Pharmacological Chaperones Attenuate the Development of Opioid Tolerance


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Repetitive Morphine Intake Induces Morphine Tolerance
2.2. Development of Morphine Tolerance Was Attenuated by Pharmacological ER Chaperone Administration
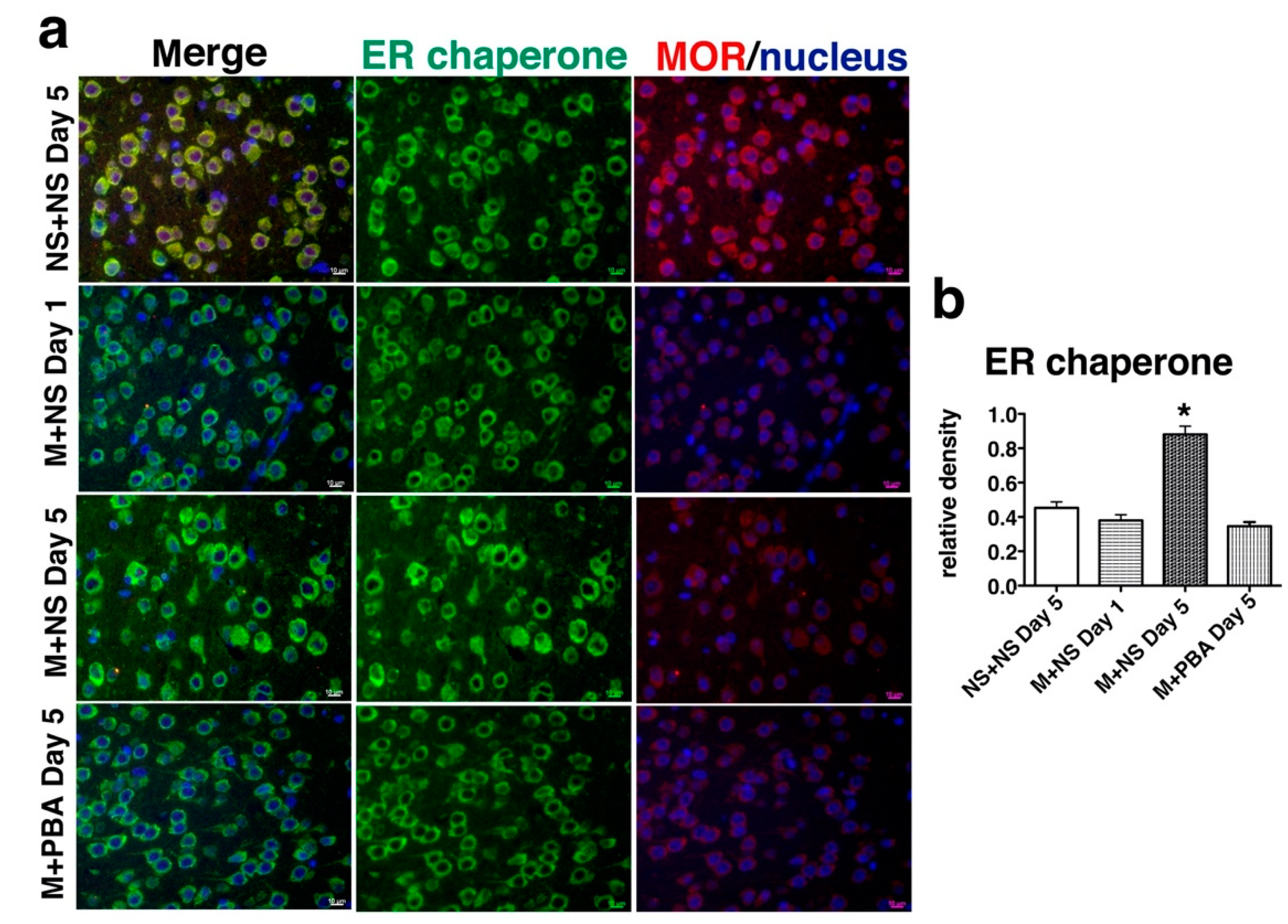
2.3. Repetitive Morphine Intake Induced ER Chaperone Expression
2.4. Pharmacological Chaperones Attenuate GSK3β Activation
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Antibodies and Reagents
4.3. Immunohistochemistry
4.4. Western Blotting
4.5. Hot Plate Test
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATF4 | Activating transcription factor 4 |
| ATF6 | Activating transcription factor 6 |
| BiP | Immunoglobulin heavy chain binding protein |
| COP | Coat protein complex |
| eIF2α | Eukaryotic translation initiation factor 2α |
| ER | Endoplasmic reticulum |
| FDA | Food and Drug Administration |
| G proteins | Heterotrimeric guanine nucleotide-binding proteins |
| GSK3β | Glycogen synthase kinase 3β |
| IRE1 | Inositol requiring enzyme-1 |
| MORs | Mu opioid receptors |
| PBA | 4-Phenylbutyric acid |
| PERK | PKR-like ER kinase |
| TUDCA | Tauroursodeoxycholic acid |
| UDCA | Ursodeoxycholic acid |
| UPR | Unfolded protein response |
| XBP1 | X-box binding protein 1 |
References
- Somogyi, A.A.; Barratt, D.T.; Coller, J.K. Pharmacogenetics of Opioids. Clin. Pharmacol. Ther. 2007, 81, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Ghelardini, C.; Mannelli, L.D.C.; Bianchi, E. The pharmacological basis of opioids. Clin. Cases Miner. Bone Metab. 2015, 12, 219–221. [Google Scholar] [CrossRef] [PubMed]
- Rutkow, L.; Vernick, J.S. Emergency Legal Authority and the Opioid Crisis. N. Engl. J. Med. 2017, 377, 2512–2514. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Collins, F.S. The Role of Science in Addressing the Opioid Crisis. N. Engl. J. Med. 2017, 377, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Stoicea, N.; Soghomonyan, S.; Bergese, S.D. Intraoperative use of remifentanil and opioid induced hyperalgesia/acute opioid tolerance: Systematic review. Front. Pharmacol. 2014, 5, 108. [Google Scholar] [CrossRef] [Green Version]
- Chan, P.; Lutfy, K. Molecular Changes in Opioid Addiction: The Role of Adenylyl Cyclase and cAMP/PKA System. Prog. Mol. Biol. Transl. Sci. 2016, 137, 203–227. [Google Scholar] [CrossRef] [Green Version]
- Volkow, N.D.; McLellan, A.T. Opioid Abuse in Chronic Pain—Misconceptions and Mitigation Strategies. N. Engl. J. Med. 2016, 374, 1253–1263. [Google Scholar] [CrossRef]
- Development of Opioid Tolerance and Endoplasmic Reticulum Stress. J. Pain Relief 2015, 4, 4. [CrossRef] [Green Version]
- Dickinson, P.; Kimber, W.L.; Kilanowski, F.M.; Webb, S.; Stevenson, B.J.; Porteous, D.J.; Dorin, J.R. Enhancing the efficiency of introducing precise mutations into the mouse genome by hit and run gene targeting. Transgenic Res. 2000, 9, 55–66. [Google Scholar] [CrossRef]
