DNA Damage Response and Immune Defense




Abstract
:1. Introduction
2. DNA Damage and DNA Damage Response
3. At the Intersection of DNA Damage and Innate Immunity
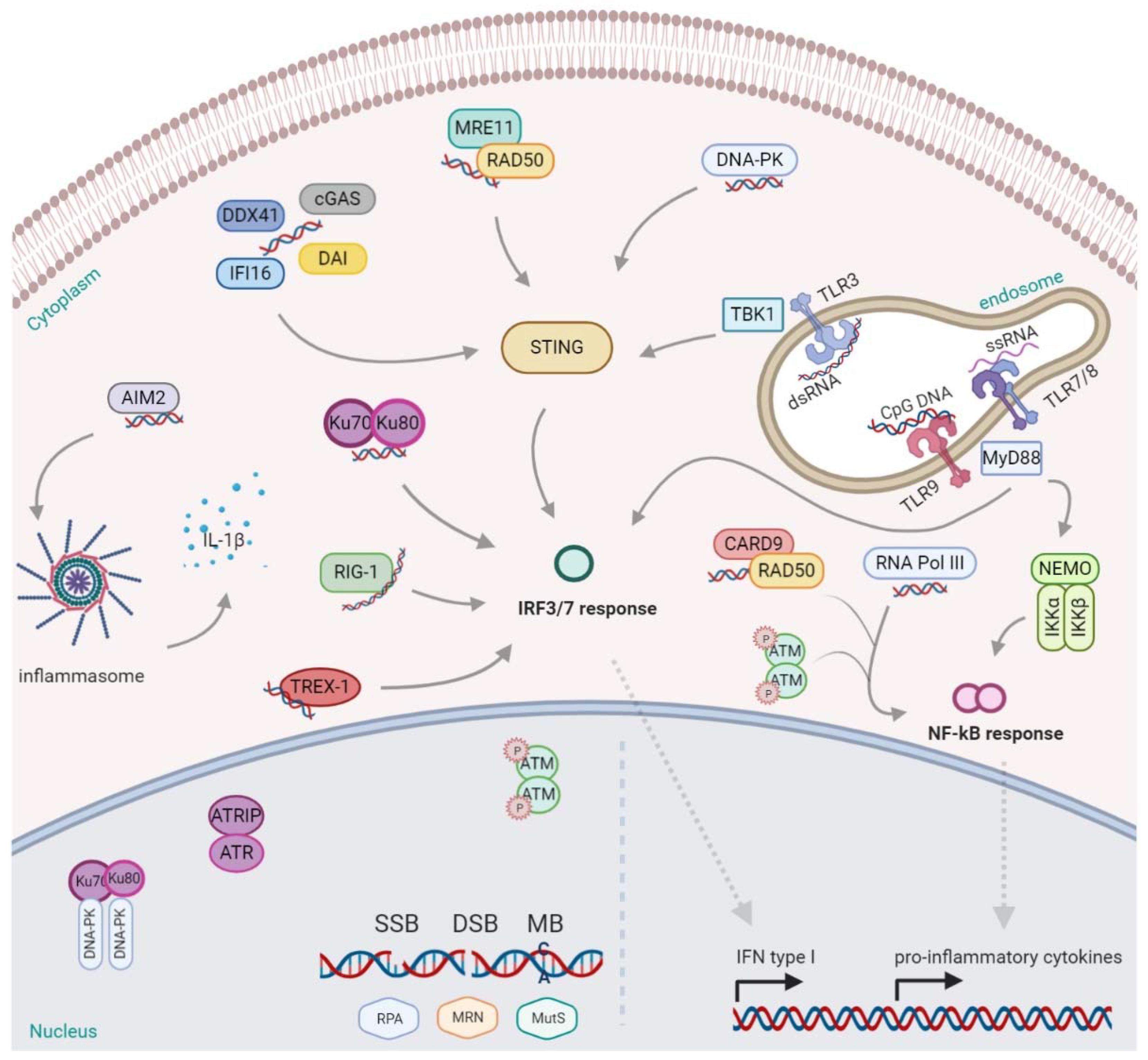
Innate DNA Sensors and Signaling
4. DNA Damage Inducers
4.1. From Carcinogens to Radiation and Chemotherapy
4.2. DDR Induced Oxidative Stress: Meaning for the Host Immune Response
5. DNA Damage and Inflammation: A Strong Interplay
6. Targeting DDR to Defeat Cancer
PARPs at the Intersection of DNA Damage and Immunity
7. Concluding Remarks and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| (8-oxodA) | 8-oxo-7,8-dihydro-20-deoxyadenosine |
| (8-oxodG) | 8-oxo-7,8-dihydro-20-deoxyguanosine |
| (AA) | Aristolochic acid; Ascorbic acid |
| (AGS) | Aicardi–Goutieres syndrome |
| (AhR) | Aryl hydrocarbon receptor |
| (AIM2) | Absent in melanoma 2 |
| (ALRs) | AIM2-like receptors |
| (AMPs) | Antimicrobial peptides |
| (AP) | Apurinic/apyrimidinic |
| (ATM) | Ataxia telangiectasia mutated |
| (ATR) | ATM- and Rad3-Related complex |
| (ATRIP) | ATR interacting proteins |
| (B reg) | B regulatory cells |
| (BRCA1/2) | Breast Related Cancer Antigen 1/2 |
| (CAD) | Caspase-activated DNase |
| (CAR-T) | Chimeric Antigen Receptor T cell therapies |
| (CCL2) | C-C Motif Chemokine Ligand 2 |
| (CD) | Crohn’s disease |
| (cGAMP) | 2′,3′-cyclic GMP-AMP |
| (cGAS) | Cyclic GMP-AMP synthase |
| (CIN) | Chromosome instability phenotype |
| (cis- UCA) | cis- urocanic acid |
| (CLRs) | C-type lectin receptors |
| (CRI) | Cancer related inflammation |
| (CTLA-4) | Cytotoxic T lymphocyte antigen 4 |
| (CXCL8) | C-X-C Motif Chemokine Ligand 8 |
| (DAI) | DNA-dependent activator of IFN regulatory factor |
| (DAMPs) | Damage-associated molecular patterns |
| (DDR) | DNA damage repair |
| (DDX41) | DExD/H-box helicase 41 |
| (DNA-PK) | DNA-dependent protein kinase |
| (DR) | Direct repair |
| (DSBs) | Double-strand breaks |
| (dsDNA) | Double-stranded DNAs |
| (DSBR) | Double-strand break repair |
| (dsRNAs) | Double-stranded RNAs |
| (Erk) | Extracellular signal-activated kinase |
| (FA) | Fanconi Anaemia repair complex |
| (FICZ) | 6-formylindolo[3,2-b]carbazole |
| (GG-NER) | Global Genomic Repair |
| (GSH) | Glutathione |
| (HAAs) | Heterocyclic aromatic amines |
| (HMGB1) | High mobility group box 1 protein |
| (HR) | Homologous Recombination |
| (HRD) | Homologous recombination deficiency |
| (ICAM-1) | Intercellular adhesion molecule-1 |
| (ICD) | Immune cell death |
| (ICIs) | Immune checkpoint inhibitors |
| (ICL) | Interstrand crosslink repair |
| (IFI16) | Interferon gamma-inducible protein 16 |
| (IFN) | Interferon |
| (IFN-γ) | Interferon-gamma |
| (IL-1) | Interleukin-1 |
| (IL-10) | Interleukin-10 |
| (IL-18) | Interleukin-18 |
| (IL-1β) | Interleukin-1β |
| (IL-4) | Interleukin-4 |
| (IL-6) | Interleukin-6 |
| (iNOS) | Inducible nitric-oxide synthase |
| (IR) | Ionizing radiation |
| (IRFs) | IFN-regulatory factors |
| (LET) | Linear Energy Transfer |
| (LOH) | Loss of heterozygosity |
| (LPS) | Lipopolysaccharide |
| (mAbs) | Monoclonal antibodies |
| (mDCs) | Myeloid dendritic cells |
| (MGMT) | O6-methylguanine DNA methyltransferase |
| (MH) | Microhomology |
| (MM) | Malignant Mesothelioma |
| (MMEJ) | Microhomology Mediated End Joining |
| (MMR) | Mismatch Repair |
| (MPM) | Malignant pleural mesothelioma |
| (mtDNA) | Mitochondrial DNA |
| (MyD88) | Myeloid differentiation primary response gene 88 |
| (NADPH) | Nicotinamide adenine dinucleotide phosphate |
| (NER) | Nucleotide Excision Repair |
| (NF-kB) | Nuclear factor kappa B |
| (NHEJ) | Non-Homologous End Joining |
| (NKT) | Natural Killer T cells |
| (NLRP3) | NOD-, LRR- and pyrin domain-containing protein 3 |
| (NLRs) | NOD-like receptors |
| (NRF2) | NFE2-related factor 2 |
| (OH·) | Hydroxyl radicals |
| (PAFs) | Platelet- activating factors |
| (PAHs) | Polycyclic aromatic hydrocarbons |
| (PAMPs) | Pathogen-associated molecular patterns |
| (PARP1) | Poly-(ADP-ribose)-polymerase 1 |
| (PARPi) | PARP inhibitors |
| (PD1) | programmed cell death protein 1 |
| (PDL1) | programmed cell death ligand 1 |
| (PRRs) | Pattern recognition receptors |
| (RANKL) | Epidermal- derived receptor activator of nuclear factor-κB ligand |
| (RIG-I) | Retinoic acid-inducible gene-I |
| (RLRs) | Retinoic acid-inducible gene-I (RIG-I)-like receptors |
| (RNS) | Reactive nitrogen species |
| (ROS) | Reactive oxygen species |
| (RPA) | Replication Protein A |
| (RT) | Radiation Therapy |
| (SASP) | Senescence associated secretory phenotype |
| (SDSA) | Synthesis-dependent strand annealing repair |
| (SLE) | Systemic lupus erythematosus |
| (SSA) | Single Strand Annealing |
| (SSBs) | Single-strand breaks |
| (ssDNA) | Single-stranded DNA |
| (STING) | cGAS- sensor protein stimulator of IFN genes |
| (STAT3) | Signal Transducer And Activator Of Transcription 3 |
| (TAI) | Tumor allelic imbalance |
| (TAMs) | Tumor-associated macrophages |
| (TBK1) | TANK-binding kinase 1 |
| (TC-NER) | Transcription-Coupled Repair |
| (TCR) | Transcription-Coupled Repair |
| (Th1) | T helper 1 cells |
| (Th17) | T helper 17 cells |
| (Th2) | T helper 2 cells |
| (TLRs) | Toll-like receptors |
| (TMB) | Tumor mutational burden |
| (TME) | Tumor microenviroment |
| (TNF-α) | Tumor necrosis factor alpha |
| (TRCs) | Transcription-replication conflicts |
| (Treg) | T regulatory cells |
| (TREX) 1 | Three prime repair exonuclease |
| (UVR) | Ultraviolet radiation |
| (Vit C) | Vitamin C |
References
