Looking for Targets to Restore the Contractile Function in Congenital Myopathy Caused by Gln147Pro Tropomyosin


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
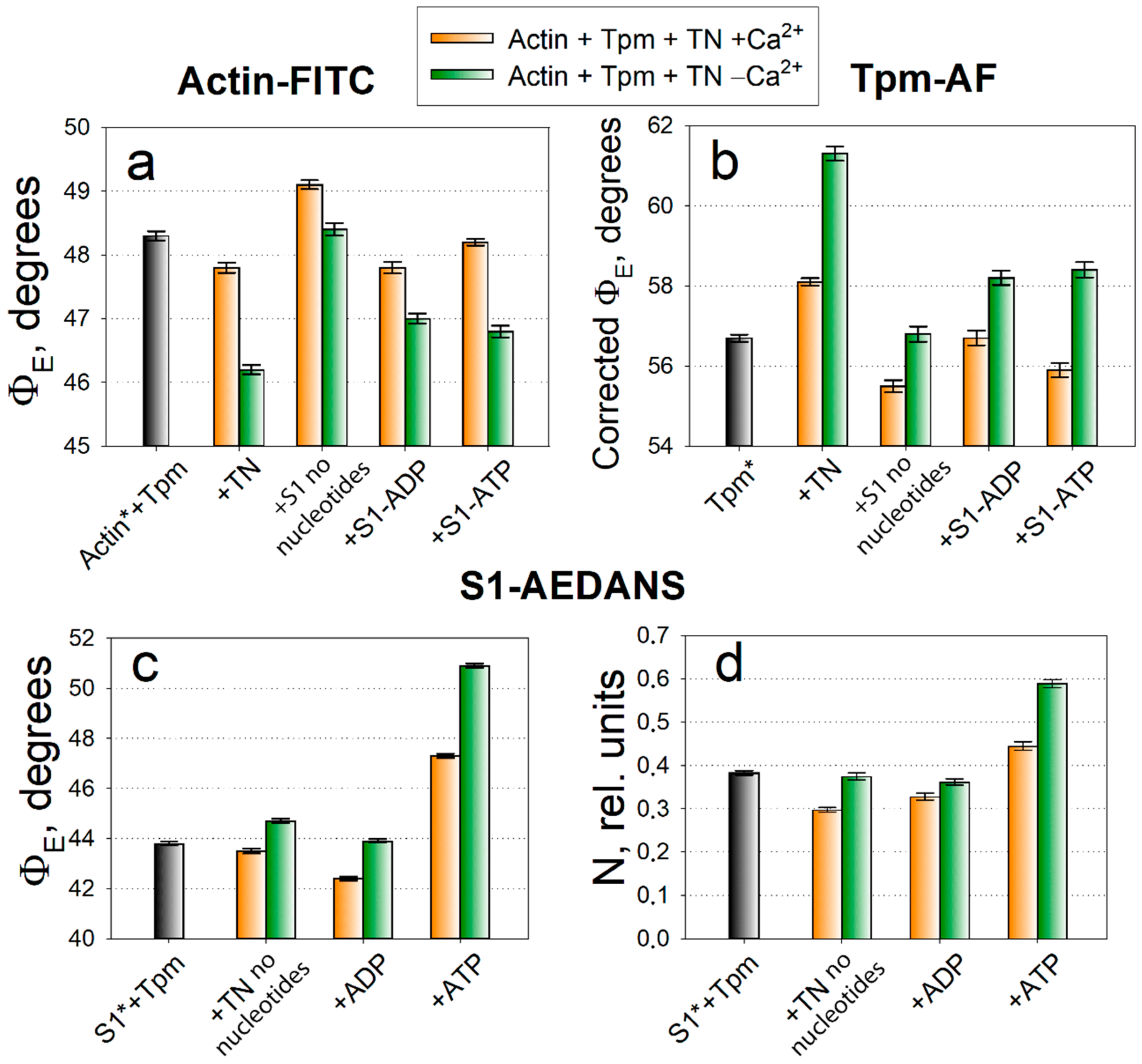
2.1. Analysis of Conformational Rearrangements of Actin and Tpm in Control
2.2. Analysis of Conformational Rearrangements of the Myosin Heads in Control
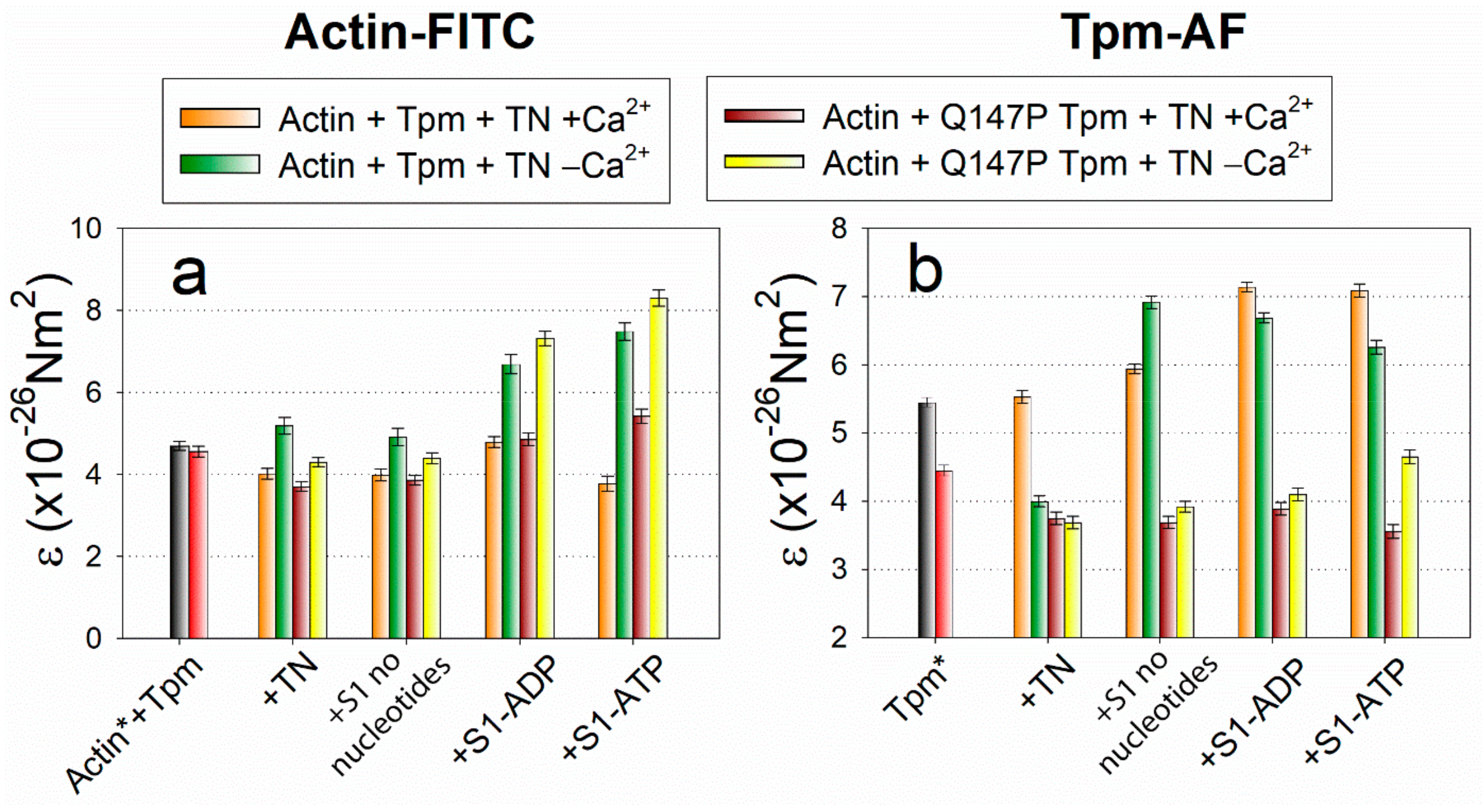
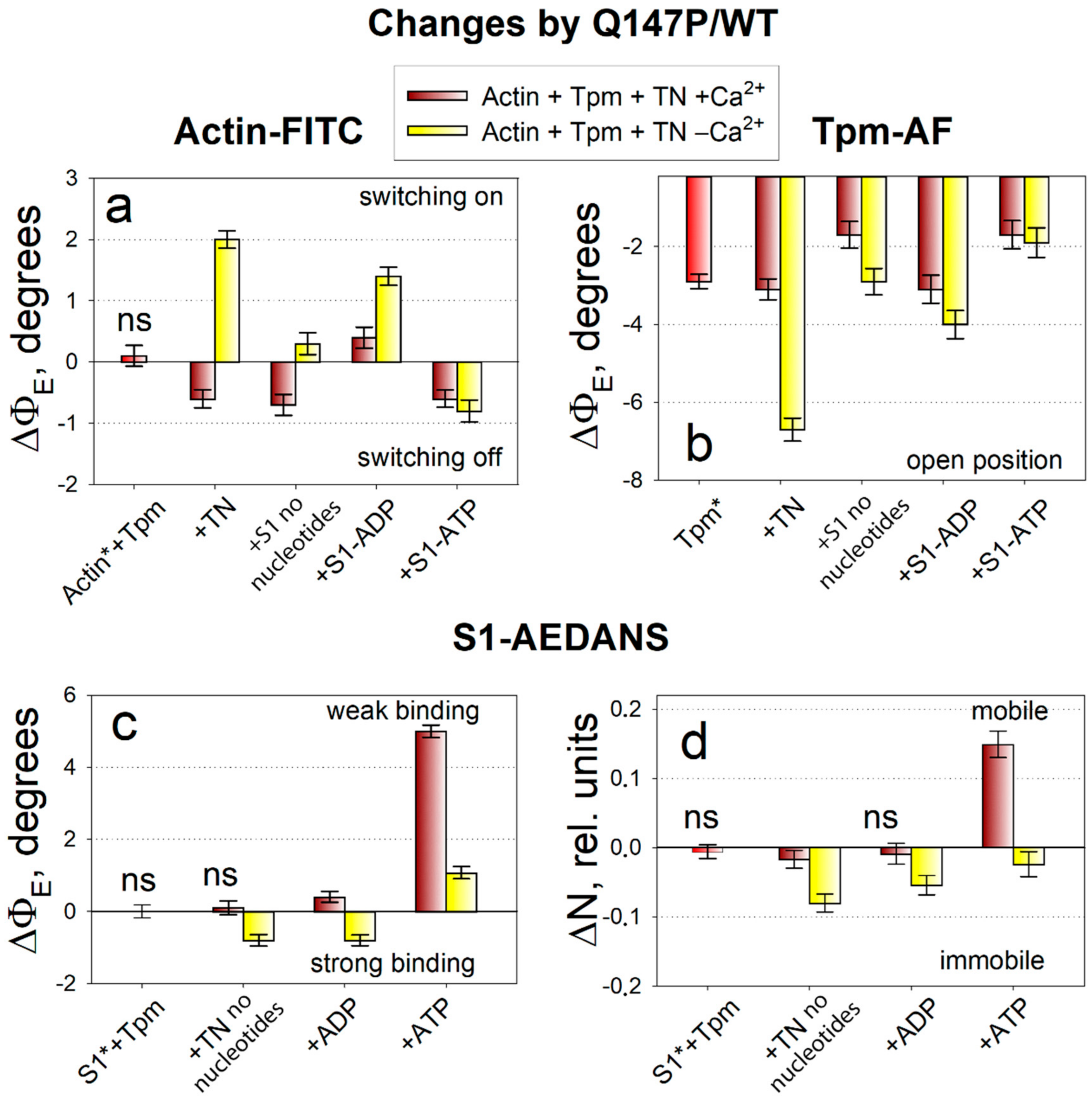
2.3. The Influence of The Q147P Substitution in Tpm on Conformational Rearrangements of Actin, Tpm and the Myosin Heads
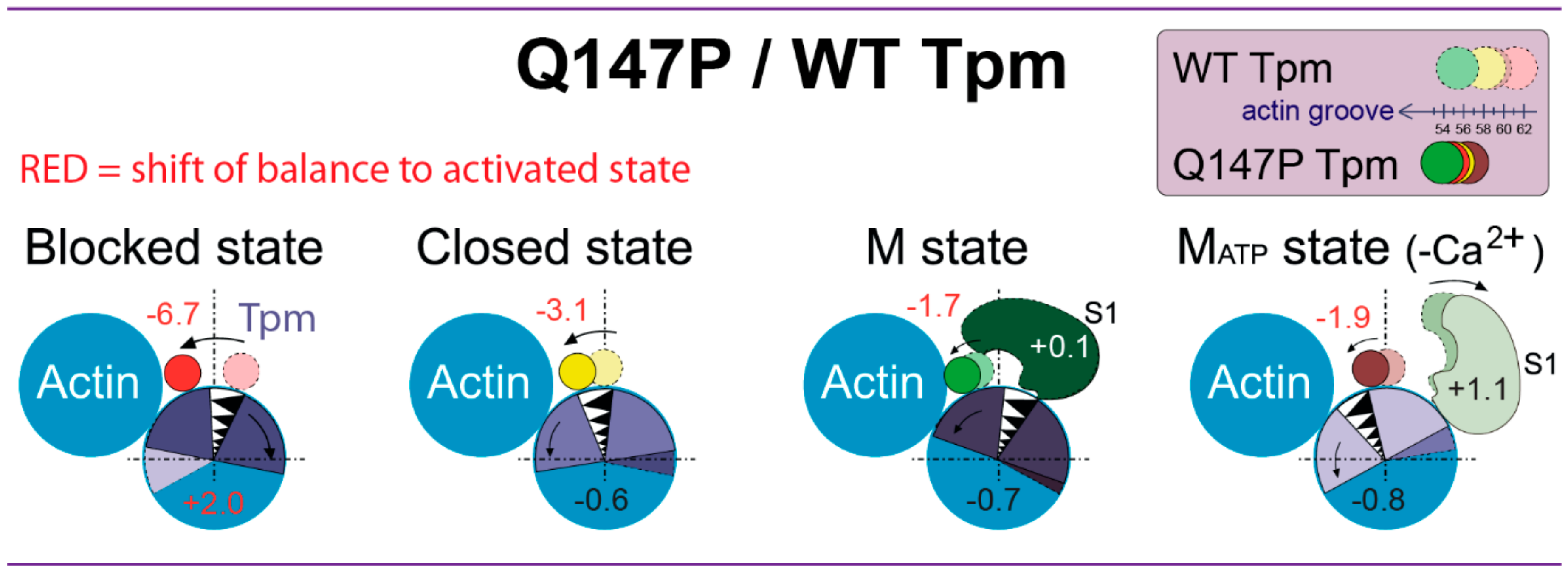
3. Discussion
4. Materials and Methods
4.1. Proteins Preparation and Its Modification by Fluorescent Probes
4.2. Ghost Muscle Fibers Preparation
4.3. Polarized Microfluorimetry Technique
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Tpm | Tropomyosin |
| WT Tpm | Wild-type tropomyosin |
| Q147P Tpm | Tropomyosin with the Gln147Pro substitution |
| TN | Troponin |
| S1 | Myosin subfragment-1 |
| 1,5-IAEDANS | N-(iodoacetaminoethyl)-1-naphthyl-amine-5-sulfonic acid |
| 5-IAF | 5-Iodoacetamidofluorescein |
| FITC | Fluorescein isothiocyanate |
| IVMA | In vitro motility assay |
| NGS | Next-generation sequencing |
References
- Boussouf, S.E.; Geeves, M.A. Tropomyosin and troponin cooperativity on the thin filament. Adv. Exp. Med. Biol. 2007, 592, 99–109. [Google Scholar]
- Geeves, M.A.; Lehrer, S.S.; Lehman, W. The mechanism of thin filament regulation: Models in conflict? J. Gen. Physiol. 2019, 151, 1265–1271. [Google Scholar] [CrossRef] [Green Version]
- Borovikov, Y.S.; Karpicheva, O.E.; Avrova, S.V.; Redwood, C.S. Modulation of the effects of tropomyosin on actin and myosin conformational changes by troponin and Ca2+. Biochim. Biophys. Acta—Proteins Proteomics 2009, 1794, 985–994. [Google Scholar] [CrossRef]
- Craig, R.; Lehman, W. Crossbridge and tropomyosin positions observed in native, interacting thick and thin filaments. J. Mol. Biol. 2001, 311, 1027–1036. [Google Scholar] [CrossRef]
- Rynkiewicz, M.J.; Schott, V.; Orzechowski, M.; Lehman, W.; Fischer, S. Electrostatic interaction map reveals a new binding position for tropomyosin on F-actin. J. Muscle Res. Cell Motil. 2015, 36, 525–533. [Google Scholar] [CrossRef] [Green Version]