- Zhang, J.; Ferguson, S.S.; Barak, L.S.; Bodduluri, S.R.; Laporte, S.A.; Law, P.Y.; Caron, M.G. Role for G protein-coupled receptor kinase in agonist-specific regulation of mu-opioid receptor responsiveness. Proc. Natl. Acad. Sci. USA 1998, 95, 7157–7162. [Google Scholar] [CrossRef] [Green Version]
- Johnson, E.E.; Christie, M.J.; Connor, M. The Role of Opioid Receptor Phosphorylation and Trafficking in Adaptations to Persistent Opioid Treatment. Neurosignals 2005, 14, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Bohn, L.M.; Lefkowitz, R.J.; Gainetdinov, R.R.; Peppel, K.; Caron, M.G.; Lin, F.T. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science 1999, 286, 2495–2498. [Google Scholar] [CrossRef]
- Bohn, L.M.; Gainetdinov, R.R.; Lin, F.-T.; Lefkowitz, R.J.; Caron, M.G. μ-Opioid receptor desensitization by β-arrestin-2 determines morphine tolerance but not dependence. Nat. Cell Biol. 2000, 408, 720–723. [Google Scholar] [CrossRef] [PubMed]
- Gintzler, A.R.; Chakrabarti, S. Post-opioid receptor adaptations to chronic morphine; Altered functionality and associations of signaling molecules. Life Sci. 2006, 79, 717–722. [Google Scholar] [CrossRef]
- Martini, L.; Whistler, J.L. The role of mu opioid receptor desensitization and endocytosis in morphine tolerance and dependence. Curr. Opin. Neurobiol. 2007, 17, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Zöllner, C.; Mousa, S.A.; Fischer, O.; Rittner, H.L.; Shaqura, M.; Brack, A.; Shakibaei, M.; Binder, W.; Urban, F.; Stein, C.; et al. Chronic morphine use does not induce peripheral tolerance in a rat model of inflammatory pain. J. Clin. Investig. 2008, 118, 1065–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granados-Soto, V.; Kalcheva, I.; Hua, X.-Y.; Newton, A.; Yaksh, T.L. Spinal PKC activity and expression: Role in tolerance produced by continuous spinal morphine infusion. Pain 2000, 85, 395–404. [Google Scholar] [CrossRef]
- A Trujillo, K.; Akil, H. Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK-801. Science 1991, 251, 85–87. [Google Scholar] [CrossRef]
- Adam, F.; Bonnet, F.; Le Bars, D. Tolerance to morphine analgesia: Evidence for stimulus intensity as a key factor and complete reversal by a glycine site-specific NMDA antagonist. Neuropharmacology 2006, 51, 191–202. [Google Scholar] [CrossRef]
- Al-Hasani, R.; Bruchas, M.R. Molecular Mechanisms of Opioid Receptor-dependent Signaling and Behavior. Anesthesiology 2011, 115, 1363–1381. [Google Scholar] [CrossRef] [Green Version]
- A Colvin, L.; Bull, F.; Hales, T.G. Perioperative opioid analgesia—When is enough too much? A review of opioid-induced tolerance and hyperalgesia. Lancet 2019, 393, 1558–1568. [Google Scholar] [CrossRef] [Green Version]