- de Lopez Saro, F.J. Regulation of Interactions with Sliding Clamps During DNA Replication and Repair. Curr. Genom. 2009, 10, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C. A brief history of the DNA repair field. Cell Res. 2008, 18, 3–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanna, A. DNA Damage in Cancer Therapeutics: A Boon or a Curse? Cancer Res. 2015, 75, 2133–2138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleaver, J.E. Defective Repair Replication of DNA in Xeroderma Pigmentosum. Nature 1968, 218, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Setlow, R.B.; Regan, J.D.; German, J.; Carrier, W.L. Evidence That Xeroderma Pigmentosum Cells do not Perform the First Step in the Repair of Ultraviolet Damage to Their DNA. Proc. Natl. Acad. Sci. USA 1969, 64, 1035–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirkin, E.V.; Mirkin, S.M. Replication Fork Stalling at Natural Impediments. Microbiol. Mol. Biol. Rev. 2007, 71, 13–35. [Google Scholar] [CrossRef] [Green Version]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Magdalou, I.; Lopez, B.S.; Pasero, P.; Lambert, S.A.E. The causes of replication stress and their consequences on genome stability and cell fate. Semin. Cell Dev. Biol. 2014, 30, 154–164. [Google Scholar] [CrossRef]
- Barnes, J.L.; Zubair, M.; John, K.; Poirier, M.C.; Martin, F.L. Carcinogens and DNA damage. Biochem. Soc. Trans. 2018, 46, 1213–1224. [Google Scholar] [CrossRef] [Green Version]
- Bernard, J.J.; Gallo, R.L.; Krutmann, J. Photoimmunology: How ultraviolet radiation affects the immune system. Nat. Rev. Immunol. 2019, 19, 688–701. [Google Scholar] [CrossRef]
- Valko, M.; Morris, H.M.; Cronin, M.T.D. Metals, Toxicity and Oxidative Stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic Cell Death in Cancer Therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Shatalin, K.; Epshtein, V.; Gottesman, M.E.; Nudler, E. Linking RNA Polymerase Backtracking to Genome Instability in E. coli. Cell 2011, 146, 533–543. [Google Scholar] [CrossRef] [Green Version]
- Merrikh, H.; Machón, C.; Grainger, W.H.; Grossman, A.D.; Soultanas, P. Co-directional replication–transcription conflicts lead to replication restart. Nature 2011, 470, 554–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sankar, T.S.; Wastuwidyaningtyas, B.D.; Dong, Y.; Lewis, S.A.; Wang, J.D. The nature of mutations induced by replication–transcription collisions. Nature 2016, 535, 178–181. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, A.; García-Muse, T. R Loops: From Transcription Byproducts to Threats to Genome Stability. Mol. Cell 2012, 46, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Paulsen, R.D.; Soni, D.V.; Wollman, R.; Hahn, A.T.; Yee, M.-C.; Guan, A.; Hesley, J.A.; Miller, S.C.; Cromwell, E.F.; Solow-Cordero, D.E.; et al. A Genome-wide siRNA Screen Reveals Diverse Cellular Processes and Pathways that Mediate Genome Stability. Mol. Cell 2009, 35, 228–239. [Google Scholar] [CrossRef] [Green Version]
- Santos-Pereira, J.M.; Aguilera, A. R loops: New modulators of genome dynamics and function. Nat. Rev. Genet. 2015, 16, 583–597. [Google Scholar] [CrossRef]
- Sollier, J.; Stork, C.T.; García-Rubio, M.L.; Paulsen, R.D.; Aguilera, A.; Cimprich, K.A. Transcription-Coupled Nucleotide Excision Repair Factors Promote R-Loop-Induced Genome Instability. Mol. Cell 2014, 56, 777–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macheret, M.; Halazonetis, T.D. DNA Replication Stress as a Hallmark of Cancer. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 425–448. [Google Scholar] [CrossRef] [Green Version]
- Pan, M.-R.; Li, K.; Lin, S.-Y.; Hung, W.-C. Connecting the Dots: From DNA Damage and Repair to Aging. Int. J. Mol. Sci. 2016, 17, 685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, M.; Kastan, M.B. The DNA Damage Response: Implications for Tumor Responses to Radiation and Chemotherapy. Annu. Rev. Med. 2015, 66, 129–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [CrossRef]
- Spivak, G. Nucleotide excision repair in humans. DNA Repair 2015, 36, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Sedgwick, B. Repairing DNA-methylation damage. Nat. Rev. Mol. Cell Biol. 2004, 5, 148–157. [Google Scholar] [CrossRef]
- Pegg, A.E. Mammalian O6-Alkylguanine-DNA Alkyltransferase: Regulation and Importance in Response to Alkylating Carcinogenic and Therapeutic Agents. Cancer Res. 1990, 50, 6119–6129. [Google Scholar]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Price, B.D.; D’Andrea, A.D. Chromatin Remodeling at DNA Double-Strand Breaks. Cell 2013, 152, 1344–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiruvella, K.K.; Liang, Z.; Wilson, T.E. Repair of Double-Strand Breaks by End Joining. Cold Spring Harb. Perspect Biol. 2013, 5, a012757. [Google Scholar] [CrossRef] [PubMed]
- Seol, J.-H.; Shim, E.Y.; Lee, S.E. Microhomology-mediated end joining: Good, bad and ugly. Mutat. Res. Fundam. Mol. Mech. Mutagen. 2018, 809, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; D’Andrea, A.D. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012, 26, 1393–1408. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Martinez, D.; Liang, C.-C.; Cohn, M.A. Cellular response to DNA interstrand crosslinks: The Fanconi anemia pathway. Cell. Mol. Life Sci. 2016, 73, 3097–3114. [Google Scholar] [CrossRef] [Green Version]
- Peña-Diaz, J.; Jiricny, J. Mammalian mismatch repair: Error-free or error-prone? Trends Biochem. Sci. 2012, 37, 206–214. [Google Scholar] [CrossRef] [Green Version]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef]
- Li, G.-M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Bhargava, R.; Onyango, D.O.; Stark, J.M. Regulation of Single Strand Annealing and its role in genome maintenance. Trends Genet 2016, 32, 566–575. [Google Scholar] [CrossRef] [Green Version]