- Li, X.E.; Lehman, W.; Fischer, S. The relationship between curvature, flexibility and persistence length in the tropomyosin coiled-coil. J. Struct. Biol. 2010, 170, 313–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehman, W.; Orzechowski, M.; Li, X.E.; Fischer, S.; Raunser, S. Gestalt-Binding of tropomyosin on actin during thin filament activation. J. Muscle Res. Cell Motil. 2013, 34, 155–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hitchcock-DeGregori, S.E. Tropomyosin: Function follows structure. Adv. Exp. Med. Biol. 2008, 644, 60–72. [Google Scholar] [PubMed]
- Mason, J.M.; Arndt, K.M. Coiled Coil Domains: Stability, Specificity, and Biological Implications. Chembiochem. 2004, 5, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Nance, J.R.; Dowling, J.J.; Gibbs, E.M.; Bönnemann, C.G. Congenital myopathies: An update. Curr. Neurol. Neurosci. Rep. 2012, 12, 165–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finsterer, J.; Stöllberger, C. Review of Cardiac Disease in Nemaline Myopathy. Pediatr. Neurol. 2015, 53, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Gonorazky, H.D.; Bonnemann, C.G.; Dowling, J.J. The genetics of congenital myopathies. Handb. Clin. Neurol. 2018, 148, 549–564. [Google Scholar]
- Jungbluth, H.; Treves, S.; Zorzato, F.; Sarkozy, A.; Ochala, J.; Sewry, C.; Phadke, R.; Gautel, M.; Muntoni, F. Congenital myopathies: Disorders of excitation-contraction coupling and muscle contraction. Nat. Rev. Neurol. 2018, 14, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.; Spendiff, S.; Roos, A.; Bourque, P.R.; Warman Chardon, J.; Kirschner, J.; Horvath, R.; Lochmüller, H. Advances in the diagnosis of inherited neuromuscular diseases and implications for therapy development. Lancet. Neurol. 2020, 19, 522–532. [Google Scholar] [CrossRef]
- Davidson, A.E.; Siddiqui, F.M.; Lopez, M.A.; Lunt, P.; Carlson, H.A.; Moore, B.E.; Love, S.; Born, D.E.; Roper, H.; Majumdar, A.; et al. Novel deletion of lysine 7 expands the clinical, histopathological and genetic spectrum of TPM2-related myopathies. Brain 2013, 136, 508–521. [Google Scholar] [CrossRef] [Green Version]
- Marttila, M.; Lehtokari, V.L.; Marston, S.; Nyman, T.A.; Barnerias, C.; Beggs, A.H.; Bertini, E.; Ceyhan-Birsoy, O.; Cintas, P.; Gerard, M.; et al. Mutation update and genotype-phenotype correlations of novel and previously described mutations in TPM2 and TPM3 causing congenital myopathies. Hum. Mutat. 2014, 35, 779–790. [Google Scholar] [CrossRef] [Green Version]