- Parkitna, J.R.; Obara, I.; Wawrzczak-Bargiela, A.; Makuch, W.; Przewlocka, B.; Przewlocki, R. Effects of glycogen synthase kinase 3beta and cyclin-dependent kinase 5 inhibitors on morphine-induced analgesia and tolerance in rats. J. Pharmacol. Exp. Ther. 2006, 319, 832–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobashi, T.; Tanabe, S.; Jin, H.; Mimura, N.; Yamamoto, T.; Nishino, T.; Aoe, T. BiP, an endoplasmic reticulum chaperone, modulates the development of morphine antinociceptive tolerance. J. Cell. Mol. Med. 2009, 14, 2816–2826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bull, F.A.; Baptista-Hon, D.T.; Sneddon, C.; Wright, L.; Walwyn, W.; Hales, T.G. Src Kinase Inhibition Attenuates Morphine Tolerance without Affecting Reinforcement or Psychomotor Stimulation. Anesthesiology 2017, 127, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Ellgaard, L.; Helenius, A. Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 2003, 4, 181–191. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The Unfolded Protein Response and Cell Fate Control. Mol. Cell 2018, 69, 169–181. [Google Scholar] [CrossRef] [Green Version]
- Ozcan, U. Endoplasmic Reticulum Stress Links Obesity, Insulin Action, and Type 2 Diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [Green Version]
- Bridges, J.P.; Xu, Y.; Na, C.-L.; Wong, H.R.; Weaver, T.E. Adaptation and increased susceptibility to infection associated with constitutive expression of misfolded SP-C. J. Cell Biol. 2006, 172, 395–407. [Google Scholar] [CrossRef]
- Jin, H.; Komita, M.; Aoe, T. The Role of BiP Retrieval by the KDEL Receptor in the Early Secretory Pathway and its Effect on Protein Quality Control and Neurodegeneration. Front. Mol. Neurosci. 2017, 10, 222. [Google Scholar] [CrossRef] [Green Version]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Raykhel, I.; Alanen, H.; Salo, K.; Jurvansuu, J.; Nguyen, V.D.; Latva-Ranta, M.; Ruddock, L.W. A molecular specificity code for the three mammalian KDEL receptors. J. Cell Biol. 2007, 179, 1193–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, K.; Fujii, R.; Toyofuku, Y.; Saito, T.; Koseki, H.; Hsu, V.W.; Aoe, T. The KDEL receptor mediates a retrieval mechanism that contributes to quality control at the endoplasmic reticulum. EMBO J. 2001, 20, 3082–3091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mimura, N.; Hamada, H.; Kashio, M.; Jin, H.; Toyama, Y.; Kimura, K.; Iida, M.; Goto, S.; Saisho, H.; Toshimori, K.; et al. Aberrant quality control in the endoplasmic reticulum impairs the biosynthesis of pulmonary surfactant in mice expressing mutant BiP. Cell Death Differ. 2007, 14, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Mimura, N.; Kashio, M.; Koseki, H.; Aoe, T. Late-Onset of Spinal Neurodegeneration in Knock-In Mice Expressing a Mutant BiP. PLoS ONE 2014, 9, e112837. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Komita, M.; Aoe, T. Decreased Protein Quality Control Promotes the Cognitive Dysfunction Associated With Aging and Environmental Insults. Front. Neurosci. 2018, 12, 753. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Hamada, H.; Shinkai, H.; Kohno, Y.; Koseki, H.; Aoe, T. The KDEL Receptor Modulates the Endoplasmic Reticulum Stress Response through Mitogen-activated Protein Kinase Signaling Cascades. J. Biol. Chem. 2003, 278, 34525–34532. [Google Scholar] [CrossRef] [Green Version]
