CagA Associates with c-Met, E-Cadherin, andp120-Catenin in a Multiproteic Complex that Suppresses Helicobacter pylori–Induced Cell-Invasive Phenotype. J. Infect. Dis. 2009, 200, 745–755. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Ju, M.K.; Jeon, H.M.; Lee, Y.J.; Kim, C.H.; Park, H.G.; Han, S.I.; Kang, H.S. Oncogenic Metabolism Acts as a Prerequisite Step for Induction of Cancer Metastasis and Cancer Stem Cell Phenotype. Oxidative Med. Cell. Longev. 2018, 2018, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–Mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2018, 20, 69–84. [Google Scholar] [CrossRef]
- Park, A.H.; Shin, J.E.; Park, H.W. The role of Hippo Pathway in Cancer Stem Cell Biology. Mol. Cells 2018, 41, 83–92. [Google Scholar] [PubMed]
- Singh, M.; Yelle, N.; Venugopal, C.; Singh, S.K. EMT: Mechanisms and therapeutic implications. Pharmacol. Ther. 2018, 182, 80–94. [Google Scholar] [CrossRef] [PubMed]
- Toh, T.B.; Lim, J.J.; Chow, E.K.H. Epigenetics in cancer stem cells. Mol. Cancer 2017, 16, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, J.C.; Carrasco-Garcia, E.; Garcia-Puga, M.; Aldaz, P.; Montes, M.; Fernandez-Reyes, M.; de Oliveira, C.C.; Lawrie, C.H.; Arauzo-Bravo, M.J.; Ribeiro, M.L.; et al. SOX9 Elevation Acts with Canonical WNT Signaling to Drive Gastric Cancer Progression. Cancer Res. 2016, 76, 6735–6746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bessède, E.; Staedel, C.; Acuña Amador, L.A.; Nguyen, P.H.; Chambonnier, L.; Hatakeyama, M.; Belleannee, G.; Megraud, F.; Varon, C. Helicobacter pylori generates cells with cancer stem cell properties via epithelial-Mesenchymal transition-Like changes. Oncogene 2014, 33, 4123–4131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.G.; Kim, H.S.; Lee, Y.S.; Kim, S.; Cha, S.Y.; Ota, I.; Kim, N.H.; Cha, Y.H.; Yand, D.H.; Lee, Y.; et al. Helicobacter pylori CagA promotes Snail-Mediated epithelial-Mesenchymal transition by reducing GSK-3 activity. Nat. Commun. 2014, 5, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Mo, J.S.; Meng, Z.; Kim, Y.C.; Park, H.W.; Hansen, C.G.; Kim, S.; Lim, D.S.; Guan, K.L. Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat. Cell Biol. 2015, 17, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.H.; Guan, K.-L. Interplay between YAP/TAZ and Metabolism. Cell Metab. 2018, 28, 196–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-A.; Lu, C.-Y.; Cheng, T.-Y.; Pan, S.-H.; Chen, H.-F.; Chang, N.-S. WW Domain-Containing Proteins YAP and TAZ in the Hippo Pathway as Key Regulators in Stemness Maintenance, Tissue Homeostasis, and Tumorigenesis. Front. Oncol. 2019, 9, 60. [Google Scholar] [CrossRef] [Green Version]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef]
- Menheniott, T.R.; Kurklu, B.; Giraud, A.S. Gastrokines: Stomach-specific proteins with putative homeostatic and tumor suppressor roles. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 304, 109–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshikawa, Y.; Mukai, H.; Hino, F.; Asada, K.; Kato, I. Isolation of two novel genes, down-regulated in gastric cancer. Jpn. J. Cancer Res. 2000, 91, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Xing, R.; Li, W.; Cui, J.; Zhang, J.; Kang, B.; Wang, Y.; Wang, Z.; Liu, S.; Lu, Y. Gastrokine 1 induces senescence through p16/Rb pathway activation in gastric cancer cells. Gut 2012, 61, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Kang, Y.H.; Choi, Y.J.; Nam, S.W.; Lee, J.Y.; Lee, Y.S.; Park, W.S. Gastrokine 1 functions as a tumor suppressor by inhibition of epithelial-mesenchymal transition in gastric cancers. J. Cancer Res. Clin. Oncol. 2011, 137, 1697–1704. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Choi, W.S.; Kim, O.; Park, W.S. The role of gastrokine 1 in gastric cancer. J. Gastric Cancer 2014, 14, 147–155. [Google Scholar] [CrossRef]
- Moss, S.F.; Lee, J.W.; Sabo, E.; Rubin, A.K.; Rommel, J.; Westley, B.R.; May, F.E.B.; Gao, J.; Meitner, P.A.; Tavares, R.; et al. Decreased expression of gastrokine 1 and the trefoil factor interacting protein TFIZ1/GKN2 in gastric cancer: Influence of tumor histology and relationship to prognosis. Clin. Cancer Res. 2008, 14, 4161–4167. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Zhang, T.; Shi, Y.; Zhang, J.; Li, M.; Zhang, J.; Chen, X.; Ding, S. Helicobacter pylori inhibits GKN1 expression via the CagA/pERK/AUF1 pathway. Helicobacter 2020, 25, e12665. [Google Scholar] [CrossRef]