- Tajsharghi, H.; Ohlsson, M.; Palm, L.; Oldfors, A. Myopathies associated with β-tropomyosin mutations. Neuromuscul. Disord. 2012, 22, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Clarke, N.F.; Kolski, H.; Dye, D.E.; Lim, E.; Smith, R.L.; Patel, R.; Fahey, M.C.; Bellance, R.; Romero, N.B.; Johnson, E.S.; et al. Mutations in TPM3 are a common cause of congenital fiber type disproportion. Ann. Neurol. 2008, 63, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Sewry, C.A.; Wallgren-Pettersson, C. Myopathology in congenital myopathies. Neuropathol. Appl. Neurobiol. 2017, 43, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Donner, K.; Ollikainen, M.; Ridanpää, M.; Christen, H.J.; Goebel, H.H.; de Visser, M.; Pelin, K.; Wallgren-Pettersson, C. Mutations in the beta-tropomyosin (TPM2) gene—A rare cause of nemaline myopathy. Neuromuscul. Disord. 2002, 12, 151–158. [Google Scholar] [CrossRef]
- Brandis, A.; Aronica, E.; Goebel, H.H. TPM2 mutation. Neuromuscul. Disord. 2008, 18, 1005. [Google Scholar] [CrossRef]
- Borovikov, Y.S.; Gusev, N.B. Effect of troponin-tropomyosin complex and Ca2+ on conformational changes in F-actin induced by myosin subfragment-1. Eur. J. Biochem. 1983, 136, 363–369. [Google Scholar] [CrossRef]
- Iwamoto, H. Effects of myosin inhibitors on the X-ray diffraction patterns of relaxed and calcium-activated rabbit skeletal muscle fibers. Biophys. Physicobiol. 2018, 15, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Mckillop, D.F.A.; Geeves, M.A. Regulation of the Interaction between Actin and Myosin Subfragment 1: Evidence for Three States of the Thin Filament. Biophys. J. 1993, 65, 693–701. [Google Scholar] [CrossRef] [Green Version]
- Avrova, S.V.; Karpicheva, O.E.; Simonyan, A.O.; Sirenko, V.V.; Redwood, C.S.; Borovikov, Y.S. The molecular mechanisms of a high Ca(2+)-sensitivity and muscle weakness associated with the Ala155Thr substitution in Tpm3.12. Biochem. Biophys. Res. Commun. 2019, 515, 372–377. [Google Scholar] [CrossRef]
- Borovikov, Y.S.; Rysev, N.A.; Karpicheva, O.E.; Sirenko, V.V.; Avrova, S.V.; Piers, A.; Redwood, C.S. Molecular mechanisms of dysfunction of muscle fibers associated with Glu139 deletion in TPM2 gene. Sci. Rep. 2017, 7, 16797. [Google Scholar] [CrossRef] [Green Version]
- Oda, T.; Namba, K.; Maeda, Y. Position and orientation of phalloidin in F-actin determined by X-ray fiber diffraction analysis. Biophys. J. 2005, 88, 2727–2736. [Google Scholar] [CrossRef] [Green Version]
- Borovikov, Y.S.; Simonyan, A.O.; Avrova, S.V.; Sirenko, V.V.; Redwood, C.S.; Karpicheva, O.E. Molecular Mechanisms of Muscle Weakness Associated with E173A Mutation in Tpm3.12. Troponin Ca(2+) Sensitivity Inhibitor W7 Can Reduce the Damaging Effect of This Mutation. Int. J. Mol. Sci. 2020, 21, 4421. [Google Scholar] [CrossRef]