- Kamileri, I.; Karakasilioti, I.; Garinis, G.A. Nucleotide excision repair: New tricks with old bricks. Trends Genet. 2012, 28, 566–573. [Google Scholar] [CrossRef]
- Schwertman, P.; Lagarou, A.; Dekkers, D.H.W.; Raams, A.; van der Hoek, A.C.; Laffeber, C.; Hoeijmakers, J.H.J.; Demmers, J.A.A.; Fousteri, M.; Vermeulen, W.; et al. UV-sensitive syndrome protein UVSSA recruits USP7 to regulate transcription-coupled repair. Nat. Genet. 2012, 44, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.E.; Bjørås, M. Base Excision Repair. Cold Spring Harb. Perspect Biol. 2013, 5, a012583. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.; Wilson, D.M. DNA Damage and Associated DNA Repair Defects in Disease and Premature Aging. Am. J. Hum. Genet. 2019, 105, 237–257. [Google Scholar] [CrossRef] [Green Version]
- da Silva, P.F.L.; Schumacher, B. DNA damage responses in ageing. Open Biol. 2019, 9, 190168. [Google Scholar] [CrossRef]
- Stein, D.; Toiber, D. DNA damage and neurodegeneration: The unusual suspect. Neural Regen. Res. 2017, 12, 1441–1442. [Google Scholar] [CrossRef]
- Abugable, A.A.; Morris, J.L.M.; Palminha, N.M.; Zaksauskaite, R.; Ray, S.; El-Khamisy, S.F. DNA repair and neurological disease: From molecular understanding to the development of diagnostics and model organisms. DNA Repair 2019, 81, 102669. [Google Scholar] [CrossRef]
- Cao, X. Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat. Rev. Immunol. 2016, 16, 35–50. [Google Scholar] [CrossRef]
- Reale, M.; Conti, L.; Velluto, D. Immune and Inflammatory-Mediated Disorders: From Bench to Bedside. J. Immunol. Res. 2018, 2018, 7197931. [Google Scholar] [CrossRef]
- Medzhitov, R.; Janeway, C. Innate immunity. N. Engl. J. Med. 2000, 343, 338–344. [Google Scholar] [CrossRef]
- Janeway, C.A.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMPs and DAMPs: Signal 0s that spur autophagy and immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Dixit, V.M. Signaling in Innate Immunity and Inflammation. Cold Spring Harb. Perspect. Biol. 2012, 4, a006049. [Google Scholar] [CrossRef]
- Cai, X.; Chiu, Y.-H.; Chen, Z.J. The cGAS-cGAMP-STING Pathway of Cytosolic DNA Sensing and Signaling. Mol. Cell 2014, 54, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen Recognition and Innate Immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasser, S.; Raulet, D.H. The DNA Damage Response Arouses the Immune System. Cancer Res. 2006, 66, 3959–3962. [Google Scholar] [CrossRef] [Green Version]
- Gasser, S.; Zhang, W.Y.L.; Tan, N.Y.J.; Tripathi, S.; Suter, M.A.; Chew, Z.H.; Khatoo, M.; Ngeow, J.; Cheung, F.S.G. Sensing of dangerous DNA. Mech. Ageing Dev. 2017, 165, 33–46. [Google Scholar] [CrossRef]
- Hong, C.; Tijhuis, A.E.; Foijer, F. The cGAS Paradox: Contrasting Roles for cGAS-STING Pathway in Chromosomal Instability. Cells 2019, 8, 1228. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Chen, P.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; Ye, L.; He, Y.; et al. cGAS-STING, an important pathway in cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 81. [Google Scholar] [CrossRef]
- Zhu, Y.; An, X.; Zhang, X.; Qiao, Y.; Zheng, T.; Li, X. STING: A master regulator in the cancer-immunity cycle. Mol. Cancer 2019, 18, 152. [Google Scholar] [CrossRef] [Green Version]
- Stetson, D.B.; Ko, J.S.; Heidmann, T.; Medzhitov, R. Trex1 Prevents Cell-Intrinsic Initiation of Autoimmunity. Cell 2008, 134, 587–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stetson, D.B.; Medzhitov, R. Recognition of Cytosolic DNA Activates an IRF3-Dependent Innate Immune Response. Immunity 2006, 24, 93–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brzostek-Racine, S.; Gordon, C.; Scoy, S.V.; Reich, N.C. The DNA Damage Response Induces IFN. J. Immunol. 2011, 187, 5336–5345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; DeOliveira, R.B.; Kalantari, P.; Parroche, P.; Goutagny, N.; Jiang, Z.; Chan, J.; Bartholomeu, D.C.; Lauw, F.; Hall, J.P.; et al. Innate Immune Recognition of an AT-Rich Stem-Loop DNA Motif in the Plasmodium falciparum Genome. Immunity 2011, 35, 194–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ablasser, A.; Bauernfeind, F.; Hartmann, G.; Latz, E.; Fitzgerald, K.A.; Hornung, V. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III–transcribed RNA intermediate. Nat. Immunol. 2009, 10, 1065–1072. [Google Scholar] [CrossRef] [Green Version]
- Whitehead, L.; Brown, G.D. Pattern Recognition Receptors. In Inflammation; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2017; pp. 175–216. ISBN 978-3-527-69215-6. [Google Scholar]
- Spies, B.; Hochrein, H.; Vabulas, M.; Huster, K.; Busch, D.H.; Schmitz, F.; Heit, A.; Wagner, H. Vaccination with Plasmid DNA Activates Dendritic Cells via Toll-Like Receptor 9 (TLR9) but Functions in TLR9-Deficient Mice. J. Immunol. 2003, 171, 5908–5912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathinam, V.A.K.; Vanaja, S.K.; Fitzgerald, K.A. Regulation of inflammasome signaling. Nat. Immunol. 2012, 13, 333–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef]
- Kuriakose, T.; Man, S.M.; Malireddi, R.K.S.; Karki, R.; Kesavardhana, S.; Place, D.E.; Neale, G.; Vogel, P.; Kanneganti, T.-D. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol. 2016, 1. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Comella, N.; Tognazzi, K.; Brown, L.F.; Dvorak, H.F.; Kocher, O. Cloning of DLM-1, a novel gene that is up-regulated in activated macrophages, using RNA differential display. Gene 1999, 240, 157–163. [Google Scholar] [CrossRef]