- Kokubun, H.; Jin, H.; Aoe, T. Pathogenic Effects of Impaired Retrieval between the Endoplasmic Reticulum and Golgi Complex. Int. J. Mol. Sci. 2019, 20, 5614. [Google Scholar] [CrossRef] [Green Version]
- Liguori, L.; Monticelli, M.; Allocca, M.; Mele, B.H.; Lukas, J.; Cubellis, M.V.; Andreotti, G. Pharmacological Chaperones: A Therapeutic Approach for Diseases Caused by Destabilizing Missense Mutations. Int. J. Mol. Sci. 2020, 21, 489. [Google Scholar] [CrossRef] [Green Version]
- Kolb, P.; Ayaub, E.; Zhou, W.; Yum, V.; Dickhout, J.; Ask, K. The therapeutic effects of 4-phenylbutyric acid in maintaining proteostasis. Int. J. Biochem. Cell Biol. 2015, 61, 45–52. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, Z.; Zhao, S.; Xiang, R. Chemical chaperones reduce ER stress and adipose tissue inflammation in high fat diet-induced mouse model of obesity. Sci. Rep. 2016, 6, 27486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusaczuk, M. Tauroursodeoxycholate—Bile Acid with Chaperoning Activity: Molecular and Cellular Effects and Therapeutic Perspectives. Cells 2019, 8, 1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, K.M.; Saunders, J.K.W.; Rutter, C.M.; Banta-Green, M.C.J.; Merrill, J.O.; Sullivan, M.D.; Weisner, D.C.M.; Silverberg, M.J.; Campbell, C.I.; Psaty, B.M.; et al. Opioid Prescriptions for Chronic Pain and Overdose. Ann. Intern. Med. 2010, 152, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.M.; Fossum, E.N.; Levine, C.S.; Ingram, S.L. Antinociceptive tolerance revealed by cumulative intracranial microinjections of morphine into the periaqueductal gray in the rat. Pharmacol. Biochem. Behav. 2006, 85, 214–219. [Google Scholar] [CrossRef]
- Tsai, Y.-L.; Ha, D.P.; Zhao, H.; Carlos, A.J.; Wei, S.; Pun, T.K.; Wu, K.; Zandi, E.; Kelly, K.; Lee, A.S. Endoplasmic reticulum stress activates SRC, relocating chaperones to the cell surface where GRP78/CD109 blocks TGF-β signaling. Proc. Natl. Acad. Sci. USA 2018, 115, E4245–E4254. [Google Scholar] [CrossRef] [Green Version]
- A Grimes, C.; Jope, R.S. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog. Neurobiol. 2001, 65, 391–426. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. THE MAMMALIAN UNFOLDED PROTEIN RESPONSE. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef]
- Schindler, A.J.; Schekman, R. In vitro reconstitution of ER-stress induced ATF6 transport in COPII vesicles. Proc. Natl. Acad. Sci. USA 2009, 106, 17775–17780. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER Stress Regulation of ATF6 Localization by Dissociation of BiP/GRP78 Binding and Unmasking of Golgi Localization Signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Tirasophon, W.; Shen, X.; Michalak, M.; Prywes, R.; Okada, T.; Yoshida, H.; Mori, K.; Kaufman, R.J. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002, 16, 452–466. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of Stress in the ER to Activation of JNK Protein Kinases by Transmembrane Protein Kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, D.; Mallucci, G.R. The unfolded protein response in neurodegenerative disorders - therapeutic modulation of the PERK pathway. FEBS J. 2018, 286, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, R.J. Orchestrating the unfolded protein response in health and disease. J. Clin. Investig. 2002, 110, 1389–1398. [Google Scholar] [CrossRef]