- Borovikov, Y.S.; Karpicheva, O.E.; Simonyan, A.O.; Avrova, S.V.; Rogozovets, E.A.; Sirenko, V.V.; Redwood, C.S. The Primary Causes of Muscle Dysfunction Associated with the Point Mutations in Tpm3.12; Conformational Analysis of Mutant Proteins as a Tool for Classification of Myopathies. Int. J. Mol. Sci. 2018, 19, 3975. [Google Scholar] [CrossRef] [Green Version]
- Karpicheva, O.E.; Simonyan, A.O.; Kuleva, N.V.; Redwood, C.S.; Borovikov, Y.S. Myopathy-causing Q147P TPM2 mutation shifts tropomyosin strands further towards the open position and increases the proportion of strong-binding cross-bridges during the ATPase cycle. Biochim. Biophys. Acta 2016, 1864, 260–267. [Google Scholar] [CrossRef]
- Moraczewska, J. Thin filament dysfunctions caused by mutations in tropomyosin Tpm3.12 and Tpm1.1. J. Muscle Res. Cell Motil. 2020, 41, 39–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marttila, M.; Lemola, E.; Wallefeld, W.; Memo, M.; Donner, K.; Laing, N.G.; Marston, S.; Grönholm, M.; Wallgren-Pettersson, C. Abnormal actin binding of aberrant β-tropomyosins is a molecular cause of muscle weakness in TPM2-related nemaline and cap myopathy. Biochem. J. 2012, 442, 231–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marston, S.B. Why is there a limit to the changes in myofilament Ca2+-sensitivity associated with myopathy causing mutations? Front. Physiol. 2016, 7, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orzechowski, M.; Fischer, S.; Moore, J.R.; Lehman, W.; Farman, G.P. Energy landscapes reveal the myopathic effects of tropomyosin mutations. Arch. Biochem. Biophys. 2014, 564, 89–99. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.H. How sequence directs bending in tropomyosin and other two-stranded alpha-helical coiled coils. Protein Sci. 2010, 19, 1366–1375. [Google Scholar] [CrossRef] [Green Version]
- Hitchcock-DeGregori, S.E.; Barua, B. Tropomyosin Structure, Function, and Interactions: A Dynamic Regulator. Subcell Biochem. 2017, 82, 253–284. [Google Scholar]
- Robaszkiewicz, K.; Dudek, E.; Kasprzak, A.A.; Moraczewska, J. Functional effects of congenital myopathy-related mutations in gamma-tropomyosin gene. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 1562–1569. [Google Scholar] [CrossRef] [Green Version]
- Donkervoort, S.; Papadaki, M.; de Winter, J.M.; Neu, M.B.; Kirschner, J.; Bolduc, V.; Yang, M.L.; Gibbons, M.A.; Hu, Y.; Dastgir, J.; et al. TPM3 deletions cause a hypercontractile congenital muscle stiffness phenotype. Ann. Neurol. 2015, 78, 985–994. [Google Scholar] [CrossRef] [Green Version]
