Cell Cycle and DNA Repair Regulation in the Damage Response: Protein Phosphatases Take Over the Reins


Abstract
:1. Introduction
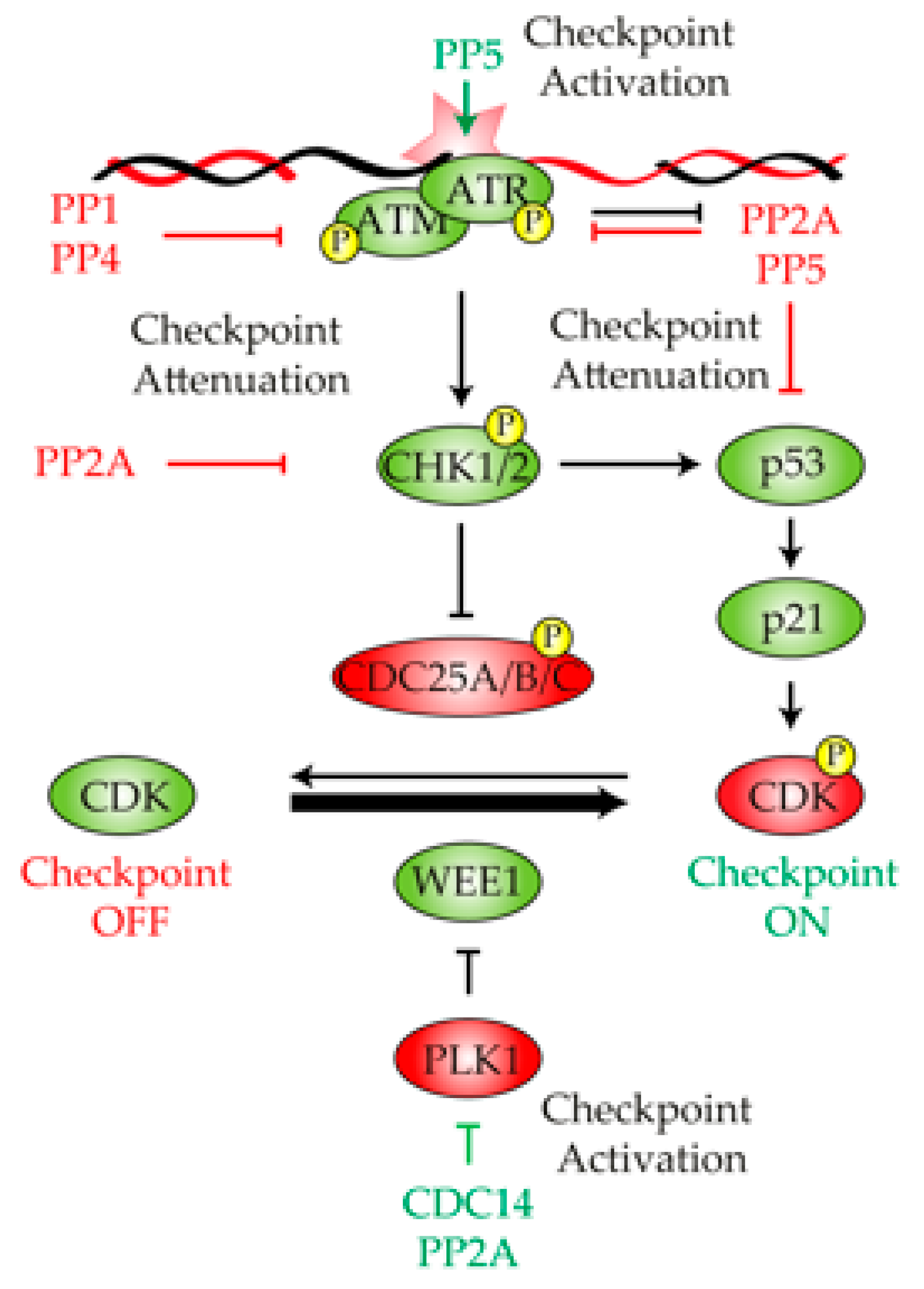
2. Protein Phosphatases in the Control of the DNA Damage Checkpoint
2.1. Protein Phosphatase 1
2.2. Protein Phosphatase 2A
2.3. Protein Phosphatase 4
2.4. Protein Phosphatase 5
2.5. CDC14
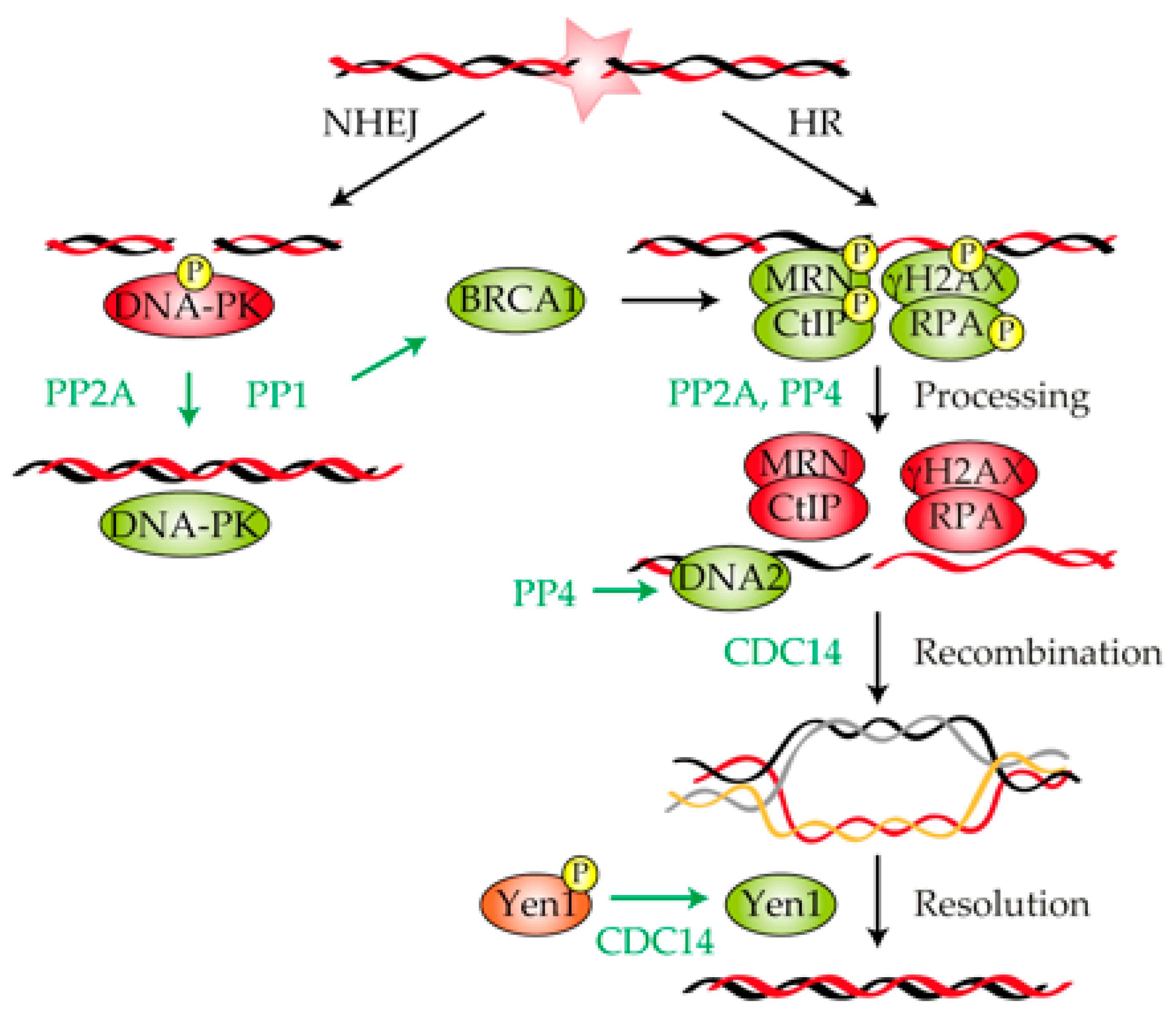
3. Role of Protein Phosphatases in the Repair of a DNA Lesion
3.1. Protein Phosphatase 1
3.2. Protein Phosphatase 2A
3.3. Protein Phosphatase 4
3.4. CDC14
4. Protein Phosphatases in DNA Damage Checkpoint Deactivation and Cell Cycle Recovery
4.1. Protein Phosphatase 1
4.2. Protein Phosphatase 2A
4.3. Protein Phosphatase 4
4.4. WIP1
5. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 4-NQO | 4-Nitroquinoline 1-oxide |
| BER | Base excision repair |
| CDK | Cyclin-dependent kinase |
| DDR | DNA damage response |
| DSB | Double-strand break |
| DUSP | Dual specificity phosphatase |
| HR | Homologous recombination |
| HU | Hydroxyurea |
| IR | Ionizing radiation |
| MEFs | Mouse embryonic fibroblasts |
| MMR | Mismatch repair |
| MMS | Methyl methanesulfonate |
| NER | Nucleotide excision repair |
| NHEJ | Non-homologous end joining |
| PIP | PP1’s interacting proteins |
| PPP | Phosphoprotein phosphatase |
| RPA | Replication protein A complex |
| SAC | Spindle assemble checkpoint |
| SPB | Spindle pole body |
| SSB | Single-strand break |
| ssDNA | Single-strand DNA |
| UV | Ultraviolet radiation |
References
- Deng, C.X.; Wang, R.H. Roles of BRCA1 in DNA damage repair: A link between development and cancer. Hum. Mol. Genet. 2003, 12, R113–R123. [Google Scholar] [CrossRef] [Green Version]
- O’Driscoll, M. Diseases associated with defective responses to DNA damage. Cold Spring Harb. Perspect. Biol. 2012, 4, a012773. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, I.; Yoshida, Y.; Suda, M.; Minamino, T. DNA damage response and metabolic disease. Cell Metab. 2014, 20, 967–977. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Shimada, M.; Nakanishi, M. Response to DNA damage: Why do we need to focus on protein phosphatases? Front. Oncol. 2013, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [CrossRef]
- Giglia-Mari, G.; Zotter, A.; Vermeulen, W. DNA damage response. Cold Spring Harb. Perspect. Biol. 2011, 3, a000745. [Google Scholar] [CrossRef]
- Bensimon, A.; Aebersold, R.; Shiloh, Y. Beyond ATM: The protein kinase landscape of the DNA damage response. FEBS Lett. 2011, 585, 1625–1639. [Google Scholar] [CrossRef] [Green Version]
- Trovesi, C.; Manfrini, N.; Falcettoni, M.; Longhese, M.P. Regulation of the DNA damage response by cyclin-dependent kinases. J. Mol. Biol. 2013, 425, 4756–4766. [Google Scholar] [CrossRef]
- Bakkenist, C.J.; Kastan, M.B. Phosphatases join kinases in DNA-damage response pathways. Trends Cell Biol. 2004, 14, 339–341. [Google Scholar] [CrossRef]
- Tercero, J.A.; Longhese, M.P.; Diffley, J.F. A central role for DNA replication forks in checkpoint activation and response. Mol. Cell 2003, 11, 1323–1336. [Google Scholar] [CrossRef]
- Ariza, R.R.; Keyse, S.M.; Moggs, J.G.; Wood, R.D. Reversible protein phosphorylation modulates nucleotide excision repair of damaged DNA by human cell extracts. Nucleic Acids Res. 1996, 24, 433–440. [Google Scholar] [CrossRef] [Green Version]
- Alonso, A.; Sasin, J.; Bottini, N.; Friedberg, I.; Friedberg, I.; Osterman, A.; Godzik, A.; Hunter, T.; Dixon, J.; Mustelin, T. Protein tyrosine phosphatases in the human genome. Cell 2004, 117, 699–711. [Google Scholar] [CrossRef] [Green Version]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [Green Version]
- Sacco, F.; Perfetto, L.; Castagnoli, L.; Cesareni, G. The human phosphatase interactome: An intricate family portrait. FEBS Lett. 2012, 586, 2732–2739. [Google Scholar] [CrossRef]
- Virshup, D.M.; Shenolikar, S. From promiscuity to precision: Protein phosphatases get a makeover. Mol. Cell 2009, 33, 537–545. [Google Scholar] [CrossRef]
- Lee, D.H.; Chowdhury, D. What goes on must come off: Phosphatases gate-crash the DNA damage response. Trends Biochem. Sci. 2011, 36, 569–577. [Google Scholar] [CrossRef] [Green Version]
- Blanco, M.G.; Matos, J.; West, S.C. Dual control of Yen1 nuclease activity and cellular localization by Cdk and Cdc14 prevents genome instability. Mol. Cell 2014, 54, 94–106. [Google Scholar] [CrossRef] [Green Version]
- Eissler, C.L.; Mazon, G.; Powers, B.L.; Savinov, S.N.; Symington, L.S.; Hall, M.C. The Cdk/cDc14 module controls activation of the Yen1 holliday junction resolvase to promote genome stability. Mol. Cell 2014, 54, 80–93. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Luis, J.; Clemente-Blanco, A.; Aragon, L.; Machin, F. Cdc14 targets the Holliday junction resolvase Yen1 to the nucleus in early anaphase. Cell Cycle 2014, 13, 1392–1399. [Google Scholar] [CrossRef] [Green Version]
- Villoria, M.T.; Gutierrez-Escribano, P.; Alonso-Rodriguez, E.; Ramos, F.; Merino, E.; Campos, A.; Montoya, A.; Kramer, H.; Aragon, L.; Clemente-Blanco, A. PP4 phosphatase cooperates in recombinational DNA repair by enhancing double-strand break end resection. Nucleic Acids Res. 2019, 47, 10706–10727. [Google Scholar] [CrossRef] [Green Version]
