Deferasirox-Dependent Iron Chelation Enhances Mitochondrial Dysfunction and Restores p53 Signaling by Stabilization of p53 Family Members in Leukemic Cells


 , ,
, ,  , ,
, ,  ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. Results
2.1. Iron Chelation Induces the Fragmentation of Mitochondrial Network and a Dysfunction in the Oxidative Phosphorylation in Acute Myeloid Leukemia Cell Lines
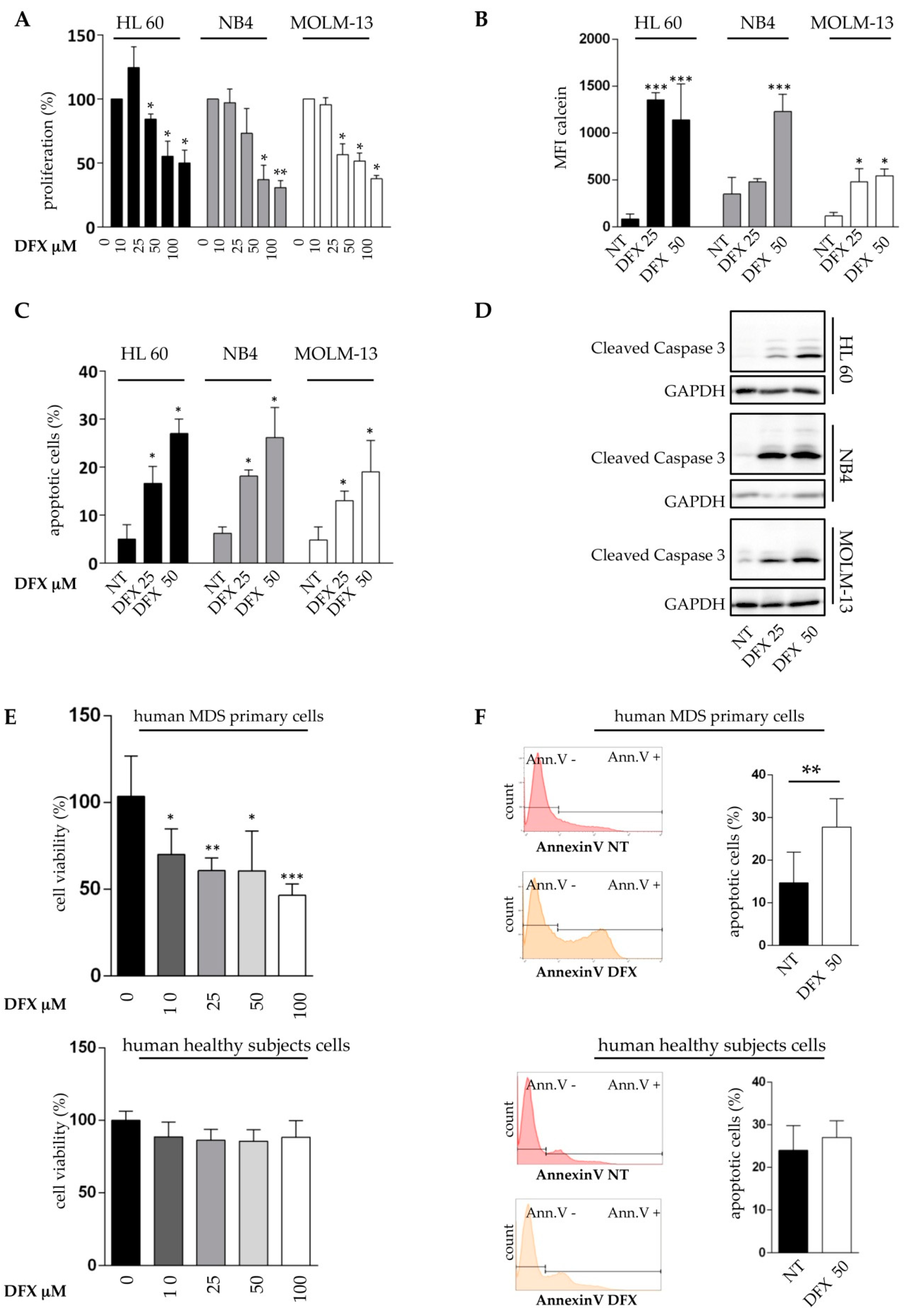
2.2. Deferasirox Exerts In Vitro Anti-Leukemic Activity on Acute Myeloid Leukemia Cell Lines and on Primary MDS Cells
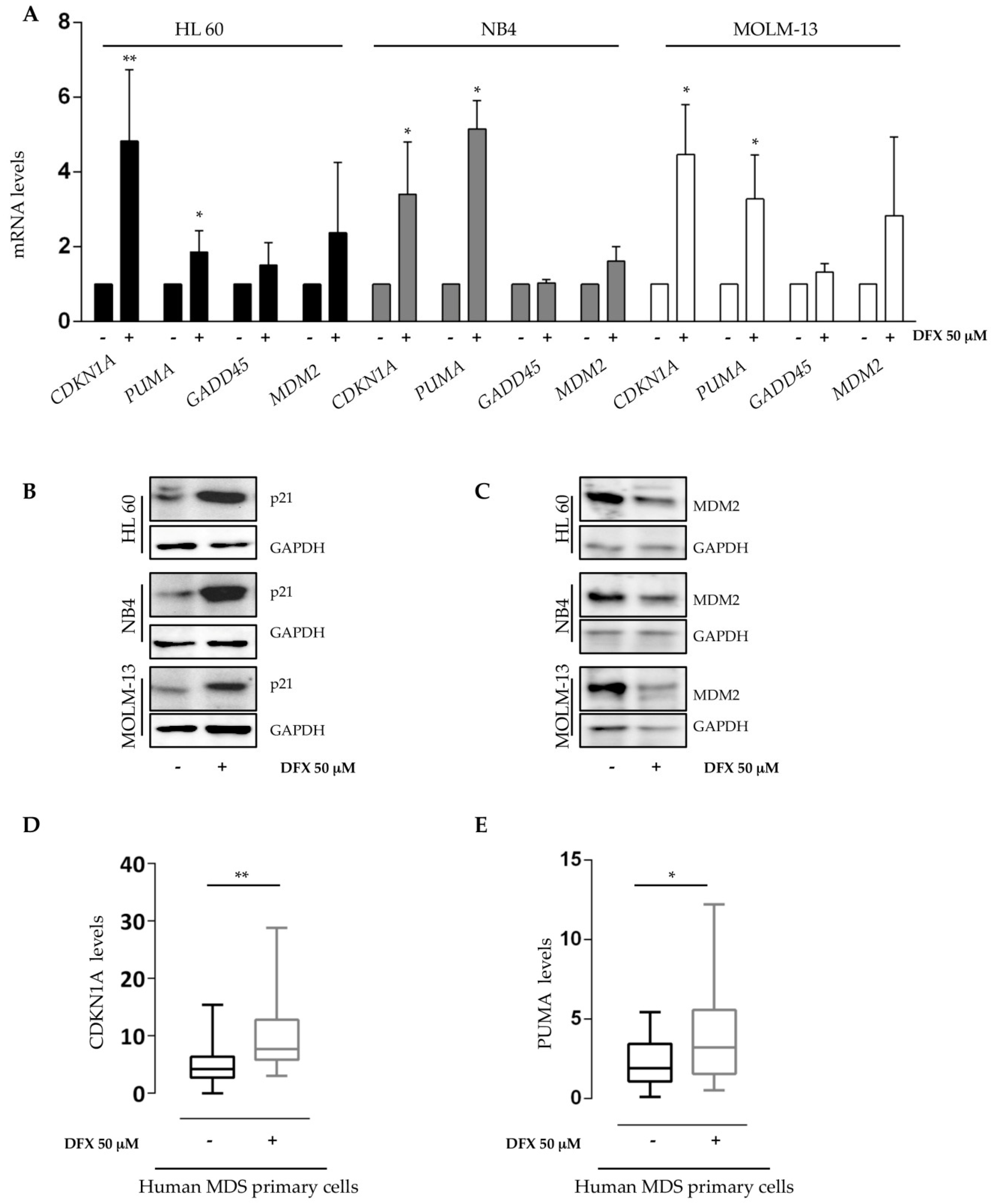
2.3. Deferasirox Activates p53 Targets on Acute Myeloid Leukemia Cell Lines and on Primary MDS/AML Cells
2.4. Deferasirox Regulates p53 and p73 Protein Stability
3. Discussion
4. Materials and Methods
4.1. Cell Culture Conditions
4.2. Patients Cohort
4.3. Cell Treatment and Calcein Fluorescence Assay
4.4. Proliferation and Apoptosis Assay
4.5. RNA Extraction and qRT-PCR Analysis
4.6. MitoTracker Staining and Morphological Analysis of Mitochondria
4.7. Immunofluorescence Assay
4.8. Protein Extraction and Immunoblotting
4.9. Gene Expression Analysis in Deferasirox-Treated Cells
4.10. Evaluation of ATP/AMP Ratio as Marker of Cellular Energy Status
4.11. Oxygen Consumption Rate (OCR), ATP Synthesis, and P/O Ratio Evaluation
4.12. Immunohistochemistry on MDS Bone Marrow Samples
4.13. Statistical analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ROS | Reactive Oxygen Species |
| MDS | Myelodysplastic Syndromes |
| AL | Acute Leukemias |
| IOL | iron overload |
| RBC | red blood cell |
| ICT | iron chelation therapy |
| OS | overall survival |
| DFX | iron chelator Deferasirox |
| DNA | DeoxyRibonucleic Acid |
| RNA | RiboNucleic Acid |
| NF-Κb | nuclear factor kappa B |
| CKI | cyclin-dependent kinase |
| CDKN1A (p21 protein) | Cyclin-Dependent Kinase Inhibitor 1A |
| PUMA (BBC3) | p53 upregulated modulator of apoptosis |
| MDM2 | mouse double minute 2 homolog |
| FBS | fetal bovine serum |
| PB | peripheral blood |
| BM | bone marrow |
| WBCs | White blood cells |
| LIP | labile iron pool |
| CA-AM | calcein acetoxymethyl ester |