- Redwood, C.; Robinson, P. Alpha-tropomyosin mutations in inherited cardiomyopathies. J. Muscle Res. Cell Motil. 2013, 34, 285–294. [Google Scholar] [CrossRef]
- Zheng, W.; Hitchcock-DeGregori, S.E.; Barua, B. Investigating the effects of tropomyosin mutations on its flexibility and interactions with filamentous actin using molecular dynamics simulation. J. Muscle Res. Cell Motil. 2016, 37, 131–147. [Google Scholar] [CrossRef]
- Moraczewska, J.; Robaszkiewicz, K.; Śliwinska, M.; Czajkowska, M.; Ly, T.; Kostyukova, A.; Wen, H.; Zheng, W. Congenital myopathy-related mutations in tropomyosin disrupt regulatory function through altered actin affinity and tropomodulin binding. FEBS J. 2019, 286, 1877–1893. [Google Scholar] [CrossRef]
- Marttila, M.; Hanif, M.; Lemola, E.; Nowak, K.J.; Laitila, J.; Grönholm, M.; Wallgren-Pettersson, C.; Pelin, K. Nebulin interactions with actin and tropomyosin are altered by disease-causing mutations. Skelet. Muscle 2014, 4, 15. [Google Scholar] [CrossRef] [Green Version]
- Ilkovski, B.; Mokbel, N.; Lewis, R.A.; Walker, K.; Nowak, K.J.; Domazetovska, A.; Laing, N.G.; Fowler, V.M.; North, K.N.; Cooper, S.T. Disease severity and thin filament regulation in M9R TPM3 nemaline myopathy. J. Neuropathol. Exp. Neurol. 2008, 67, 867–877. [Google Scholar] [CrossRef] [Green Version]
- Ly, T.; Moroz, N.; Pappas, C.T.; Novak, S.M.; Tolkatchev, D.; Wooldridge, D.; Mayfield, R.M.; Helms, G.; Gregorio, C.C.; Kostyukova, A.S. The N-terminal tropomyosin- and actin-binding sites are important for leiomodin 2′s function. Mol. Biol. Cell 2016, 27, 2565–2575. [Google Scholar] [CrossRef] [Green Version]
- Robaszkiewicz, K.; Ostrowska, Z.; Marchlewicz, K.; Moraczewska, J. Tropomyosin isoforms differentially modulate the regulation of actin filament polymerization and depolymerization by cofilins. FEBS J. 2016, 283, 723–737. [Google Scholar] [CrossRef] [Green Version]
- Matyushenko, A.M.; Shchepkin, D.V.; Susorov, D.S.; Nefedova, V.V.; Kopylova, G.V.; Berg, V.Y.; Kleymenov, S.Y.; Levitsky, D.I. Structural and functional properties of αβ-heterodimers of tropomyosin with myopathic mutations Q147P and K49del in the β-chain. Biochem. Biophys. Res. Commun. 2019, 508, 934–939. [Google Scholar] [CrossRef]
- Clarke, N.F.; Domazetovska, A.; Waddell, L.; Kornberg, A.; McLean, C.; North, K.N. Cap disease due to mutation of the beta-tropomyosin gene (TPM2). Neuromuscul. Disord. 2009, 19, 348–351. [Google Scholar] [CrossRef]