- Villoria, M.T.; Ramos, F.; Duenas, E.; Faull, P.; Cutillas, P.R.; Clemente-Blanco, A. Stabilization of the metaphase spindle by Cdc14 is required for recombinational DNA repair. EMBO J. 2017, 36, 79–101. [Google Scholar] [CrossRef]
- Kuntziger, T.; Landsverk, H.B.; Collas, P.; Syljuasen, R.G. Protein phosphatase 1 regulators in DNA damage signaling. Cell Cycle 2011, 10, 1356–1362. [Google Scholar] [CrossRef] [Green Version]
- Durocher, D.; Jackson, S.P. DNA-PK, ATM and ATR as sensors of DNA damage: Variations on a theme? Curr. Opin. Cell Biol. 2001, 13, 225–231. [Google Scholar] [CrossRef]
- Zou, L.; Elledge, S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003, 300, 1542–1548. [Google Scholar] [CrossRef] [Green Version]
- Ira, G.; Pellicioli, A.; Balijja, A.; Wang, X.; Fiorani, S.; Carotenuto, W.; Liberi, G.; Bressan, D.; Wan, L.; Hollingsworth, N.M.; et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 2004, 431, 1011–1017. [Google Scholar] [CrossRef] [Green Version]
- Raleigh, J.M.; O’Connell, M.J. The G(2) DNA damage checkpoint targets both Wee1 and Cdc25. J. Cell Sci. 2000, 113 Pt 10, 1727–1736. [Google Scholar]
- Lee, J.; Kumagai, A.; Dunphy, W.G. Positive regulation of Wee1 by Chk1 and 14-3-3 proteins. Mol. Biol. Cell 2001, 12, 551–563. [Google Scholar] [CrossRef] [Green Version]
- O’Connell, M.J.; Raleigh, J.M.; Verkade, H.M.; Nurse, P. Chk1 is a wee1 kinase in the G2 DNA damage checkpoint inhibiting cdc2 by Y15 phosphorylation. EMBO J. 1997, 16, 545–554. [Google Scholar] [CrossRef] [Green Version]
- Mailand, N.; Podtelejnikov, A.V.; Groth, A.; Mann, M.; Bartek, J.; Lukas, J. Regulation of G(2)/M events by Cdc25A through phosphorylation-dependent modulation of its stability. EMBO J. 2002, 21, 5911–5920. [Google Scholar] [CrossRef]
- Goldstone, S.; Pavey, S.; Forrest, A.; Sinnamon, J.; Gabrielli, B. Cdc25-dependent activation of cyclin A/cdk2 is blocked in G2 phase arrested cells independently of ATM/ATR. Oncogene 2001, 20, 921–932. [Google Scholar] [CrossRef] [Green Version]
- Heroes, E.; Lesage, B.; Gornemann, J.; Beullens, M.; Van Meervelt, L.; Bollen, M. The PP1 binding code: A molecular-lego strategy that governs specificity. FEBS J. 2013, 280, 584–595. [Google Scholar] [CrossRef]
- Bollen, M.; Peti, W.; Ragusa, M.J.; Beullens, M. The extended PP1 toolkit: Designed to create specificity. Trends Biochem. Sci. 2010, 35, 450–458. [Google Scholar] [CrossRef] [Green Version]
- Egloff, M.P.; Johnson, D.F.; Moorhead, G.; Cohen, P.T.; Cohen, P.; Barford, D. Structural basis for the recognition of regulatory subunits by the catalytic subunit of protein phosphatase 1. EMBO J. 1997, 16, 1876–1887. [Google Scholar] [CrossRef]
- Cannon, J.F. Function of protein phosphatase-1, Glc7, in Saccharomyces cerevisiae. Adv. Appl. Microbiol. 2010, 73, 27–59. [Google Scholar] [CrossRef]
- Pinsky, B.A.; Kotwaliwale, C.V.; Tatsutani, S.Y.; Breed, C.A.; Biggins, S. Glc7/protein phosphatase 1 regulatory subunits can oppose the Ipl1/aurora protein kinase by redistributing Glc7. Mol. Cell Biol. 2006, 26, 2648–2660. [Google Scholar] [CrossRef] [Green Version]
- Marquina, M.; Queralt, E.; Casamayor, A.; Arino, J. Lack of the Glc7 phosphatase regulatory subunit Ypi1 activates the morphogenetic checkpoint. Int. J. Biochem. Cell Biol. 2012, 44, 1862–1871. [Google Scholar] [CrossRef]
- Trinkle-Mulcahy, L.; Andersen, J.; Lam, Y.W.; Moorhead, G.; Mann, M.; Lamond, A.I. Repo-Man recruits PP1 gamma to chromatin and is essential for cell viability. J. Cell Biol. 2006, 172, 679–692. [Google Scholar] [CrossRef] [Green Version]
- Vagnarelli, P.; Hudson, D.F.; Ribeiro, S.A.; Trinkle-Mulcahy, L.; Spence, J.M.; Lai, F.; Farr, C.J.; Lamond, A.I.; Earnshaw, W.C. Condensin and Repo-Man-PP1 co-operate in the regulation of chromosome architecture during mitosis. Nat. Cell Biol. 2006, 8, 1133–1142. [Google Scholar] [CrossRef]
- Peng, A.; Yamamoto, T.M.; Goldberg, M.L.; Maller, J.L. A novel role for greatwall kinase in recovery from DNA damage. Cell Cycle 2010, 9, 4364–4369. [Google Scholar] [CrossRef] [Green Version]
- Peng, A.; Lewellyn, A.L.; Schiemann, W.P.; Maller, J.L. Repo-man controls a protein phosphatase 1-dependent threshold for DNA damage checkpoint activation. Curr. Biol. 2010, 20, 387–396. [Google Scholar] [CrossRef] [Green Version]
- Allen, P.B.; Kwon, Y.G.; Nairn, A.C.; Greengard, P. Isolation and characterization of PNUTS, a putative protein phosphatase 1 nuclear targeting subunit. J. Biol. Chem. 1998, 273, 4089–4095. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Lee, J.K.; Maeng, Y.S.; Kim, Y.M.; Kwon, Y.G. Langerhans cell protein 1 (LCP1) binds to PNUTS in the nucleus: Implications for this complex in transcriptional regulation. Exp. Mol. Med. 2009, 41, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Rose, M.; Dutting, E.; Schroder, N.; Sticht, H.; Brandstatter, J.H.; Enz, R. PNUTS forms a trimeric protein complex with GABA(C) receptors and protein phosphatase 1. Mol. Cell Neurosci. 2008, 37, 808–819. [Google Scholar] [CrossRef]