- Li, N.; Feng, Y.; Hu, Y.; He, C.; Xie, C.; Ouyang, Y.; Artim, S.C.; Huang, D.; Zhu, Y.; Luo, Z.; et al. Helicobacter pylori CagA promotes epithelial mesenchymal transition in gastric carcinogenesis via triggering oncogenic YAP pathway. J. Exp. Clin. Cancer Res. 2018, 37, 280. [Google Scholar] [CrossRef] [Green Version]
- Buti, L.; Ruiz-Puig, C.; Sangberg, D.; Leissing, T.M.; Brewer, R.C.; Owen, R.P.; Sgromo, B.; Royer, C.; Ebner, D.; Lu, X. CagA–ASPP2 complex mediates loss of cell polarity and favors H. pylori colonization of human gastric organoids. Proc. Natl. Acad. Sci. USA 2020, 117, 2645–2655. [Google Scholar] [CrossRef]
- Palrasu, M.; Zaika, E.; El-Rifai, W.; Garcia-Buitrago, M.; Piazuelo, M.B.; Wilson, K.T.; Peek, R.M., Jr.; Zaika, A.I. Bacterial CagA protein compromises tumor suppressor mechanisms in gastric epithelial cells. J. Clin. Investig. 2020, 130, 2422–2434. [Google Scholar] [CrossRef]
- Wroblewski, L.E.; Choi, E.; Petersen, C.; Delgado, A.G.; Piazuelo, M.B.; Romero-Gallo, J.; Lantz, T.L.; Zavros, Y.; Coffey, R.J.; Goldenring, J.R.; et al. Targeted mobilization of Lrig1 gastric epithelial stem cell populations by a carcinogenic Helicobacter pylori type IV secretion system. Proc. Natl. Acad. Sci. USA 2019, 116, 19652–19658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roncarati, D.; Scarlato, V. The Interplay between Two Transcriptional Repressors and Chaperones Orchestrates Helicobacter pylori Heat-Shock Response. Int. J. Mol. Sci. 2018, 19, 1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, B.J.; Gorrell, R.J.; Tafreshi, M.; Hatakeyama, M.; Kwok, T.; Price, J.T. The Helicobacter pylori cytotoxin CagA is essential for suppressing host heat shock protein expression. Cell Stress Chaperones 2016, 21, 523–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Axsen, W.S.; Styer, C.M.; Solnick, J.V. Inhibition of heat shock protein expression by Helicobacter pylori. Microb. Pathog. 2009, 47, 231–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Li, X.; Sun, F.; Tong, X.; Bai, Y.; Jin, K.; Liu, L.; Dai, F.; Li, N. HP-CagA+ Regulates the Expression of CDK4/CyclinD1 via reg3 to Change Cell Cycle and Promote Cell Proliferation. Int. J. Mol. Sci. 2020, 21, 224. [Google Scholar] [CrossRef] [Green Version]
- Waldum, H.L.; Qvigstad, G.; Sandvik, A.K. Reg protein in gastric cancer tumour cells. FEBS Lett. 2003, 553, 464–465. [Google Scholar] [CrossRef] [Green Version]
- Grainger, S.; Hryniuk, A.; Lohnes, D. Cdx1 and Cdx2 exhibit transcriptional specificity in the intestine. PLoS ONE 2013, 8, e54757. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.I.; Yoon, C.; Park, M.R.; Lee, D.; Kook, M.C.; Lin, J.X.; Kang, J.H.; Ashktorab, H.; Smoot, D.T.; Yoon, S.S.; et al. CDX1 expression induced by CagA-expressing Helicobacter pylori promotes gastric tumorigenesis. Mol. Cancer Res. 2019, 17, 2169–2183. [Google Scholar] [CrossRef] [Green Version]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Dalerba, P.; Dylla, S.J.; Park, I.K.; Liu, R.; Wang, X.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M.; et al. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10158–10163. [Google Scholar] [CrossRef] [Green Version]
- Ishimoto, T.; Nagano, O.; Yae, T.; Tamada, M.; Motohara, T.; Oshima, H.; Oshima, M.; Ikeda, T.; Asaba, R.; Yagi, H.; et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell 2011, 19, 387–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyoshi, S.; Tsugawa, H.; Matsuzaki, J.; Hirata, K.; Mori, H.; Saya, H.; Kanai, T.; Suzuki, H. Inhibiting xCT improves 5-fluorouracil resistance of gastric cancer induced by CD44 variant 9 expression. Anticancer Res. 2018, 38, 6163–6170. [Google Scholar] [CrossRef] [PubMed]