- Li, T.; Diner, B.A.; Chen, J.; Cristea, I.M. Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc. Natl. Acad. Sci. USA 2012, 109, 10558–10563. [Google Scholar] [CrossRef] [Green Version]
- Kerur, N.; Veettil, M.V.; Sharma-Walia, N.; Bottero, V.; Sadagopan, S.; Otageri, P.; Chandran, B. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi sarcoma associated herpesvirus infection. Cell Host Microbe 2011, 9, 363–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, A.; Dutta, D.; Iqbal, J.; Pisano, G.; Gjyshi, O.; Ansari, M.A.; Kumar, B.; Chandran, B. Nuclear Innate Immune DNA Sensor IFI16 Is Degraded during Lytic Reactivation of Kaposi’s Sarcoma-Associated Herpesvirus (KSHV): Role of IFI16 in Maintenance of KSHV Latency. J. Virol. 2016, 90, 8822–8841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bürckstümmer, T.; Baumann, C.; Blüml, S.; Dixit, E.; Dürnberger, G.; Jahn, H.; Planyavsky, M.; Bilban, M.; Colinge, J.; Bennett, K.L.; et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 2009, 10, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.-H.; MacMillan, J.B.; Chen, Z.J. RNA Polymerase III Detects Cytosolic DNA and Induces Type I Interferons through the RIG-I Pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luecke, S.; Holleufer, A.; Christensen, M.H.; Jønsson, K.L.; Boni, G.A.; Sørensen, L.K.; Johannsen, M.; Jakobsen, M.R.; Hartmann, R.; Paludan, S.R. cGAS is activated by DNA in a length-dependent manner. EMBO Rep. 2017, 18, 1707–1715. [Google Scholar] [CrossRef] [PubMed]
- Berger, G.; Marloye, M.; Lawler, S.E. Pharmacological Modulation of the STING Pathway for Cancer Immunotherapy. Trends Mol. Med. 2019, 25, 412–427. [Google Scholar] [CrossRef] [Green Version]
- Kwon, J.; Bakhoum, S.F. The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov 2020, 10, 26–39. [Google Scholar] [CrossRef]
- Zhang, Z.; Yuan, B.; Bao, M.; Lu, N.; Kim, T.; Liu, Y.-J. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat. Immunol. 2011, 12, 959–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.-G.; Lindahl, T.; Barnes, D.E. Trex1 Exonuclease Degrades ssDNA to Prevent Chronic Checkpoint Activation and Autoimmune Disease. Cell 2007, 131, 873–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, P.J.; Cheng, T.-F.; Cooper, L. Do all of the neurologic diseases in patients with DNA repair gene mutations result from the accumulation of DNA damage? DNA Repair 2008, 7, 834–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crow, Y.J.; Hayward, B.E.; Parmar, R.; Robins, P.; Leitch, A.; Ali, M.; Black, D.N.; van Bokhoven, H.; Brunner, H.G.; Hamel, B.C.; et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nat. Genet. 2006, 38, 917–920. [Google Scholar] [CrossRef]
- Huang, Y.; Li, L. DNA crosslinking damage and cancer a tale of friend and foe. Transl. Cancer Res. 2013, 2, 144–154. [Google Scholar] [CrossRef]
- Ravanat, J.-L.; Douki, T. UV and ionizing radiations induced DNA damage, differences and similarities. Radiat. Phys. Chem. 2016, 128, 92–102. [Google Scholar] [CrossRef]
- Singh, N.; Manshian, B.; Gareth, J.S.J.; Sioned, M.G.; Thierry, G.G.M.; Chris, J.W.; Shareen, H. Doak NanoGenotoxicology: The DNA damaging potential of engineered nanomaterials. Biomaterials 2009, 30, 3891–3914. [Google Scholar] [CrossRef]
- Musk, A.W.; de Klerk, N.; Reid, A.; Hui, J.; Franklin, P.; Brims, F. Asbestos-related diseases. Int. J. Tuberc. Lung Dis. 2020, 24, 562–567. [Google Scholar] [CrossRef]
- Kondo, N.; Takahashi, A.; Ono, K.; Ohnishi, T. DNA Damage Induced by Alkylating Agents and Repair Pathways. Available online: https://www.hindawi.com/journals/jna/2010/543531/ (accessed on 6 July 2020).
- Cohen, S.M.; Arnold, L.L. Chemical Carcinogenesis. Toxicol. Sci. 2011, 120, S76–S92. [Google Scholar] [CrossRef]
- Fu, D.; Calvo, J.A.; Samson, L.D. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 2012, 12, 104–120. [Google Scholar] [CrossRef] [Green Version]
- Chew, S.H.; Toyokuni, S. Malignant mesothelioma as an oxidative stress-induced cancer: An update. Free Radic. Biol. Med. 2015, 86, 166–178. [Google Scholar] [CrossRef]
- Benedetti, S.; Nuvoli, B.; Catalani, S.; Galati, R. Reactive oxygen species a double-edged sword for mesothelioma. Oncotarget 2015, 6, 16848–16865. [Google Scholar] [CrossRef] [Green Version]
- Ceresoli, G.L.; Bombardieri, E.; D’Incalci, M. Mesothelioma from Research to Clinical Practice; Springer Nature Switzerland AG: Cham, Switzerland, 2019. [Google Scholar]
- Walsh, A.A.; Szklarz, G.D.; Scott, E.E. Human Cytochrome P450 1A1 Structure and Utility in Understanding Drug and Xenobiotic Metabolism. J. Biol. Chem. 2013, 288, 12932–12943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith Martyn, T.; Guyton Kathryn, Z.; Gibbons Catherine, F.; Fritz Jason, M.; Portier Christopher, J.; Rusyn, I.; DeMarini David, M.; Caldwell Jane, C.; Kavlock Robert, J.; Lambert Paul, F.; et al. Key Characteristics of Carcinogens as a Basis for Organizing Data on Mechanisms of Carcinogenesis. Environ. Health Perspect. 2016, 124, 713–721. [Google Scholar] [CrossRef] [Green Version]
- Wohak, L.E.; Krais, A.M.; Kucab, J.E.; Stertmann, J.; Øvrebø, S.; Seidel, A.; Phillips, D.H.; Arlt, V.M. Carcinogenic polycyclic aromatic hydrocarbons induce CYP1A1 in human cells via a p53-dependent mechanism. Arch Toxicol. 2016, 90, 291–304. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yamamoto, J.F.; Caberto, C.; Saltzman, B.; Decker, R.; Vogt, T.M.; Yokochi, L.; Chanock, S.; Wilkens, L.R.; Le Marchand, L. Genetic variation in the bioactivation pathway for polycyclic hydrocarbons and heterocyclic amines in relation to risk of colorectal neoplasia. Carcinogenesis 2011, 32, 203–209. [Google Scholar] [CrossRef]