- Landsverk, H.B.; Kirkhus, M.; Bollen, M.; Kuntziger, T.; Collas, P. PNUTS enhances in vitro chromosome decondensation in a PP1-dependent manner. Biochem. J. 2005, 390, 709–717. [Google Scholar] [CrossRef] [Green Version]
- Landsverk, H.B.; Mora-Bermudez, F.; Landsverk, O.J.; Hasvold, G.; Naderi, S.; Bakke, O.; Ellenberg, J.; Collas, P.; Syljuasen, R.G.; Kuntziger, T. The protein phosphatase 1 regulator PNUTS is a new component of the DNA damage response. EMBO Rep. 2010, 11, 868–875. [Google Scholar] [CrossRef] [Green Version]
- Eichhorn, P.J.; Creyghton, M.P.; Bernards, R. Protein phosphatase 2A regulatory subunits and cancer. Biochim. Biophys. Acta 2009, 1795, 1–15. [Google Scholar] [CrossRef]
- Seshacharyulu, P.; Pandey, P.; Datta, K.; Batra, S.K. Phosphatase: PP2A structural importance, regulation and its aberrant expression in cancer. Cancer Lett. 2013, 335, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Janssens, V.; Goris, J. Protein phosphatase 2A: A highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J. 2001, 353, 417–439. [Google Scholar] [CrossRef]
- Janssens, V.; Longin, S.; Goris, J. PP2A holoenzyme assembly: In cauda venenum (the sting is in the tail). Trends Biochem. Sci. 2008, 33, 113–121. [Google Scholar] [CrossRef]
- Li, H.H.; Cai, X.; Shouse, G.P.; Piluso, L.G.; Liu, X. A specific PP2A regulatory subunit, B56gamma, mediates DNA damage-induced dephosphorylation of p53 at Thr55. EMBO J. 2007, 26, 402–411. [Google Scholar] [CrossRef]
- McCright, B.; Rivers, A.M.; Audlin, S.; Virshup, D.M. The B56 family of protein phosphatase 2A (PP2A) regulatory subunits encodes differentiation-induced phosphoproteins that target PP2A to both nucleus and cytoplasm. J. Biol. Chem. 1996, 271, 22081–22089. [Google Scholar] [CrossRef] [Green Version]
- Goodarzi, A.A.; Jonnalagadda, J.C.; Douglas, P.; Young, D.; Ye, R.; Moorhead, G.B.; Lees-Miller, S.P.; Khanna, K.K. Autophosphorylation of ataxia-telangiectasia mutated is regulated by protein phosphatase 2A. EMBO J. 2004, 23, 4451–4461. [Google Scholar] [CrossRef] [Green Version]
- Bialojan, C.; Takai, A. Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Specificity and kinetics. Biochem. J. 1988, 256, 283–290. [Google Scholar] [CrossRef]
- Awasthi, P.; Foiani, M.; Kumar, A. ATM and ATR signaling at a glance. J. Cell Sci. 2015, 128, 4255–4262. [Google Scholar] [CrossRef] [Green Version]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef]
- Goder, A.; Emmerich, C.; Nikolova, T.; Kiweler, N.; Schreiber, M.; Kuhl, T.; Imhof, D.; Christmann, M.; Heinzel, T.; Schneider, G.; et al. HDAC1 and HDAC2 integrate checkpoint kinase phosphorylation and cell fate through the phosphatase-2A subunit PR130. Nat. Commun. 2018, 9, 764. [Google Scholar] [CrossRef] [Green Version]
- Shouse, G.P.; Nobumori, Y.; Panowicz, M.J.; Liu, X. ATM-mediated phosphorylation activates the tumor-suppressive function of B56gamma-PP2A. Oncogene 2011, 30, 3755–3765. [Google Scholar] [CrossRef] [Green Version]
- Margolis, S.S.; Perry, J.A.; Forester, C.M.; Nutt, L.K.; Guo, Y.; Jardim, M.J.; Thomenius, M.J.; Freel, C.D.; Darbandi, R.; Ahn, J.H.; et al. Role for the PP2A/B56delta phosphatase in regulating 14-3-3 release from Cdc25 to control mitosis. Cell 2006, 127, 759–773. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Cao, P.T.; Greer, P.M.; Nagengast, E.S.; Kolb, R.H.; Mumby, M.C.; Cowan, K.H. Protein phosphatase 2A has an essential role in the activation of gamma-irradiation-induced G2/M checkpoint response. Oncogene 2010, 29, 4317–4329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dozier, C.; Bonyadi, M.; Baricault, L.; Tonasso, L.; Darbon, J.M. Regulation of Chk2 phosphorylation by interaction with protein phosphatase 2A via its B’ regulatory subunit. Biol. Cell 2004, 96, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Freeman, A.K.; Dapic, V.; Monteiro, A.N. Negative regulation of CHK2 activity by protein phosphatase 2A is modulated by DNA damage. Cell Cycle 2010, 9, 736–747. [Google Scholar] [CrossRef]
- Liang, X.; Reed, E.; Yu, J.J. Protein phosphatase 2A interacts with Chk2 and regulates phosphorylation at Thr-68 after cisplatin treatment of human ovarian cancer cells. Int. J. Mol. Med. 2006, 17, 703–708. [Google Scholar] [CrossRef] [Green Version]
- Dohoney, K.M.; Guillerm, C.; Whiteford, C.; Elbi, C.; Lambert, P.F.; Hager, G.L.; Brady, J.N. Phosphorylation of p53 at serine 37 is important for transcriptional activity and regulation in response to DNA damage. Oncogene 2004, 23, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Jang, Y.J.; Ji, J.H.; Choi, Y.C.; Ryu, C.J.; Ko, S.Y. Regulation of Polo-like kinase 1 by DNA damage in mitosis. Inhibition of mitotic PLK-1 by protein phosphatase 2A. J. Biol. Chem. 2007, 282, 2473–2482. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Guo, Q.; Fisher, L.A.; Liu, D.; Peng, A. Regulation of polo-like kinase 1 by DNA damage and PP2A/B55alpha. Cell Cycle 2015, 14, 157–166. [Google Scholar] [CrossRef] [Green Version]
- Brewis, N.D.; Cohen, P.T. Protein phosphatase X has been highly conserved during mammalian evolution. Biochim. Biophys. Acta 1992, 1171, 231–233. [Google Scholar] [CrossRef]