| MFI | Mean Fluorescence Intensity |
| MTT | bromide 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium |
| FITC | fluorescein isothiocyanate |
| DMSO | dimethyl sulfoxide |
| MiNA | Mitochondrial Network Analysis |
| SDS | Sodium dodecyl sulfate |
| DOC | deoxycolate |
| SDS-PAGE | Sodium Dodecyl Sulphate-PolyAcrylamide Gel Electrophoresis |
| BSA | Bovine serum albumin |
| TBS | Tris Buffered Saline |
| RT | retrotrascriptin |
| ON | over night |
| GAPDH | glyceraldheyde-3-phosphate dehydrogenase |
| GO | Gene Ontology |
| PID | Pathway Interaction Database |
| FACS | Fluorescence-activated cell sorting |
| DRP1 | Dynamin-related protein 1 |
| OPA1 | Mitochondrial Dynamin-Like GTPase |
| GADD45 | Growth Arrest and DNA Damage 45 |
| PMAIP1(NOXA) | Phorbol-12-Myristate-13-Acetate-Induced Protein 1 |
| MCL1 | myeloid cell leukemia 1 |
| GDF15 | Growth Differentiation Factor 15 |
References
- Pantopoulos, K.; Porwal, S.K.; Tartakoff, A.; Devireddy, L. Mechanisms of mammalian iron homeostasis. Biochemistry 2012, 51, 5705–5724. [Google Scholar] [CrossRef] [PubMed]
- Torti, S.V.; Torti, F.M. Iron and cancer: More ore to be mined. Nat. Rev. Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyokuni, S. Role of iron in carcinogenesis: Cancer as a ferrotoxic disease. Cancer Sci. 2009, 100, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Pilo, F.; Angelucci, E. A storm in the niche: Iron, oxidative stress and haemopoiesis. Blood Rev. 2018, 32, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Buss, J.L.; Greene, B.T.; Turner, J.; Torti, F.M.; Torti, S.V. Iron chelators in cancer chemotherapy. Curr. Top. Med. Chem. 2004, 4, 1623–1635. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.; Koumenis, C.; Kute, T.E.; Planalp, R.P.; Brechbiel, M.W.; Beardsley, D.; Cody, B.; Brown, K.D.; Torti, F.M.; Torti, S.V. Tachpyridine, a metal chelator, induces G2 cell-cycle arrest, activates checkpoint kinases, and sensitizes cells to ionizing radiation. Blood 2005, 106, 3191–3199. [Google Scholar] [CrossRef] [Green Version]
- Zeidan, A.M.; Griffiths, E.A. To chelate or not to chelate in MDS: That is the question! Blood Rev. 2018, 32, 368–377. [Google Scholar] [CrossRef]