- Zhou, L. Ahr function in lymphocytes: Emerging concepts. Trends Immunol. 2016, 37, 17–31. [Google Scholar] [CrossRef] [Green Version]
- Esser, C.; Rannug, A. The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharm. Rev. 2015, 67, 259–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauben, E.; Gregori, S.; Draghici, E.; Migliavacca, B.; Olivieri, S.; Woisetschläger, M.; Roncarolo, M.G. Activation of the aryl hydrocarbon receptor promotes allograft-specific tolerance through direct and dendritic cell–mediated effects on regulatory T cells. Blood 2008, 112, 1214–1222. [Google Scholar] [CrossRef]
- Bruhs, A.; Haarmann-Stemmann, T.; Frauenstein, K.; Krutmann, J.; Schwarz, T.; Schwarz, A. Activation of the arylhydrocarbon receptor causes immunosuppression primarily by modulating dendritic cells. J. Investig. Derm. 2015, 135, 435–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Uchi, H.; Hashimoto-Hachiya, A.; Furue, M. Tryptophan Photoproduct FICZ Upregulates IL1A, IL1B, and IL6 Expression via Oxidative Stress in Keratinocytes. Oxid. Med. Cell Longev. 2018, 2018, 9298052. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.H.; Grimbaldeston, M.A.; Swift, G.J.; Hosszu, E.K.; Finlay-Jones, J.J. A critical role for dermal mast cells in cis-urocanic acid-induced systemic suppression of contact hypersensitivity responses in mice. Photochem. Photobiol. 1999, 70, 807–812. [Google Scholar] [CrossRef]
- Chacón-Salinas, R.; Chen, L.; Chávez-Blanco, A.D.; Limón-Flores, A.Y.; Ma, Y.; Ullrich, S.E. An essential role for platelet-activating factor in activating mast cell migration following ultraviolet irradiation. J. Leukoc. Biol. 2014, 95, 139–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halliday, G.M. Inflammation, gene mutation and photoimmunosuppression in response to UVR-induced oxidative damage contributes to photocarcinogenesis. Mutat. Res. 2005, 571, 107–120. [Google Scholar] [CrossRef]
- Gallo, R.L.; Bernard, J.J. Innate immune sensors stimulate inflammatory and immunosuppressive responses to UVB radiation. J. Investig. Derm. 2014, 134, 1508–1511. [Google Scholar] [CrossRef] [Green Version]
- Teunissen, M.B.M.; Piskin, G.; di Nuzzo, S.; Sylva-Steenland, R.M.R.; de Rie, M.A.; Bos, J.D. Ultraviolet B Radiation Induces a Transient Appearance of IL-4+ Neutrophils, Which Support the Development of Th2 Responses. J. Immunol. 2002, 168, 3732–3739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piskin, G.; Bos, J.D.; Teunissen, M.B.M. Neutrophils infiltrating ultraviolet B-irradiated normal human skin display high IL-10 expression. Arch. Derm. Res. 2005, 296, 339–342. [Google Scholar] [CrossRef]
- Grewe, M.; Gyufko, K.; Krutmann, J. Interleukin-10 production by cultured human keratinocytes: Regulation by ultraviolet B and ultraviolet A1 radiation. J. Investig. Derm. 1995, 104, 3–6. [Google Scholar] [CrossRef] [Green Version]
- Fukunaga, A.; Khaskhely, N.M.; Ma, Y.; Sreevidya, C.S.; Taguchi, K.; Nishigori, C.; Ullrich, S.E. Langerhans Cells Serve as Immunoregulatory Cells by Activating NKT Cells. J. Immunol. 2010, 185, 4633–4640. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, Y.; Byrne, S.N.; Nghiem, D.X.; Miyahara, Y.; Ullrich, S.E. A role for inflammatory mediators in the induction of immunoregulatory B cells. J. Immunol. 2006, 177, 4810–4817. [Google Scholar] [CrossRef] [Green Version]
- Mizoguchi, A.; Mizoguchi, E.; Takedatsu, H.; Blumberg, R.S.; Bhan, A.K. Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d upregulation. Immunity 2002, 16, 219–230. [Google Scholar] [CrossRef] [Green Version]
- Cheung-Ong, K.; Giaever, G.; Nislow, C. DNA-Damaging Agents in Cancer Chemotherapy: Serendipity and Chemical Biology. Chem. Biol. 2013, 20, 648–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chabner, B.A.; Roberts, T.G. Chemotherapy and the war on cancer. Nat. Rev. Cancer 2005, 5, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Allouch, A.; Martins, I.; Brenner, C.; Modjtahedi, N.; Deutsch, E.; Perfettini, J.-L. Modulating Both Tumor Cell Death and Innate Immunity Is Essential for Improving Radiation Therapy Effectiveness. Front. Immunol. 2017, 8, 613. [Google Scholar] [CrossRef]
- de Andrade Carvalho, H.; Villar, R.C. Radiotherapy and immune response: The systemic effects of a local treatment. Clinics 2018, 73. [Google Scholar] [CrossRef]
- Cadet, J.; Davies, K.J.A. Oxidative DNA damage & repair: An introduction. Free Radic. Biol. Med. 2017, 107, 2–12. [Google Scholar] [CrossRef]
- Lee, H.-T.; Bose, A.; Lee, C.-Y.; Opresko, P.L.; Myong, S. Molecular mechanisms by which oxidative DNA damage promotes telomerase activity. Nucleic Acids Res 2017, 45, 11752–11765. [Google Scholar] [CrossRef] [Green Version]
- Conklin, K.A. Chemotherapy-associated oxidative stress: Impact on chemotherapeutic effectiveness. Integr. Cancer Ther. 2004, 3, 294–300. [Google Scholar] [CrossRef]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef]
- Mikhed, Y.; Daiber, A.; Steven, S. Mitochondrial Oxidative Stress, Mitochondrial DNA Damage and Their Role in Age-Related Vascular Dysfunction. Int. J. Mol. Sci. 2015, 16, 15918–15953. [Google Scholar] [CrossRef] [Green Version]
- Shokolenko, I.; Venediktova, N.; Bochkareva, A.; Wilson, G.L.; Alexeyev, M.F. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 2009, 37, 2539–2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlgren, C.; Karlsson, A. Respiratory burst in human neutrophils. J. Immunol. Methods 1999, 232, 3–14. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Available online: https://www.hindawi.com/journals/omcl/2016/4350965/ (accessed on 17 July 2020).