- Da Cruz e Silva, O.B.; da Cruz e Silva, E.F.; Cohen, P.T. Identification of a novel protein phosphatase catalytic subunit by cDNA cloning. FEBS Lett. 1988, 242, 106–110. [Google Scholar] [CrossRef] [Green Version]
- Ramos, F.; Villoria, M.T.; Alonso-Rodriguez, E.; Clemente-Blanco, A. Role of protein phosphatases PP1, PP2A, PP4 and Cdc14 in the DNA damage response. Cell Stress 2019, 3, 70–85. [Google Scholar] [CrossRef] [Green Version]
- Paciotti, V.; Clerici, M.; Scotti, M.; Lucchini, G.; Longhese, M.P. Characterization of mec1 kinase-deficient mutants and of new hypomorphic mec1 alleles impairing subsets of the DNA damage response pathway. Mol. Cell Biol. 2001, 21, 3913–3925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hustedt, N.; Seeber, A.; Sack, R.; Tsai-Pflugfelder, M.; Bhullar, B.; Vlaming, H.; van Leeuwen, F.; Guenole, A.; van Attikum, H.; Srivas, R.; et al. Yeast PP4 interacts with ATR homolog Ddc2-Mec1 and regulates checkpoint signaling. Mol. Cell 2015, 57, 273–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinds, T.D., Jr.; Sanchez, E.R. Protein phosphatase 5. Int. J. Biochem. Cell Biol. 2008, 40, 2358–2362. [Google Scholar] [CrossRef] [PubMed]
- Becker, W.; Kentrup, H.; Klumpp, S.; Schultz, J.E.; Joost, H.G. Molecular cloning of a protein serine/threonine phosphatase containing a putative regulatory tetratricopeptide repeat domain. J. Biol. Chem. 1994, 269, 22586–22592. [Google Scholar]
- Chen, M.X.; McPartlin, A.E.; Brown, L.; Chen, Y.H.; Barker, H.M.; Cohen, P.T. A novel human protein serine/threonine phosphatase, which possesses four tetratricopeptide repeat motifs and localizes to the nucleus. EMBO J. 1994, 13, 4278–4290. [Google Scholar] [CrossRef]
- Chinkers, M. Targeting of a distinctive protein-serine phosphatase to the protein kinase-like domain of the atrial natriuretic peptide receptor. Proc. Natl. Acad. Sci. USA 1994, 91, 11075–11079. [Google Scholar] [CrossRef] [Green Version]
- Oberoi, J.; Dunn, D.M.; Woodford, M.R.; Mariotti, L.; Schulman, J.; Bourboulia, D.; Mollapour, M.; Vaughan, C.K. Structural and functional basis of protein phosphatase 5 substrate specificity. Proc. Natl. Acad. Sci. USA 2016, 113, 9009–9014. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.; Zhang, J.; Bao, S.; Liu, I.; Otterness, D.; Dean, N.M.; Abraham, R.T.; Wang, X.F. Requirement of protein phosphatase 5 in DNA-damage-induced ATM activation. Genes Dev. 2004, 18, 249–254. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Bao, S.; Furumai, R.; Kucera, K.S.; Ali, A.; Dean, N.M.; Wang, X.F. Protein phosphatase 5 is required for ATR-mediated checkpoint activation. Mol. Cell Biol. 2005, 25, 9910–9919. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.; Cheong, H.M.; Lee, J.H.; Song, P.I.; Lee, K.H.; Kim, S.Y.; Jun, J.Y.; You, H.J. Protein phosphatase 5 is necessary for ATR-mediated DNA repair. Biochem. Biophys. Res. Commun. 2011, 404, 476–481. [Google Scholar] [CrossRef]
- Yong, W.; Bao, S.; Chen, H.; Li, D.; Sanchez, E.R.; Shou, W. Mice lacking protein phosphatase 5 are defective in ataxia telangiectasia mutated (ATM)-mediated cell cycle arrest. J. Biol. Chem. 2007, 282, 14690–14694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Shen, T.; Zhu, W.; Dou, L.; Gu, H.; Zhang, L.; Yang, Z.; Chen, H.; Zhou, Q.; Sanchez, E.R.; et al. Protein phosphatase 5 and the tumor suppressor p53 down-regulate each other’s activities in mice. J. Biol. Chem. 2018, 293, 18218–18229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amable, L.; Grankvist, N.; Largen, J.W.; Ortsater, H.; Sjoholm, A.; Honkanen, R.E. Disruption of serine/threonine protein phosphatase 5 (PP5:PPP5c) in mice reveals a novel role for PP5 in the regulation of ultraviolet light-induced phosphorylation of serine/threonine protein kinase Chk1 (CHEK1). J. Biol. Chem. 2011, 286, 40413–40422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wechsler, T.; Chen, B.P.; Harper, R.; Morotomi-Yano, K.; Huang, B.C.; Meek, K.; Cleaver, J.E.; Chen, D.J.; Wabl, M. DNA-PKcs function regulated specifically by protein phosphatase 5. Proc. Natl. Acad. Sci. USA 2004, 101, 1247–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bremmer, S.C.; Hall, H.; Martinez, J.S.; Eissler, C.L.; Hinrichsen, T.H.; Rossie, S.; Parker, L.L.; Hall, M.C.; Charbonneau, H. Cdc14 phosphatases preferentially dephosphorylate a subset of cyclin-dependent kinase (Cdk) sites containing phosphoserine. J. Biol. Chem. 2012, 287, 1662–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meitinger, F.; Palani, S.; Pereira, G. The power of MEN in cytokinesis. Cell Cycle 2012, 11, 219–228. [Google Scholar] [CrossRef] [Green Version]
- Meitinger, F.; Petrova, B.; Lombardi, I.; Bertazzi, D.; Hub, B.; Zentgraf, H.; Pereira, G. Targeted localization of Inn1, Cyk3 and Chs2 by the mitotic-exit network regulates cytokinesis in budding yeast. J. Cell Sci. 2010, 123, 1851–1861. [Google Scholar] [CrossRef] [Green Version]
- Clemente-Blanco, A.; Mayán-Santos, M.; Schneider, D.; Machín, F.; Jarmuz, A.; Tschochner, H.; Aragón, L. Cdc14 inhibits transcription by RNA polymerase I during anaphase. Nature 2009, 458, 219–222. [Google Scholar] [CrossRef]
- Mocciaro, A.; Schiebel, E. Cdc14: A highly conserved family of phosphatases with non-conserved functions? J. Cell Sci. 2010, 123, 2867–2876. [Google Scholar] [CrossRef] [Green Version]