- Malcovati, L.; Della Porta, M.G.; Cazzola, M. Predicting survival and leukemic evolution in patients with myelodysplastic syndrome. Haematologica 2006, 91, 1588–1590. [Google Scholar]
- Rose, C.; Brechignac, S.; Vassilief, D.; Pascal, L.; Stamatoullas, A.; Guerci, A.; Larbaa, D.; Dreyfus, F.; Beyne-Rauzy, O.; Chaury, M.P.; et al. Does iron chelation therapy improve survival in regularly transfused lower risk MDS patients? A multicenter study by the GFM (Groupe Francophone des Myelodysplasies). Leuk. Res. 2010, 34, 864–870. [Google Scholar] [CrossRef]
- Raptis, A.; Duh, M.S.; Wang, S.T.; Dial, E.; Fanourgiakis, I.; Fortner, B.; Paley, C.; Mody-Patel, N.; Corral, M.; Scott, J. Treatment of transfusional iron overload in patients with myelodysplastic syndrome or severe anemia: Data from multicenter clinical practices. Transfusion 2010, 50, 190–199. [Google Scholar] [CrossRef]
- Neukirchen, J.; Fox, F.; Kundgen, A.; Nachtkamp, K.; Strupp, C.; Haas, R.; Germing, U.; Gattermann, N. Improved survival in MDS patients receiving iron chelation therapy—A matched pair analysis of 188 patients from the Dusseldorf MDS registry. Leuk. Res. 2012, 36, 1067–1070. [Google Scholar] [CrossRef] [PubMed]
- Delforge, M.; Selleslag, D.; Beguin, Y.; Triffet, A.; Mineur, P.; Theunissen, K.; Graux, C.; Trullemans, F.; Boulet, D.; Van Eygen, K.; et al. Adequate iron chelation therapy for at least six months improves survival in transfusion-dependent patients with lower risk myelodysplastic syndromes. Leuk. Res. 2014, 38, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Hoeks, M.; Yu, G.; Langemeijer, S.; Crouch, S.; de Swart, L.; Fenaux, P.; Symeonidis, A.; Cermak, J.; Hellstrom-Lindberg, E.; Sanz, G.; et al. Impact of treatment with iron chelation therapy in patients with lower-risk myelodysplastic syndromes participating in the European MDS registry. Haematologica 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Lu, W.Y.; Zhao, M.F.; Cao, X.L.; Jiang, Y.Y.; Jin, X.; Xu, P.; Yuan, T.T.; Zhang, Y.C.; Chai, X.; et al. Reactive oxygen species mediated T lymphocyte abnormalities in an iron-overloaded mouse model and iron-overloaded patients with myelodysplastic syndromes. Ann. Hematol. 2017, 96, 1085–1095. [Google Scholar] [CrossRef]