- Ottenheijm, C.A.C.; Lawlor, M.W.; Stienen, G.J.M.; Granzier, H.; Beggs, A.H. Changes in cross-bridge cycling underlie muscle weakness in patients with tropomyosin 3-based myopathy. Hum. Mol. Genet. 2011, 20, 2015–2025. [Google Scholar] [CrossRef] [Green Version]
- Borovikov, Y.S. Conformational changes of contractile proteins and their role in muscle contraction. Int. Rev. Cytol. 1999, 189, 267–301. [Google Scholar]
- Borovikov, Y.S.; Dedova, I.V.; dos Remedios, C.G.; Vikhoreva, N.N.; Vikhorev, P.G.; Avrova, S.V.; Hazlett, T.L.; Van Der Meer, B.W. Fluorescence depolarization of actin filaments in reconstructed myofibers: The effect of S1 or pPDM-S1 on movements of distinct areas of actin. Biophys. J. 2004, 86, 3020–3029. [Google Scholar] [CrossRef] [Green Version]
- Lehman, W.; Li, X.; Kiani, F.A.; Moore, J.R.; Campbell, S.G.; Fischer, S.; Rynkiewicz, M.J. Precise Binding of Tropomyosin on Actin Involves Sequence-Dependent Variance in Coiled-Coil Twisting. Biophys. J. 2018, 115, 1082–1092. [Google Scholar] [CrossRef] [Green Version]
- Tasca, G.; Fattori, F.; Ricci, E.; Monforte, M.; Rizzo, V.; Mercuri, E.; Bertini, E.; Silvestri, G. Somatic mosaicism in TPM2-related myopathy with nemaline rods and cap structures. Acta Neuropathol. 2013, 125, 169–171. [Google Scholar] [CrossRef]
- Kiphuth, I.C.; Krause, S.; Huttner, H.B.; Dekomien, G.; Struffert, T.; Schröder, R. Autosomal dominant nemaline myopathy caused by a novel α-tropomyosin 3 mutation. J. Neurol. 2010, 257, 658–660. [Google Scholar] [CrossRef]
- Sewry, C.A.; Laitila, J.M.; Wallgren-Pettersson, C. Nemaline myopathies: A current view. J. Muscle Res. Cell Motil. 2019, 40, 111–126. [Google Scholar] [CrossRef] [Green Version]
- Malfatti, E.; Schaeffer, U.; Chapon, F.; Yang, Y.; Eymard, B.; Xu, R.; Laporte, J.; Romero, N.B. Combined cap disease and nemaline myopathy in the same patient caused by an autosomal dominant mutation in the TPM3 gene. Neuromuscul. Disord. 2013, 23, 992–997. [Google Scholar] [CrossRef]
- Lehtokari, V.L.; Ceuterick-de Groote, C.; de Jonghe, P.; Marttila, M.; Laing, N.G.; Pelin, K.; Wallgren-Pettersson, C. Cap disease caused by heterozygous deletion of the beta-tropomyosin gene TPM2. Neuromuscul. Disord. 2007, 17, 433–442. [Google Scholar] [CrossRef]
- Singh, A.; Hitchcock-degregori, S.E. Local Destabilization of the Tropomyosin Coiled Coil Gives the Molecular Flexibility Required for Actin Binding. Biochemistry 2003, 42, 14114–14121. [Google Scholar] [CrossRef]