- Clemente-Blanco, A.; Sen, N.; Mayan-Santos, M.; Sacristan, M.P.; Graham, B.; Jarmuz, A.; Giess, A.; Webb, E.; Game, L.; Eick, D.; et al. Cdc14 phosphatase promotes segregation of telomeres through repression of RNA polymerase II transcription. Nat. Cell Biol. 2011, 13, 1450–1456. [Google Scholar] [CrossRef] [Green Version]
- Guillamot, M.; Manchado, E.; Chiesa, M.; Gomez-Lopez, G.; Pisano, D.G.; Sacristan, M.P.; Malumbres, M. Cdc14b regulates mammalian RNA polymerase II and represses cell cycle transcription. Sci. Rep. 2011, 1, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clement, A.; Solnica-Krezel, L.; Gould, K.L. The Cdc14B phosphatase contributes to ciliogenesis in zebrafish. Development 2011, 138, 291–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz-Cuervo, H.; Bueno, A. Cds1 controls the release of Cdc14-like phosphatase Flp1 from the nucleolus to drive full activation of the checkpoint response to replication stress in fission yeast. Mol. Biol. Cell 2008, 19, 2488–2499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassermann, F.; Frescas, D.; Guardavaccaro, D.; Busino, L.; Peschiaroli, A.; Pagano, M. The Cdc14B-Cdh1-Plk1 axis controls the G2 DNA-damage-response checkpoint. Cell 2008, 134, 256–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mocciaro, A.; Berdougo, E.; Zeng, K.; Black, E.; Vagnarelli, P.; Earnshaw, W.; Gillespie, D.; Jallepalli, P.; Schiebel, E. Vertebrate cells genetically deficient for Cdc14A or Cdc14B retain DNA damage checkpoint proficiency but are impaired in DNA repair. J. Cell Biol. 2010, 189, 631–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Z.; Peddibhotla, S.; Lin, H.; Fang, X.; Li, M.; Rosen, J.M.; Zhang, P. Early-onset aging and defective DNA damage response in Cdc14b-deficient mice. Mol. Cell Biol. 2011, 31, 1470–1477. [Google Scholar] [CrossRef] [Green Version]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2015, 26, 52–64. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Huang, J. DNA End Resection: Facts and Mechanisms. Genom. Proteom. Bioinform. 2016, 14, 126–130. [Google Scholar] [CrossRef] [Green Version]
- Longhese, M.P.; Bonetti, D.; Manfrini, N.; Clerici, M. Mechanisms and regulation of DNA end resection. EMBO J. 2010, 29, 2864–2874. [Google Scholar] [CrossRef] [Green Version]
- Symington, L.S. Mechanism and regulation of DNA end resection in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 195–212. [Google Scholar] [CrossRef] [Green Version]
- Haber, J.E. DNA Repair: The Search for Homology. Bioessays 2018, 40, e1700229. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, S.N.; Kachnic, L.A. Roles of BRCA1 and BRCA2 in homologous recombination, DNA replication fidelity and the cellular response to ionizing radiation. Oncogene 2003, 22, 5784–5791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Densham, R.M.; Morris, J.R. Moving Mountains-The BRCA1 Promotion of DNA Resection. Front. Mol. Biosci. 2019, 6, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huen, M.S.; Sy, S.M.; Chen, J. BRCA1 and its toolbox for the maintenance of genome integrity. Nat. Rev. Mol. Cell Biol. 2010, 11, 138–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Chen, J. DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol. Cell Biol. 2004, 24, 9478–9486. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Nievera, C.J.; Lee, A.Y.; Wu, X. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J. Biol. Chem. 2008, 283, 7713–7720. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Virshup, D.M.; White, R.L.; Hsu, L.C. Regulation of BRCA1 phosphorylation by interaction with protein phosphatase 1alpha. Cancer Res. 2002, 62, 6357–6361. [Google Scholar]
- Yu, Y.M.; Pace, S.M.; Allen, S.R.; Deng, C.X.; Hsu, L.C. A PP1-binding motif present in BRCA1 plays a role in its DNA repair function. Int. J. Biol. Sci. 2008, 4, 352–361. [Google Scholar] [CrossRef]
- Hsu, L.C. Identification and functional characterization of a PP1-binding site in BRCA1. Biochem. Biophys. Res. Commun. 2007, 360, 507–512. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Fisher, L.A.; Bessho, T.; Peng, A. Protein phosphatase 1 and phosphatase 1 nuclear targeting subunit-dependent regulation of DNA-dependent protein kinase and non-homologous end joining. Nucleic Acids Res. 2017, 45, 10583–10594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, C.; Rouget, R.; Wu, D.; Beullens, M.; Van Eynde, A.; Bollen, M. Overexpression of PP1-NIPP1 limits the capacity of cells to repair DNA double-strand breaks. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, P.; Moorhead, G.B.; Ye, R.; Lees-Miller, S.P. Protein phosphatases regulate DNA-dependent protein kinase activity. J. Biol. Chem. 2001, 276, 18992–18998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merigliano, C.; Marzio, A.; Renda, F.; Somma, M.P.; Gatti, M.; Verni, F. A Role for the Twins Protein Phosphatase (PP2A-B55) in the Maintenance of Drosophila Genome Integrity. Genetics 2017, 205, 1151–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, D.; Keogh, M.C.; Ishii, H.; Peterson, C.L.; Buratowski, S.; Lieberman, J. gamma-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol. Cell 2005, 20, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Nan, A.; Xiao, Y.; Chen, Y.; Lai, Y. PP2A-B56 complex is involved in dephosphorylation of gamma-H2AX in the repair process of CPT-induced DNA double-strand breaks. Toxicology 2015, 331, 57–65. [Google Scholar] [CrossRef]