- Zhao, L.; Ackerman, S.L. Endoplasmic reticulum stress in health and disease. Curr. Opin. Cell Biol. 2006, 18, 444–452. [Google Scholar] [CrossRef]
- Hamada, H.; Suzuki, M.; Yuasa, S.; Mimura, N.; Shinozuka, N.; Takada, Y.; Suzuki, M.; Nishino, T.; Nakaya, H.; Koseki, H.; et al. Dilated Cardiomyopathy Caused by Aberrant Endoplasmic Reticulum Quality Control in Mutant KDEL Receptor Transgenic Mice. Mol. Cell. Biol. 2004, 24, 8007–8017. [Google Scholar] [CrossRef] [Green Version]
- Kimura, K.; Jin, H.; Ogawa, M.; Aoe, T. Dysfunction of the ER chaperone BiP accelerates the renal tubular injury. Biochem. Biophys. Res. Commun. 2008, 366, 1048–1053. [Google Scholar] [CrossRef]
- Gintzler, A.R.; Chakrabarti, S. Opioid Tolerance and the Emergence of New Opioid Receptor-Coupled Signaling. Mol. Neurobiol. 2000, 21, 021–034. [Google Scholar] [CrossRef]
- Araldi, D.; Khomula, E.V.; Ferrari, L.F.; Levine, J.D. Fentanyl Induces Rapid Onset Hyperalgesic Priming: Type I at Peripheral and Type II at Central Nociceptor Terminals. J. Neurosci. 2018, 38, 2226–2245. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; De Sarno, P.; Jope, R.S. Central role of glycogen synthase kinase-3beta in endoplasmic reticulum stress-induced caspase-3 activation. J. Biol. Chem. 2002, 277, 44701–44708. [Google Scholar] [CrossRef] [Green Version]
- Qu, L.; Huang, S.; Baltzis, D.; Rivas-Estilla, A.M.; Pluquet, O.; Hatzoglou, M.; Koumenis, C.; Taya, Y.; Yoshimura, A.; Koromilas, A.E. Endoplasmic reticulum stress induces p53 cytoplasmic localization and prevents p53-dependent apoptosis by a pathway involving glycogen synthase kinase-3beta. Genes Dev. 2004, 18, 261–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jope, R.S.; Yuskaitis, C.J.; Beurel, E. Glycogen Synthase Kinase-3 (GSK3): Inflammation, Diseases, and Therapeutics. Neurochem. Res. 2006, 32, 577–595. [Google Scholar] [CrossRef] [Green Version]
- Özcan, U.; Yilmaz, E.; Özcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Görgün, C.Z.; Hotamisligil, G.S. Chemical Chaperones Reduce ER Stress and Restore Glucose Homeostasis in a Mouse Model of Type 2 Diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [Green Version]
- Kars, M.; Yang, L.; Gregor, M.F.; Mohammed, B.S.; Pietka, T.A.; Finck, B.N.; Patterson, B.W.; Horton, J.D.; Mittendorfer, B.; Hotamisligil, G.S.; et al. Tauroursodeoxycholic Acid May Improve Liver and Muscle but Not Adipose Tissue Insulin Sensitivity in Obese Men and Women. Diabetes 2010, 59, 1899–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elia, A.E.; Lalli, S.; Monsurrò, M.R.; Sagnelli, A.; Taiello, A.C.; Reggiori, B.; La Bella, V.; Tedeschi, G.; Albanese, A. Tauroursodeoxycholic acid in the treatment of patients with amyotrophic lateral sclerosis. Eur. J. Neurol. 2015, 23, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Vang, S.; Longley, K.; Steer, C.J.; Low, W.C. The Unexpected Uses of Urso- and Tauroursodeoxycholic Acid in the Treatment of Non-liver Diseases. Glob. Adv. Heal. Med. 2014, 3, 58–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusaczuk, M.; Bartoszewicz, M.; Cechowska-Pasko, M. Phenylbutyric Acid: Simple structure—Multiple effects. Curr. Pharm. Des. 2015, 21, 2147–2166. [Google Scholar] [CrossRef]