- Kwok, S.C.; Hodges, R.S. Stabilizing and Destabilizing Clusters in the Hydrophobic Core of Long Two-stranded α-Helical Coiled-coils. J. Biol. Chem. 2004, 279, 21576–21588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marston, S.; Memo, M.; Messer, A.; Papadaki, M.; Nowak, K.; McNamara, E.; Ong, R.; El-Mezgueldi, M.; Li, X.; Lehman, W. Mutations in repeating structural motifs of TM cause gain of function in skeletal muscle myopathy patients. Hum. Mol. Genet. 2013, 22, 4978–4987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpicheva, O.E.; Sirenko, V.V.; Rysev, N.A.; Simonyan, A.O.; Borys, D.; Moraczewska, J.; Borovikov, Y.S. Deviations in conformational rearrangements of thin filaments and myosin caused by the Ala155Thr substitution in hydrophobic core of tropomyosin. Biochim. Biophys. Acta - Proteins Proteomics 2017, 1865, 1790–1799. [Google Scholar] [CrossRef] [PubMed]
- Borovikov, Y.S.; Karpicheva, O.E.; Avrova, S.V.; Simonyan, A.O.; Sirenko, V.V.; Redwood, C.S. The molecular mechanism of muscle dysfunction associated with the R133W mutation in Tpm2.2. Biochem. Biophys. Res. Commun. 2020, 523, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.; Lipscomb, S.; Preston, L.C.; Altin, E.; Watkins, H.; Ashley, C.C.; Redwood, C. Mutations in fast skeletal troponin I, troponin T, and beta-tropomyosin that cause distal arthrogryposis all increase contractile function. FASEB J. 2007, 21, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, P.B.; Lataro, R.C.; Ferro, J.A.; de C Reinach, F. Functional alpha-tropomyosin produced in Escherichia coli. A dipeptide extension can substitute the amino-terminal acetyl group. J. Biol. Chem. 1994, 269, 10461–10466. [Google Scholar] [PubMed]
- Margossian, S.; Lowey, S. Preparation of myosin and its subfragments from rabbit skeletal muscle. Methods Enzym. 1982, 85, 55–71. [Google Scholar]
- Okamoto, Y.; Sekine, T. A streamlined method of subfragment one preparation from myosin. J. Biochem. 1985, 98, 1143–1145. [Google Scholar] [CrossRef] [PubMed]
- Potter, J.D. Preparation of TN and its subunits. Methods Enzymol. 1982, 85, 241–263. [Google Scholar] [PubMed]
- Borovikov, Y.S.; Avrova, S.V.; Rysev, N.A.; Sirenko, V.V.; Simonyan, A.O.; Chernev, A.A.; Karpicheva, O.E.; Piers, A.; Redwood, C.S. Aberrant movement of β-tropomyosin associated with congenital myopathy causes defective response of myosin heads and actin during the ATPase cycle. Arch. Biochem. Biophys. 2015, 577–578, 13–23. [Google Scholar] [CrossRef]
- Lamkin, M.; Tao, T.; Lehrer, S.S. Tropomyosin-troponin and tropomyosin-actin interactions: A fluorescence quenching study. Biochemistry 1983, 22, 3053–3058. [Google Scholar] [CrossRef]