- Feng, J.; Wakeman, T.; Yong, S.; Wu, X.; Kornbluth, S.; Wang, X.F. Protein phosphatase 2A-dependent dephosphorylation of replication protein A is required for the repair of DNA breaks induced by replication stress. Mol. Cell Biol. 2009, 29, 5696–5709. [Google Scholar] [CrossRef] [Green Version]
- Kalev, P.; Simicek, M.; Vazquez, I.; Munck, S.; Chen, L.; Soin, T.; Danda, N.; Chen, W.; Sablina, A. Loss of PPP2R2A inhibits homologous recombination DNA repair and predicts tumor sensitivity to PARP inhibition. Cancer Res. 2012, 72, 6414–6424. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, B.M.; Szyjka, S.J.; Lis, E.T.; Bailey, A.O.; Yates, J.R., 3rd; Aparicio, O.M.; Romesberg, F.E. Pph3-Psy2 is a phosphatase complex required for Rad53 dephosphorylation and replication fork restart during recovery from DNA damage. Proc. Natl. Acad. Sci. USA 2007, 104, 9290–9295. [Google Scholar] [CrossRef] [Green Version]
- Keogh, M.C.; Kim, J.A.; Downey, M.; Fillingham, J.; Chowdhury, D.; Harrison, J.C.; Onishi, M.; Datta, N.; Galicia, S.; Emili, A.; et al. A phosphatase complex that dephosphorylates gammaH2AX regulates DNA damage checkpoint recovery. Nature 2006, 439, 497–501. [Google Scholar] [CrossRef]
- Kim, J.A.; Hicks, W.M.; Li, J.; Tay, S.Y.; Haber, J.E. Protein phosphatases pph3, ptc2, and ptc3 play redundant roles in DNA double-strand break repair by homologous recombination. Mol. Cell Biol. 2011, 31, 507–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakada, S.; Chen, G.I.; Gingras, A.C.; Durocher, D. PP4 is a gamma H2AX phosphatase required for recovery from the DNA damage checkpoint. EMBO Rep. 2008, 9, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, D.; Xu, X.; Zhong, X.; Ahmed, F.; Zhong, J.; Liao, J.; Dykxhoorn, D.M.; Weinstock, D.M.; Pfeifer, G.P.; Lieberman, J. A PP4-phosphatase complex dephosphorylates gamma-H2AX generated during DNA replication. Mol. Cell 2008, 31, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.H.; Pan, Y.; Kanner, S.; Sung, P.; Borowiec, J.A.; Chowdhury, D. A PP4 phosphatase complex dephosphorylates RPA2 to facilitate DNA repair via homologous recombination. Nat. Struct. Mol. Biol. 2010, 17, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Dubrana, K.; van Attikum, H.; Hediger, F.; Gasser, S.M. The processing of double-strand breaks and binding of single-strand-binding proteins RPA and Rad51 modulate the formation of ATR-kinase foci in yeast. J. Cell Sci. 2007, 120, 4209–4220. [Google Scholar] [CrossRef] [Green Version]
- Sigurdsson, S.; Van Komen, S.; Bussen, W.; Schild, D.; Albala, J.S.; Sung, P. Mediator function of the human Rad51B-Rad51C complex in Rad51/RPA-catalyzed DNA strand exchange. Genes Dev. 2001, 15, 3308–3318. [Google Scholar] [CrossRef] [Green Version]
- Sung, P. Function of yeast Rad52 protein as a mediator between replication protein A and the Rad51 recombinase. J. Biol. Chem. 1997, 272, 28194–28197. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Ha, K.; Lu, G.; Fang, X.; Cheng, R.; Zuo, Q.; Zhang, P. Cdc14A and Cdc14B Redundantly Regulate DNA Double-Strand Break Repair. Mol. Cell Biol. 2015, 35, 3657–3668. [Google Scholar] [CrossRef] [Green Version]
- Broadus, M.R.; Gould, K.L. Multiple protein kinases influence the redistribution of fission yeast Clp1/Cdc14 phosphatase upon genotoxic stress. Mol. Biol. Cell 2012, 23, 4118–4128. [Google Scholar] [CrossRef]
- Mamely, I.; van Vugt, M.A.; Smits, V.A.; Semple, J.I.; Lemmens, B.; Perrakis, A.; Medema, R.H.; Freire, R. Polo-like kinase-1 controls proteasome-dependent degradation of Claspin during checkpoint recovery. Curr. Biol. 2006, 16, 1950–1955. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, N.; Arai, H.; Nishihara, Y.; Taniguchi, M.; Watanabe, N.; Hunter, T.; Osada, H. M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFbeta-TrCP. Proc. Natl. Acad. Sci. USA 2004, 101, 4419–4424. [Google Scholar] [CrossRef] [Green Version]
- Den Elzen, N.R.; O’Connell, M.J. Recovery from DNA damage checkpoint arrest by PP1-mediated inhibition of Chk1. EMBO J. 2004, 23, 908–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazzi, M.; Mantiero, D.; Trovesi, C.; Lucchini, G.; Longhese, M.P. Dephosphorylation of gamma H2A by Glc7/protein phosphatase 1 promotes recovery from inhibition of DNA replication. Mol. Cell Biol. 2010, 30, 131–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, G.; Wan, J.; Mu, C.; Liu, Q.; Wang, Y.; Sang, J. Sds22 participates in Glc7 mediated Rad53 dephosphorylation in MMS-induced DNA damage in Candida albicans. Fungal Genet. Biol. 2016, 93, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Li, D.W.; Liu, J.P.; Schmid, P.C.; Schlosser, R.; Feng, H.; Liu, W.B.; Yan, Q.; Gong, L.; Sun, S.M.; Deng, M.; et al. Protein serine/threonine phosphatase-1 dephosphorylates p53 at Ser-15 and Ser-37 to modulate its transcriptional and apoptotic activities. Oncogene 2006, 25, 3006–3022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haneda, M.; Kojima, E.; Nishikimi, A.; Hasegawa, T.; Nakashima, I.; Isobe, K. Protein phosphatase 1, but not protein phosphatase 2A, dephosphorylates DNA-damaging stress-induced phospho-serine 15 of p53. FEBS Lett. 2004, 567, 171–174. [Google Scholar] [CrossRef] [Green Version]