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barlan, A.U.; Griffin, T.M.; Mcguire, K.A.; Wiethoff, C.M. Adenovirus Membrane Penetration Activates the NLRP3 Inflammasome. J. Virol. 2011, 85, 146–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Zhou, Z.; Min, W. Mitochondria, Oxidative Stress and Innate Immunity. Front. Physiol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Cruz, C.M.; Rinna, A.; Forman, H.J.; Ventura, A.L.M.; Persechini, P.M.; Ojcius, D.M. ATP Activates a Reactive Oxygen Species-dependent Oxidative Stress Response and Secretion of Proinflammatory Cytokines in Macrophages. J. Biol. Chem. 2007, 282, 2871–2879. [Google Scholar] [CrossRef] [Green Version]
- Dostert, C.; Pétrilli, V.; Bruggen, R.V.; Steele, C.; Mossman, B.T.; Tschopp, J. Innate Immune Activation through Nalp3 Inflammasome Sensing of Asbestos and Silica. Science 2008, 320, 674–677. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.; Piao, S.; Nagar, H.; Jung, S.; Kim, S.; Lee, I.; Kim, S.; Song, H.-J.; Shin, N.; Kim, D.W.; et al. Isocitrate dehydrogenase 2 deficiency induces endothelial inflammation via p66sh-mediated mitochondrial oxidative stress. Biochem. Biophys. Res. Commun. 2018, 503, 1805–1811. [Google Scholar] [CrossRef]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Golubnitschaja, O.; Zhan, X. Chronic inflammation: Key player and biomarker-set to predict and prevent cancer development and progression based on individualized patient profiles. EPMA J. 2019, 10, 365–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, R.C.; Pettijohn, K.; Fike, F.; Wang, J.; Nahas, S.A.; Tunuguntla, R.; Hu, H.; Gatti, R.A.; McCurdy, D. Defective DNA double-strand break repair in pediatric systemic lupus erythematosus. Arthritis Rheum. 2012, 64, 568–578. [Google Scholar] [CrossRef] [PubMed]
- Karakasilioti, I.; Kamileri, I.; Chatzinikolaou, G.; Kosteas, T.; Vergadi, E.; Robinson, A.R.; Tsamardinos, I.; Rozgaja, T.A.; Siakouli, S.; Tsatsanis, C.; et al. DNA damage triggers a chronic auto-inflammatory response leading to fat depletion in NER progeria. Cell Metab. 2013, 18, 403–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, S4–S9. [Google Scholar] [CrossRef]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [Green Version]
- Bartsch, H.; Nair, J. Chronic inflammation and oxidative stress in the genesis and perpetuation of cancer: Role of lipid peroxidation, DNA damage, and repair. Langenbecks Arch. Surg. 2006, 391, 499–510. [Google Scholar] [CrossRef]
- Hnatyszyn, A.; Hryhorowicz, S.; Kaczmarek-Ryś, M.; Lis, E.; Słomski, R.; Scott, R.J.; Pławski, A. Colorectal carcinoma in the course of inflammatory bowel diseases. Hered. Cancer Clin. Pract. 2019, 17. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, T.; Morimoto, K. Crocidolite asbestos increased 8-hydroxydeoxyguanosine levels in cellular DNA of a human promyelocytic leukemia cell line, HL60. Carcinogenesis 1994, 15, 635–639. [Google Scholar] [CrossRef]
- Beggs, R.; Yang, E.S. Targeting DNA repair in precision medicine. Adv. Protein. Chem. Struct. Biol. 2019, 115, 135–155. [Google Scholar] [CrossRef] [PubMed]
- Gorodetska, I.; Kozeretska, I.; Dubrovska, A. BRCA Genes: The Role in Genome Stability, Cancer Stemness and Therapy Resistance. J. Cancer 2019, 10, 2109–2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mylavarapu, S.; Das, A.; Roy, M. Role of BRCA Mutations in the Modulation of Response to Platinum Therapy. Front. Oncol. 2018, 8, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Incalci, M.; Galmarini, C.M. A review of trabectedin (ET-743): A unique mechanism of action. Mol. Cancer 2010, 9, 2157–2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.R.; Coleman, R.L.; González-Martín, A.; Moore, K.N.; Colombo, N.; Ray-Coquard, I.; Pignata, S. The forefront of ovarian cancer therapy: Update on PARP inhibitors. Ann. Oncol. 2020, 31, 1148–1159. [Google Scholar] [CrossRef]
- de Luca, X.M.; Newell, F.; Kazakoff, S.H.; Hartel, G.; McCart Reed, A.E.; Holmes, O.; Xu, Q.; Wood, S.; Leonard, C.; Pearson, J.V.; et al. Using whole-genome sequencing data to derive the homologous recombination deficiency scores. NPJ Breast Cancer 2020, 6, 33. [Google Scholar] [CrossRef]
- Pellegrino, B.; Musolino, A.; Llop-Guevara, A.; Serra, V.; De Silva, P.; Hlavata, Z.; Sangiolo, D.; Willard-Gallo, K.; Solinas, C. Homologous Recombination Repair Deficiency and the Immune Response in Breast Cancer: A Literature Review. Transl. Oncol. 2020, 13, 410–422. [Google Scholar] [CrossRef]
- Fuso Nerini, I.; Roca, E.; Mannarino, L.; Grosso, F.; Frapolli, R.; D’Incalci, M. Is DNA repair a potential target for effective therapies against malignant mesothelioma? Cancer Treat. Rev. 2020, 102101. [Google Scholar] [CrossRef]
- Grignani, G.; D’Ambrosio, L.; Pignochino, Y.; Palmerini, E.; Zucchetti, M.; Boccone, P.; Aliberti, S.; Stacchiotti, S.; Bertulli, R.; Piana, R.; et al. Trabectedin and olaparib in patients with advanced and non-resectable bone and soft-tissue sarcomas (TOMAS): An open-label, phase 1b study from the Italian Sarcoma Group. Lancet Oncol. 2018, 19, 1360–1371. [Google Scholar] [CrossRef]
- Mei, L.; Zhang, J.; He, K.; Zhang, J. Ataxia telangiectasia and Rad3-related inhibitors and cancer therapy: Where we stand. J. Hematol. Oncol. 2019, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Klinakis, A.; Karagiannis, D.; Rampias, T. Targeting DNA repair in cancer: Current state and novel approaches. Cell. Mol. Life Sci. 2020, 77, 677–703. [Google Scholar] [CrossRef] [PubMed]
- Riches, L.C.; Trinidad, A.G.; Hughes, G.; Jones, G.N.; Hughes, A.M.; Thomason, A.G.; Gavine, P.; Cui, A.; Ling, S.; Stott, J.; et al. Pharmacology of the ATM Inhibitor AZD0156: Potentiation of Irradiation and Olaparib Responses Preclinically. Mol. Cancer 2020, 19, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damia, G. Targeting DNA-PK in cancer. Mutat. Res. 2020, 821, 111692. [Google Scholar] [CrossRef] [PubMed]
- Rogers, R.F.; Walton, M.I.; Cherry, D.L.; Collins, I.; Clarke, P.A.; Garrett, M.D.; Workman, P. CHK1 Inhibition Is Synthetically Lethal with Loss of B-Family DNA Polymerase Function in Human Lung and Colorectal Cancer Cells. Cancer Res. 2020, 80, 1735–1747. [Google Scholar] [CrossRef] [Green Version]
- Vilar, E.; Gruber, S.B. Microsatellite instability in colorectal cancer—the stable evidence. Nat. Rev. Clin. Oncol. 2010, 7, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Woerner, S.M.; Tosti, E.; Yuan, Y.P.; Kloor, M.; Bork, P.; Edelmann, W.; Gebert, J. Detection of coding microsatellite frameshift mutations in DNA mismatch repair-deficient mouse intestinal tumors. Mol. Carcinog. 2015, 54, 1376–1386. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germano, G.; Lamba, S.; Rospo, G.; Barault, L.; Magrì, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 2017, 552, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Schijns, V.; Fernández-Tejada, A.; Barjaktarović, Ž.; Bouzalas, I.; Brimnes, J.; Chernysh, S.; Gizurarson, S.; Gursel, I.; Jakopin, Ž.; Lawrenz, M.; et al. Modulation of immune responses using adjuvants to facilitate therapeutic vaccination. Immunol. Rev. 2020, 296, 169–190. [Google Scholar] [CrossRef] [PubMed]
- Parakh, S.; King, D.; Gan, H.K.; Scott, A.M. Current Development of Monoclonal Antibodies in Cancer Therapy. In Current Immunotherapeutic Strategies in Cancer; Recent Results in Cancer Research; Theobald, M., Ed.; Springer International Publishing: Cham, Switzerland, 2020; pp. 1–70. ISBN 978-3-030-23765-3. [Google Scholar]