- Szent-Gyorgyi, A. Free-energy relations and contraction of actomyosin. Biol. Bull. Rev. 1949, 96, 140–161. [Google Scholar] [CrossRef]
- Gordon, A.M.; Homsher, E.; Regnier, M. Regulation of contraction in striated muscle. Physiol. Rev. 2000, 80, 853–924. [Google Scholar] [CrossRef]
- Tregear, R.T.; Mendelson, R.A. Polarization from a helix of fluorophores and its relation to that obtained from muscle. Biophys. J. 1975, 15, 455–467. [Google Scholar] [CrossRef] [Green Version]
- Borejdo, J.; Putnam, S. Polarization of flourescence from single skinned glycerinated rabbit psoas fibers in rigor and relaxation. Biochim. Biophys. Acta 1977, 459, 578–595. [Google Scholar] [CrossRef]
- Roopnarine, O.; Thomas, D.D. Orientation of intermediate nucleotide states of indane dione spin-labeled myosin heads in muscle fibers. Biophys. J. 1996, 70, 2795–2806. [Google Scholar] [CrossRef] [Green Version]
- Yanagida, T.; Oosawa, F. Polarized fluorescence from ε-ADP incorporated into F-actin in a myosin-free single fiber: Conformation of F-actin and changes induced in it by heavy meromyosin. J. Mol. Biol. 1978, 126, 507–524. [Google Scholar] [CrossRef]
- Rysev, N.A.; Karpicheva, O.E.; Redwood, C.S.; Borovikov, Y.S. The effect of the Asp175Asn and Glu180Gly TPM1 mutations on actin-myosin interaction during the ATPase cycle. Biochim. Biophys. Acta 2012, 1824, 366–373. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karpicheva, O.E.; Simonyan, A.O.; Rysev, N.A.; Redwood, C.S.; Borovikov, Y.S. Looking for Targets to Restore the Contractile Function in Congenital Myopathy Caused by Gln147Pro Tropomyosin. Int. J. Mol. Sci. 2020, 21, 7590. https://doi.org/10.3390/ijms21207590
Karpicheva OE, Simonyan AO, Rysev NA, Redwood CS, Borovikov YS. Looking for Targets to Restore the Contractile Function in Congenital Myopathy Caused by Gln147Pro Tropomyosin. International Journal of Molecular Sciences. 2020; 21(20):7590. https://doi.org/10.3390/ijms21207590
Chicago/Turabian StyleKarpicheva, Olga E., Armen O. Simonyan, Nikita A. Rysev, Charles S. Redwood, and Yurii S. Borovikov. 2020. "Looking for Targets to Restore the Contractile Function in Congenital Myopathy Caused by Gln147Pro Tropomyosin" International Journal of Molecular Sciences 21, no. 20: 7590. https://doi.org/10.3390/ijms21207590
APA StyleKarpicheva, O. E., Simonyan, A. O., Rysev, N. A., Redwood, C. S., & Borovikov, Y. S. (2020). Looking for Targets to Restore the Contractile Function in Congenital Myopathy Caused by Gln147Pro Tropomyosin. International Journal of Molecular Sciences, 21(20), 7590. https://doi.org/10.3390/ijms21207590