- Helps, N.R.; Barker, H.M.; Elledge, S.J.; Cohen, P.T. Protein phosphatase 1 interacts with p53BP2, a protein which binds to the tumour suppressor p53. FEBS Lett. 1995, 377, 295–300. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Lim, C.J.; Min, J.K.; Lee, J.K.; Kim, Y.M.; Lee, J.Y.; Won, M.H.; Kwon, Y.G. Protein phosphatase 1 nuclear targeting subunit is a hypoxia inducible gene: Its role in post-translational modification of p53 and MDM2. Cell Death Differ. 2007, 14, 1106–1116. [Google Scholar] [CrossRef] [Green Version]
- Margolis, S.S.; Perry, J.A.; Weitzel, D.H.; Freel, C.D.; Yoshida, M.; Haystead, T.A.; Kornbluth, S. A role for PP1 in the Cdc2/Cyclin B-mediated positive feedback activation of Cdc25. Mol. Biol. Cell 2006, 17, 1779–1789. [Google Scholar] [CrossRef] [Green Version]
- Millar, J.B.; Blevitt, J.; Gerace, L.; Sadhu, K.; Featherstone, C.; Russell, P. p55CDC25 is a nuclear protein required for the initiation of mitosis in human cells. Proc. Natl. Acad. Sci. USA 1991, 88, 10500–10504. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, N.; Ohkura, H.; Yanagida, M. Distinct, essential roles of type 1 and 2A protein phosphatases in the control of the fission yeast cell division cycle. Cell 1990, 63, 405–415. [Google Scholar] [CrossRef]
- Wang, Y.; Ng, T.Y. Phosphatase 2A negatively regulates mitotic exit in Saccharomyces cerevisiae. Mol. Biol. Cell 2006, 17, 80–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumagai, A.; Dunphy, W.G. Regulation of the cdc25 protein during the cell cycle in Xenopus extracts. Cell 1992, 70, 139–151. [Google Scholar] [CrossRef]
- Harvey, S.L.; Enciso, G.; Dephoure, N.; Gygi, S.P.; Gunawardena, J.; Kellogg, D.R. A phosphatase threshold sets the level of Cdk1 activity in early mitosis in budding yeast. Mol. Biol. Cell 2011, 22, 3595–3608. [Google Scholar] [CrossRef]
- Lucena, R.; Alcaide-Gavilan, M.; Anastasia, S.D.; Kellogg, D.R. Wee1 and Cdc25 are controlled by conserved PP2A-dependent mechanisms in fission yeast. Cell Cycle 2017, 16, 428–435. [Google Scholar] [CrossRef] [Green Version]
- Vigneron, S.; Brioudes, E.; Burgess, A.; Labbe, J.C.; Lorca, T.; Castro, A. Greatwall maintains mitosis through regulation of PP2A. EMBO J. 2009, 28, 2786–2793. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Zhao, Y.; Li, Z.; Galas, S.; Goldberg, M.L. Greatwall kinase participates in the Cdc2 autoregulatory loop in Xenopus egg extracts. Mol. Cell 2006, 22, 83–91. [Google Scholar] [CrossRef]
- Gharbi-Ayachi, A.; Labbe, J.C.; Burgess, A.; Vigneron, S.; Strub, J.M.; Brioudes, E.; Van-Dorsselaer, A.; Castro, A.; Lorca, T. The substrate of Greatwall kinase, Arpp19, controls mitosis by inhibiting protein phosphatase 2A. Science 2010, 330, 1673–1677. [Google Scholar] [CrossRef]
- Mochida, S.; Maslen, S.L.; Skehel, M.; Hunt, T. Greatwall phosphorylates an inhibitor of protein phosphatase 2A that is essential for mitosis. Science 2010, 330, 1670–1673. [Google Scholar] [CrossRef]
- Medema, R.H.; Macurek, L. Checkpoint recovery in cells: How a molecular understanding can help in the fight against cancer. F1000 Biol. Rep. 2011, 3, 10. [Google Scholar] [CrossRef]
- Kim, S.Y.; Hyun, S.Y.; Jang, Y.J. Dephosphorylation of Plk1 occurs through PP2A-B55/ENSA/Greatwall pathway during mitotic DNA damage recovery. Cell Cycle 2019, 18, 1154–1167. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.Y.; Ma, H.T.; Lee, H.J.; Poon, R.Y. MASTL(Greatwall) regulates DNA damage responses by coordinating mitotic entry after checkpoint recovery and APC/C activation. Sci. Rep. 2016, 6, 22230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stucki, M.; Clapperton, J.A.; Mohammad, D.; Yaffe, M.B.; Smerdon, S.J.; Jackson, S.P. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 2005, 123, 1213–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiscella, M.; Zhang, H.; Fan, S.; Sakaguchi, K.; Shen, S.; Mercer, W.E.; Vande Woude, G.F.; O’Connor, P.M.; Appella, E. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc. Natl. Acad. Sci. USA 1997, 94, 6048–6053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takekawa, M.; Adachi, M.; Nakahata, A.; Nakayama, I.; Itoh, F.; Tsukuda, H.; Taya, Y.; Imai, K. p53-inducible wip1 phosphatase mediates a negative feedback regulation of p38 MAPK-p53 signaling in response to UV radiation. EMBO J. 2000, 19, 6517–6526. [Google Scholar] [CrossRef]
- Lu, X.; Nguyen, T.A.; Donehower, L.A. Reversal of the ATM/ATR-mediated DNA damage response by the oncogenic phosphatase PPM1D. Cell Cycle 2005, 4, 1060–1064. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Nannenga, B.; Donehower, L.A. PPM1D dephosphorylates Chk1 and p53 and abrogates cell cycle checkpoints. Genes Dev. 2005, 19, 1162–1174. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, H.; Onishi, N.; Kato, N.; Takekawa, M.; Xu, X.Z.; Kosugi, A.; Kondo, T.; Imamura, M.; Oishi, I.; Yoda, A.; et al. Regulation of the antioncogenic Chk2 kinase by the oncogenic Wip1 phosphatase. Cell Death Differ. 2006, 13, 1170–1180. [Google Scholar] [CrossRef]