- Li, D.; Li, X.; Zhou, W.-L.; Huang, Y.; Liang, X.; Jiang, L.; Yang, X.; Sun, J.; Li, Z.; Han, W.-D.; et al. Genetically engineered T cells for cancer immunotherapy. Signal Transduct. Target. Ther. 2019, 4, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.-K.; Hsu, J.-M.; Hsu, J.L.; Yu, W.-H.; Du, Y.; Lee, H.-H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Kim, H.-J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunphy, G.; Flannery, S.M.; Almine, J.F.; Connolly, D.J.; Paulus, C.; Jønsson, K.L.; Jakobsen, M.R.; Nevels, M.M.; Bowie, A.G.; Unterholzner, L. Non-canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-κB Signaling after Nuclear DNA Damage. Mol. Cell 2018, 71, 745–760.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, N.A.; Besson, V.C.; Boulares, A.H.; Bürkle, A.; Chiarugi, A.; Clark, R.S.; Curtin, N.J.; Cuzzocrea, S.; Dawson, T.M.; Dawson, V.L.; et al. Opportunities for the repurposing of PARP inhibitors for the therapy of non-oncological diseases. Br. J. Pharmacol. 2018, 175, 192–222. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Wu, H.-J.; Cao, L.-Q.; Li, H.-J.; He, C.-X.; Zhao, D.; Xing, L.; Li, P.-Q.; Jin, X.; Cao, H.-L. Research Progress on PARP14 as a Drug Target. Front. Pharm. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Alemasova, E.E.; Lavrik, O.I. Poly(ADP-ribosyl)ation by PARP1: Reaction mechanism and regulatory proteins. Nucleic Acids Res. 2019, 47, 3811–3827. [Google Scholar] [CrossRef] [Green Version]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol 2017, 18, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Robu, M.; Shah, R.G.; Petitclerc, N.; Brind’Amour, J.; Kandan-Kulangara, F.; Shah, G.M. Role of poly(ADP-ribose) polymerase-1 in the removal of UV-induced DNA lesions by nucleotide excision repair. Proc. Natl. Acad. Sci. USA 2013, 110, 1658–1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luijsterburg, M.S.; de Krijger, I.; Wiegant, W.W.; Shah, R.G.; Smeenk, G.; de Groot, A.J.L.; Pines, A.; Vertegaal, A.C.O.; Jacobs, J.J.L.; Shah, G.M.; et al. PARP1 Links CHD2-Mediated Chromatin Expansion and H3.3 Deposition to DNA Repair by Non-homologous End-Joining. Mol. Cell 2016, 61, 547–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, A.; Eckelmann, B.; Adhikari, S.; Ahmed, K.M.; Sengupta, S.; Pandey, A.; Hegde, P.M.; Tsai, M.-S.; Tainer, J.A.; Weinfeld, M.; et al. Microhomology-mediated end joining is activated in irradiated human cells due to phosphorylation-dependent formation of the XRCC1 repair complex. Nucleic Acids Res. 2017, 45, 2585–2599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hochegger, H.; Dejsuphong, D.; Fukushima, T.; Morrison, C.; Sonoda, E.; Schreiber, V.; Zhao, G.Y.; Saberi, A.; Masutani, M.; Adachi, N.; et al. Parp-1 protects homologous recombination from interference by Ku and Ligase IV in vertebrate cells. EMBO J. 2006, 25, 1305–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronson, G.E.; Piberger, A.L.; Higgs, M.R.; Olsen, A.L.; Stewart, G.S.; McHugh, P.J.; Petermann, E.; Lakin, N.D. PARP1 and PARP2 stabilise replication forks at base excision repair intermediates through Fbh1-dependent Rad51 regulation. Nat. Commun. 2018, 9, 746. [Google Scholar] [CrossRef] [Green Version]
- Tao, Z.; Gao, P.; Liu, H. Identification of the ADP-Ribosylation Sites in the PARP-1 Automodification Domain: Analysis and Implications. J. Am. Chem. Soc. 2009, 131, 14258–14260. [Google Scholar] [CrossRef]
- Kauppinen, T.M.; Chan, W.Y.; Suh, S.W.; Wiggins, A.K.; Huang, E.J.; Swanson, R.A. Direct phosphorylation and regulation of poly(ADP-ribose) polymerase-1 by extracellular signal-regulated kinases 1/2. Proc. Natl. Acad. Sci. USA 2006, 103, 7136–7141. [Google Scholar] [CrossRef] [Green Version]
- Stilmann, M.; Hinz, M.; Arslan, S.C.; Zimmer, A.; Schreiber, V.; Scheidereit, C. A nuclear poly(ADP-ribose)-dependent signalosome confers DNA damage-induced IkappaB kinase activation. Mol. Cell 2009, 36, 365–378. [Google Scholar] [CrossRef]
- Hinz, M.; Stilmann, M.; Arslan, S.Ç.; Khanna, K.K.; Dittmar, G.; Scheidereit, C. A Cytoplasmic ATM-TRAF6-cIAP1 Module Links Nuclear DNA Damage Signaling to Ubiquitin-Mediated NF-κB Activation. Mol. Cell 2010, 40, 63–74. [Google Scholar] [CrossRef]
- Rosado, M.M.; Bennici, E.; Novelli, F.; Pioli, C. Beyond DNA repair, the immunological role of PARP-1 and its siblings. Immunology 2013, 139, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Huang, X.; Li, Y.; Guo, K.; Ning, P.; Zhang, Y. PARP-1 Inhibitor, DPQ, Attenuates LPS-Induced Acute Lung Injury through Inhibiting NF-κB-Mediated Inflammatory Response. PLoS ONE 2013, 8, e79757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dharwal, V.; Sandhir, R.; Naura, A.S. PARP-1 inhibition provides protection against elastase-induced emphysema by mitigating the expression of matrix metalloproteinases. Mol. Cell Biochem. 2019, 457, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Farez, M.F.; Quintana, F.J.; Gandhi, R.; Izquierdo, G.; Lucas, M.; Weiner, H.L. Toll-like receptor 2 and poly(ADP-ribose) polymerase 1 promote central nervous system neuroinflammation in progressive EAE. Nat. Immunol. 2009, 10, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Iwata, H.; Goettsch, C.; Sharma, A.; Ricchiuto, P.; Goh, W.W.B.; Halu, A.; Yamada, I.; Yoshida, H.; Hara, T.; Wei, M.; et al. PARP9 and PARP14 cross-regulate macrophage activation via STAT1 ADP-ribosylation. Nat. Commun. 2016, 7, 12849. [Google Scholar] [CrossRef]
- Fehr, A.R.; Singh, S.A.; Kerr, C.M.; Mukai, S.; Higashi, H.; Aikawa, M. The impact of PARPs and ADP-ribosylation on inflammation and host–pathogen interactions. Genes Dev. 2020, 34, 341–359. [Google Scholar] [CrossRef]
- Aldinucci, A.; Gerlini, G.; Fossati, S.; Cipriani, G.; Ballerini, C.; Biagioli, T.; Pimpinelli, N.; Borgognoni, L.; Massacesi, L.; Moroni, F.; et al. A key role for poly(ADP-ribose) polymerase-1 activity during human dendritic cell maturation. J. Immunol. 2007, 179, 305–312. [Google Scholar] [CrossRef]
- Nasta, F.; Laudisi, F.; Sambucci, M.; Rosado, M.M.; Pioli, C. Increased Foxp3+ Regulatory T Cells in Poly(ADP-Ribose) Polymerase-1 Deficiency. J. Immunol. 2010, 184, 3470–3477. [Google Scholar] [CrossRef]
- Saenz, L.; Lozano, J.J.; Valdor, R.; Baroja-Mazo, A.; Ramirez, P.; Parrilla, P.; Aparicio, P.; Sumoy, L.; Yélamos, J. Transcriptional regulation by Poly(ADP-ribose) polymerase-1 during T cell activation. BMC Genom. 2008, 9, 171. [Google Scholar] [CrossRef] [Green Version]
- Yélamos, J.; Moreno-Lama, L.; Jimeno, J.; Ali, S.O. Immunomodulatory Roles of PARP-1 and PARP-2: Impact on PARP-Centered Cancer Therapies. Cancers 2020, 12, 392. [Google Scholar] [CrossRef] [Green Version]
- Galindo-Campos, M.A.; Bedora-Faure, M.; Farrés, J.; Lescale, C.; Moreno-Lama, L.; Martínez, C.; Martín-Caballero, J.; Ampurdanés, C.; Aparicio, P.; Dantzer, F.; et al. Coordinated signals from the DNA repair enzymes PARP-1 and PARP-2 promotes B-cell development and function. Cell Death Differ. 2019, 26, 2667–2681. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.; Banerjee, S.; Friggeri, A.; Bell, C.; Abraham, E.; Zerfaoui, M. Poly(ADP-ribosyl)ation of high mobility group box 1 (HMGB1) protein enhances inhibition of efferocytosis. Mol. Med. 2012, 18, 359–369. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Type of DNA Damage | Causes | DNA Repair Mechanisms | Mechanism Involved |
|---|---|---|---|