- Leitch, H.A.; Parmar, A.; Wells, R.A.; Chodirker, L.; Zhu, N.; Nevill, T.J.; Yee, K.W.L.; Leber, B.; Keating, M.M.; Sabloff, M.; et al. Overall survival in lower IPSS risk MDS by receipt of iron chelation therapy, adjusting for patient-related factors and measuring from time of first red blood cell transfusion dependence: An MDS-CAN analysis. Br. J. Haematol. 2017, 179, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, T.; Kawabata, H.; Nakamura, T.; Iwao, H.; Nakajima, A.; Miki, M.; Sakai, T.; Sawaki, T.; Fujita, Y.; Tanaka, M.; et al. Iron chelation therapy with deferasirox induced complete remission in a patient with chemotherapy-resistant acute monocytic leukemia. Anticancer Res. 2011, 31, 1741–1744. [Google Scholar]
- Kikuchi, S.; Kobune, M.; Iyama, S.; Sato, T.; Murase, K.; Kawano, Y.; Takada, K.; Ono, K.; Kaneko, Y.; Miyanishi, K.; et al. Improvement of iron-mediated oxidative DNA damage in patients with transfusion-dependent myelodysplastic syndrome by treatment with deferasirox. Free Radic. Biol. Med. 2012, 53, 643–648. [Google Scholar] [CrossRef]
- Ohyashiki, J.H.; Kobayashi, C.; Hamamura, R.; Okabe, S.; Tauchi, T.; Ohyashiki, K. The oral iron chelator deferasirox represses signaling through the mTOR in myeloid leukemia cells by enhancing expression of REDD1. Cancer Sci. 2009, 100, 970–977. [Google Scholar] [CrossRef]
- Messa, E.; Carturan, S.; Maffe, C.; Pautasso, M.; Bracco, E.; Roetto, A.; Messa, F.; Arruga, F.; Defilippi, I.; Rosso, V.; et al. Deferasirox is a powerful NF-kappaB inhibitor in myelodysplastic cells and in leukemia cell lines acting independently from cell iron deprivation by chelation and reactive oxygen species scavenging. Haematologica 2010, 95, 1308–1316. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.X.; Richardson, D.R. The effect of potent iron chelators on the regulation of p53: Examination of the expression, localization and DNA-binding activity of p53 and the transactivation of WAF1. Carcinogenesis 2003, 24, 1601–1614. [Google Scholar] [CrossRef] [Green Version]