- Hirata, K.; Suzuki, H.; Imaeda, H.; Matsuzaki, J.; Tsugawa, H.; Nagano, O.; Asakura, K.; Saya, H.; Hibi, T. CD44 variant 9 expression in primary early gastric cancer as a predictive marker for recurrence. Br. J. Cancer 2013, 109, 379–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodama, H.; Murata, S.; Ishida, M.; Yamamoto, H.; Yamaguchi, T.; Kaida, S.; Miyake, T.; Takebayashi, K.; Kushima, R.; Tani, M. Prognostic impact of CD44-positive cancer stem-like cells at the invasive front of gastric cancer. Br. J. Cancer 2017, 116, 186–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamakawa, Y.; Kusuhara, M.; Terashima, M.; Kinugasa, Y.; Sugino, T.; Abe, M.; Mochizuki, T.; Hatakeyama, K.; Kami, K.; Yamaguchi, K. CD44 variant 9 expression as a predictor for gastric cancer recurrence: Immunohistochemical and metabolomic analysis of surgically resected tissues. Biomed. Res. 2017, 38, 41–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagnoli, F.; Buti, L.; Tompkins, L.; Covacci, A.; Amieva, M.R. Helicobacter pylori CagA induces a transition from polarized to invasive phenotypes in MDCK cells. Proc. Natl. Acad. Sci. USA 2005, 102, 16339–16344. [Google Scholar] [CrossRef] [Green Version]
- Tsugawa, H.; Suzuki, H.; Saya, H.; Hatakeyama, M.; Hirayama, T.; Hirata, K.; Nagano, O.; Matsuzaki, J.; Hibi, T. Reactive oxygen species-induced autophagic degradation of Helicobacter pylori CagA is specifically suppressed in cancer stem-like cells. Cell Host Microbe 2012, 12, 764–777. [Google Scholar] [CrossRef] [Green Version]
- Tsugawa, H.; Kato, C.; Mori, H.; Matsuzaki, J.; Kameyama, K.; Saya, H.; Hatakeyama, M.; Suematsu, M.; Suzuki, H. Cancer stem-cell marker CD44v9-positive cells arise from Helicobacter pylori- infected CAPZA1-overexpressing cells. Cell Mol. Gastroenterol. Hepatol. 2019, 8, 319–334. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Factors Involved | Mechanism | Outcomes | References |
|---|---|---|---|
| Induced EMT process | Cells undergoing EMT acquire cancer stem cell (CSC) properties | Enhances tumorigenesis | [89,90,91,92,93,94] |
| Snail1 protein reduces the glycogen synthase kinase-3 (GSK-3) activity | Enhances carcinogenesis | [95] | |
| YAP and TAZ | Failure in maintaining the organ size, tissue homeostasis, cell proliferation, and stem cell properties | Transformation of epithelial cells | [97,98,99,100] |
| Gastrokine 1 (GKN1) | CagA-induced activation of ERK pathway and AUF1 upregulation decreases the expression of GKN1 | Induces cell cycle progression and inhibits apoptosis, causing invasion and metastasis of the gastric cancer cell | [103,104,105,106] |
| Apoptosis-stimulating protein p53 2 | In association with CagA, disrupts the cellular polarity, damaging the mucosal barrier | Destroys the first-line defense mechanism causing survival of bacteria | [109] |
| Siva1 protein | Causes the activation of the PI3K/Akt pathway and XIAP E3 ubiquitin ligase | Tumorigenesis via the inhibition of apoptotic cell death, promoting the survival of damaged epithelial cells | [110] |
| Lrig1 | Precise mechanism is unknown | Promotes gastric carcinogenesis | [111] |
| Heat shock protein 1 (HSP1) | CagA mediates downregulation of HSP1 | Promotes persistent infection | [113,114] |
| Reg3 | In association with CagA, alters the cell cycle, reducing its control on development | Gastric carcinogenesis | [115,116] |
| Caudal type homeobox 1 (CDX1) | CagA induces expression of CDX1, which promotes the cell proliferation and replacement of gastric epithelial cells with intestine-specific cells | Gastric carcinogenesis and failure of common gastric cancer chemotherapies | [117,118] |
| CD44v9-positive cancer stem-like cells | Protects the accumulated CagA after its translocation from autophagic degradation | Causes the reprogramming and de-differentiation of the cells into cancer progenitor cells | [95,126,127] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ansari, S.; Yamaoka, Y. Helicobacter pylori Virulence Factor Cytotoxin-Associated Gene A (CagA)-Mediated Gastric Pathogenicity. Int. J. Mol. Sci. 2020, 21, 7430. https://doi.org/10.3390/ijms21197430
Ansari S, Yamaoka Y. Helicobacter pylori Virulence Factor Cytotoxin-Associated Gene A (CagA)-Mediated Gastric Pathogenicity. International Journal of Molecular Sciences. 2020; 21(19):7430. https://doi.org/10.3390/ijms21197430
Chicago/Turabian StyleAnsari, Shamshul, and Yoshio Yamaoka. 2020. "Helicobacter pylori Virulence Factor Cytotoxin-Associated Gene A (CagA)-Mediated Gastric Pathogenicity" International Journal of Molecular Sciences 21, no. 19: 7430. https://doi.org/10.3390/ijms21197430
APA StyleAnsari, S., & Yamaoka, Y. (2020). Helicobacter pylori Virulence Factor Cytotoxin-Associated Gene A (CagA)-Mediated Gastric Pathogenicity. International Journal of Molecular Sciences, 21(19), 7430. https://doi.org/10.3390/ijms21197430