- Oliva-Trastoy, M.; Berthonaud, V.; Chevalier, A.; Ducrot, C.; Marsolier-Kergoat, M.C.; Mann, C.; Leteurtre, F. The Wip1 phosphatase (PPM1D) antagonizes activation of the Chk2 tumour suppressor kinase. Oncogene 2007, 26, 1449–1458. [Google Scholar] [CrossRef] [Green Version]
- Shreeram, S.; Demidov, O.N.; Hee, W.K.; Yamaguchi, H.; Onishi, N.; Kek, C.; Timofeev, O.N.; Dudgeon, C.; Fornace, A.J.; Anderson, C.W.; et al. Wip1 phosphatase modulates ATM-dependent signaling pathways. Mol. Cell 2006, 23, 757–764. [Google Scholar] [CrossRef]
- Lu, X.; Ma, O.; Nguyen, T.A.; Jones, S.N.; Oren, M.; Donehower, L.A. The Wip1 Phosphatase acts as a gatekeeper in the p53-Mdm2 autoregulatory loop. Cancer Cell 2007, 12, 342–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindqvist, A.; de Bruijn, M.; Macurek, L.; Bras, A.; Mensinga, A.; Bruinsma, W.; Voets, O.; Kranenburg, O.; Medema, R.H. Wip1 confers G2 checkpoint recovery competence by counteracting p53-dependent transcriptional repression. EMBO J. 2009, 28, 3196–3206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batchelor, E.; Mock, C.S.; Bhan, I.; Loewer, A.; Lahav, G. Recurrent initiation: A mechanism for triggering p53 pulses in response to DNA damage. Mol. Cell 2008, 30, 277–289. [Google Scholar] [CrossRef] [Green Version]
- Macurek, L.; Lindqvist, A.; Voets, O.; Kool, J.; Vos, H.R.; Medema, R.H. Wip1 phosphatase is associated with chromatin and dephosphorylates gammaH2AX to promote checkpoint inhibition. Oncogene 2010, 29, 2281–2291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, S.H.; Lin, L.; Zhang, X.; Nguyen, T.A.; Darlington, Y.; Waldman, A.S.; Lu, X.; Donehower, L.A. Wild-type p53-induced phosphatase 1 dephosphorylates histone variant gamma-H2AX and suppresses DNA double strand break repair. J. Biol. Chem. 2010, 285, 12935–12947. [Google Scholar] [CrossRef] [Green Version]
- Cha, H.; Lowe, J.M.; Li, H.; Lee, J.S.; Belova, G.I.; Bulavin, D.V.; Fornace, A.J., Jr. Wip1 directly dephosphorylates gamma-H2AX and attenuates the DNA damage response. Cancer Res. 2010, 70, 4112–4122. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wan, G.; Mlotshwa, S.; Vance, V.; Berger, F.G.; Chen, H.; Lu, X. Oncogenic Wip1 phosphatase is inhibited by miR-16 in the DNA damage signaling pathway. Cancer Res. 2010, 70, 7176–7186. [Google Scholar] [CrossRef] [Green Version]
- Choi, D.W.; Na, W.; Kabir, M.H.; Yi, E.; Kwon, S.; Yeom, J.; Ahn, J.W.; Choi, H.H.; Lee, Y.; Seo, K.W.; et al. WIP1, a homeostatic regulator of the DNA damage response, is targeted by HIPK2 for phosphorylation and degradation. Mol. Cell 2013, 51, 374–385. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Nguyen, T.A.; Moon, S.H.; Darlington, Y.; Sommer, M.; Donehower, L.A. The type 2C phosphatase Wip1: An oncogenic regulator of tumor suppressor and DNA damage response pathways. Cancer Metastasis Rev. 2008, 27, 123–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazhar, S.; Taylor, S.E.; Sangodkar, J.; Narla, G. Targeting PP2A in cancer: Combination therapies. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhao, A.; Sun, L.; Zhong, X.; Zhong, J.; Wang, H.; Cai, M.; Li, J.; Xu, Y.; Liao, J.; et al. Protein phosphatase PP4 is overexpressed in human breast and lung tumors. Cell Res. 2008, 18, 974–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Function | PPase | Target |
|---|---|---|
| DNA damage checkpoint activation | PP1 | hATM, hH2AX, h53BP1, hRPA, hRAD51, hCHK1 |
| PP2A | hATR, hATM, hDNA–PK, hCHK1, hCHK2, hPLK1, hP53 | |
| PP4 | scMec1 | |
| PP5 | hATM, hATR, hDNA–PK, mP53, mCHK1 | |
| CDC14 | spCds1, hCDH1 | |
| DNA repair | PP1 | hBRCA1 |
| PP2A | hDNA–PK, hRPA, hH2AX, hATM, hCHK2 | |
| PP4 | ScRad53, ScH2A, hRPA, hH2AX | |
| CDC14 | scSpc110, scYen1 | |
| DNA damage checkpoint deactivation | PP1 | xCDC25C, hP53, caRad53, scH2A, ScRad53, spChk1 |
| PP2A | hPLK1 | |
| PP4 | scRad53, scH2A, hH2AX, dH2AX | |
| Wip1 | hP53, hCHK1, hCHK2, hH2AX, mATM |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campos, A.; Clemente-Blanco, A. Cell Cycle and DNA Repair Regulation in the Damage Response: Protein Phosphatases Take Over the Reins. Int. J. Mol. Sci. 2020, 21, 446. https://doi.org/10.3390/ijms21020446
Campos A, Clemente-Blanco A. Cell Cycle and DNA Repair Regulation in the Damage Response: Protein Phosphatases Take Over the Reins. International Journal of Molecular Sciences. 2020; 21(2):446. https://doi.org/10.3390/ijms21020446
Chicago/Turabian StyleCampos, Adrián, and Andrés Clemente-Blanco. 2020. "Cell Cycle and DNA Repair Regulation in the Damage Response: Protein Phosphatases Take Over the Reins" International Journal of Molecular Sciences 21, no. 2: 446. https://doi.org/10.3390/ijms21020446
APA StyleCampos, A., & Clemente-Blanco, A. (2020). Cell Cycle and DNA Repair Regulation in the Damage Response: Protein Phosphatases Take Over the Reins. International Journal of Molecular Sciences, 21(2), 446. https://doi.org/10.3390/ijms21020446