| Stalled replication forks, DSBs | Exposure to ionizing irradiation, UV, ROS or errors during DNA replication and replication-fork collapse | Homologous recombination (HR) | HR is largely restricted to S phase and G2 phase of the cell cycle, relies on the MRN complex, and repairs via double-strand break repair (DSBR) or synthesis-dependent strand annealing repair (SDSA). After incision, the 3′-end ssDNA coated with Replication Protein A (RPA) and Rad51 invades into a homologous DNA duplex. During DSBR, two Holliday junctions are formed, each between four strands of DNA that are then converted into recombination products. SDSA gives rise to non-crossover products. DNA polymerases fill in the gaps at the end of the invading DNA strand [30,31]. |
| DSBs | Exposure to ionizing irrradiation, ultraviolet radiations (UV), ROS or errors during DNA replication and replication-fork collapse | Nonhomologous end joining (NHEJ) | NHEJ is initiated by the heterodimer of Ku70-Ku80 complex that recognizes and binds the broken DNA ends. The Ku70-Ku80 is an abundant nuclear complex and has high affinity for DNA ends that are either blunt or possess short ssDNA overhangs. To generate the two DNA blunt ends, this complex aligns the DNA ends, followed by (i) the activity of the DNA polymerases that fill in and (ii) the nucleases that trim off the DNA single-stranded overhangs. Then, the XRCC4/DNA ligase IV ligation complex is recruited to join the DNA ends together and promote end joining [30,32]. |
| DSBs | UV, chemotherapy, ROS | Microhomology Mediated End Joining (MMEJ) | MMEJ includes three discrete steps, pre-annealing, annealing, and post-annealing of the microhomology (MH) flanking a DSB. PARP1 binds to DSB ends and facilitates the recruitment of resection factors [CtIP and Mre11 complex (Mre11/Rad50/Nbs1)] to expose MHs flanking DSBs. Those MHs that are placed far from the break usually require extensive resection by BLM/EXO1 to facilitate MMEJ. Annealing of MHs, which is inhibited by single strand binding RPA complex, induces the formation of non-homologous tails/flaps. These latter are then removed by XPF/ERCC1 nuclease before filling-in synthesis by Polθ and ligation by LigI/III [33]. |
| Two nucleotide residues from opposite strands are covalently connected | Exogenous alkylating agents, cisplatin, mitomycin C or endogenous aldehydes, nitrous acid | Interstrand crosslink repair (ICL) or Fanconi Anemia (FA) repair complex | FA complementation group M protein detects DNA ICLs and induces the recruitment of the core FA complex at sites of damage. After the initial incision event, translesion DNA polymerases resume DNA replication in one strand and the resulting DNA DSB is processed by HR. In the G1 phase of the cell cycle, incision by ERCC1-XPF is followed by translesion DNA synthesis and the DNA ICL is looped out [34,35]. |
| Nucleotide misincorporation | ROS and reactive nitrogen species (RNS) or endogenous problems during DNA replication leading to nucleotide misincorporation that creates base-base mismatches | Mismatch repair (MMR) | AG or TC mismatches are recognized by two heterodimers, MUTSa or MUTSb, that discriminate between the old and the newly synthesized strand, remove the mismatched nucleotide, and allow the replication machinery to use the original DNA template to restore the damaged DNA strand to its native form [36,37,38]. |
| DSBs | Single Strand Annealing (SSA) | SSA involves annealing of homologous repeat sequences that flank a DSB, which causes a deletion rearrangement between the repeats. It is distinct from other HR pathways as it is independent from Rad51 recombinase and, instead, depends on Rad59 (which is indispensable to SSA when strand annealing is mediated by shorter (>30 bp) repeats). The successful annealing of repeat sequences forms unique recombination intermediates that contain one or two 3′ flaps; their cleavage is a key step in SSA as it produces DNA ends with the 3′ OH, suitable for repair synthesis by DNA polymerases. An endonuclease complex, XPF/ERCC1, catalyses the 3′ flap removal. Additionally, SSA requires proteins to stabilize the annealed intermediate and confer cleavage specificity [39]. | |
| Helix-distorting DNA lesions, base modifications, bulky adducts, intra-strand cross-links and thymidine dimers | UV, chemotherapy, ROS | Nucleotide excision repair (NER) | NER is divided into global genome NER (GG-NER) and TC-NER. In GG-NER, damage detection involves the XPC– RAD23B–Centrin2 complex. XPA, RPA, XPB, and XPD stabilize the damaged DNA and XPG and ERCC1-XPF structure-specific endonucleases cleave the 3′ and 5′ sides of the nucleotide fragment containing the damaged DNA. The single-strand gap is then filled by DNA polymerases and the nascent DNA fragment is sealed by DNA ligase III-XRCC1 and DNA ligase I [40]. Damage recognition in TC-NER involves the stalling of RNA polymerase II on the actively transcribed strand of a gene. RNA polymerase II is stabilized by the interaction with UVSSA, USP7 and CSB protein, which together recruit other factors like CSA. Once the remodeling of RNA polymerase II is completed, the TFIIH complex with XPA and RPA are recruited and that is where GG-NER and TC-NER converge [27,41]. |
| Non-helix-distorting lesions. Base excision that leads to an AP site (apurinic/apyrimidinic site) when deoxyribose is cleaved from its nitrogenous base | Modification due to enzymatic activity, oxidation, deamination and alkylation; exposure to hydroxyl radicals that attack that weaken the glycosyl bond | Base excision repair (BER) | BER is a two-step process initiated by DNA glycosylases that detect and remove non-helix distorting DNA lesions through hydrolysis. The resulting abasic sites are cleaved by an apurinic/apyrimidinic endonuclease, exposing DNA SSBs that are repaired by either a short- or a long-patch repair mechanism depending on the number of replaced nucleotides. DNA ligase III and X-ray repair cross complementing protein 1 catalyses the nick-sealing step in short-patch BER, while DNA ligase I ligates the DNA SSB in long-patch BER. DNA polymerase b is typically involved during the DNA synthesis step [42]. |
| Damaged base | Chemotherapeutic agents like dacarbazine and temozolomide | Direct repair (DR) | DR is the direct reversal of a damaged base to its native state without excision and de novo DNA synthesis. The DNA damage repaired in such way are of three types: photoreactivation by photolysases, O-methylation (in O6-Guanine, O4-Thymine and phosphates) by O6-methylguanine DNA methyltransferase (MGMT) and oxidative demethylation of N-methyl groups by AlkB family proteins. The self-methylated DNA methyltransferases are referred to as suicidal DNA repair proteins, as they are irreversibly inactivated during this stoichiometric repair reaction [28]. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nastasi, C.; Mannarino, L.; D’Incalci, M. DNA Damage Response and Immune Defense. Int. J. Mol. Sci. 2020, 21, 7504. https://doi.org/10.3390/ijms21207504
Nastasi C, Mannarino L, D’Incalci M. DNA Damage Response and Immune Defense. International Journal of Molecular Sciences. 2020; 21(20):7504. https://doi.org/10.3390/ijms21207504
Chicago/Turabian StyleNastasi, Claudia, Laura Mannarino, and Maurizio D’Incalci. 2020. "DNA Damage Response and Immune Defense" International Journal of Molecular Sciences 21, no. 20: 7504. https://doi.org/10.3390/ijms21207504
APA StyleNastasi, C., Mannarino, L., & D’Incalci, M. (2020). DNA Damage Response and Immune Defense. International Journal of Molecular Sciences, 21(20), 7504. https://doi.org/10.3390/ijms21207504