- Chou, R.; Turner, J.A.; Devine, E.B.; Hansen, R.N.; Sullivan, S.D.; Blazina, I.; Dana, T.; Bougatsos, C.; Deyo, R.A. The Effectiveness and Risks of Long-Term Opioid Therapy for Chronic Pain: A Systematic Review for a National Institutes of Health Pathways to Prevention Workshop. Ann. Intern. Med. 2015, 162, 276–286. [Google Scholar] [CrossRef] [Green Version]
- Krebs, E.E.; Gravely, A.; Nugent, S.; Jensen, A.C.; DeRonne, B.; Goldsmith, E.S.; Kroenke, K.; Bair, M.J.; Noorbaloochi, S. Effect of Opioid vs Nonopioid Medications on Pain-Related Function in Patients With Chronic Back Pain or Hip or Knee Osteoarthritis Pain. JAMA 2018, 319, 872–882. [Google Scholar] [CrossRef]
- Morasco, B.J.; Smith, N.; Dobscha, S.K.; Deyo, R.A.; Hyde, S.; Yarborough, B.J.H. Outcomes of prescription opioid dose escalation for chronic pain. Pain 2020, 161, 1332–1340. [Google Scholar] [CrossRef]
- Ekholm, O.; Kurita, G.P.; Højsted, J.; Juel, K.; Sjøgren, P. Chronic pain, opioid prescriptions, and mortality in Denmark: A population-based cohort study. Pain 2014, 155, 2486–2490. [Google Scholar] [CrossRef] [PubMed]
- Kaplovitch, E.; Gomes, T.; Camacho, X.; Dhalla, I.A.; Mamdani, M.M.; Juurlink, D.N. Sex Differences in Dose Escalation and Overdose Death during Chronic Opioid Therapy: A Population-Based Cohort Study. PLoS ONE 2015, 10, e0134550. [Google Scholar] [CrossRef] [Green Version]
- Babu, K.M.; Brent, J.; Juurlink, D.N. Prevention of Opioid Overdose. N. Engl. J. Med. 2019, 380, 2246–2255. [Google Scholar] [CrossRef] [PubMed]
- Wakeman, S.E.; Barnett, M.L. Primary Care and the Opioid-Overdose Crisis—Buprenorphine Myths and Realities. N. Engl. J. Med. 2018, 379, 1–4. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okuyama, Y.; Jin, H.; Kokubun, H.; Aoe, T. Pharmacological Chaperones Attenuate the Development of Opioid Tolerance. Int. J. Mol. Sci. 2020, 21, 7536. https://doi.org/10.3390/ijms21207536
Okuyama Y, Jin H, Kokubun H, Aoe T. Pharmacological Chaperones Attenuate the Development of Opioid Tolerance. International Journal of Molecular Sciences. 2020; 21(20):7536. https://doi.org/10.3390/ijms21207536
Chicago/Turabian StyleOkuyama, Youta, Hisayo Jin, Hiroshi Kokubun, and Tomohiko Aoe. 2020. "Pharmacological Chaperones Attenuate the Development of Opioid Tolerance" International Journal of Molecular Sciences 21, no. 20: 7536. https://doi.org/10.3390/ijms21207536
APA StyleOkuyama, Y., Jin, H., Kokubun, H., & Aoe, T. (2020). Pharmacological Chaperones Attenuate the Development of Opioid Tolerance. International Journal of Molecular Sciences, 21(20), 7536. https://doi.org/10.3390/ijms21207536