- Fukuchi, K.; Tomoyasu, S.; Watanabe, H.; Kaetsu, S.; Tsuruoka, N.; Gomi, K. Iron deprivation results in an increase in p53 expression. Biol. Chem. Hoppe Seyler 1995, 376, 627–630. [Google Scholar] [CrossRef]
- Ba, Q.; Hao, M.; Huang, H.; Hou, J.; Ge, S.; Zhang, Z.; Yin, J.; Chu, R.; Jiang, H.; Wang, F.; et al. Iron deprivation suppresses hepatocellular carcinoma growth in experimental studies. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 7625–7633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Kong, X.; Zhang, Y.; Sun, W.; Wang, J.; Chen, M.; Chen, X. FDXR regulates TP73 tumor suppressor via IRP2 to modulate aging and tumor suppression. J. Pathol. 2020. [Google Scholar] [CrossRef]
- Zhang, Y.; Qian, Y.; Zhang, J.; Yan, W.; Jung, Y.S.; Chen, M.; Huang, E.; Lloyd, K.; Duan, Y.; Wang, J.; et al. Ferredoxin reductase is critical for p53-dependent tumor suppression via iron regulatory protein 2. Genes Dev. 2017, 31, 1243–1256. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Feng, X.; Zhang, J.; Chen, M.; Huang, E.; Chen, X. Iron regulatory protein 2 is a suppressor of mutant p53 in tumorigenesis. Oncogene 2019, 38, 6256–6269. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, M.; Taya, Y.; Vousden, K.H. Stress signals utilize multiple pathways to stabilize p53. Mol. Cell. Biol. 2000, 20, 3224–3233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Sheng, X.; Chang, Z.; Wu, Q.; Wang, S.; Xuan, Z.; Li, D.; Wu, Y.; Shang, Y.; Kong, X.; et al. Iron metabolism regulates p53 signaling through direct heme-p53 interaction and modulation of p53 localization, stability, and function. Cell Rep. 2014, 7, 180–193. [Google Scholar] [CrossRef] [Green Version]
- Sionov, R.V.; Haupt, Y. The cellular response to p53: The decision between life and death. Oncogene 1999, 18, 6145–6157. [Google Scholar] [CrossRef] [Green Version]
- Prives, C.; Hall, P.A. The p53 pathway. J. Pathol. 1999, 187, 112–126. [Google Scholar] [CrossRef]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef] [Green Version]
- Wallingford, J.B.; Seufert, D.W.; Virta, V.C.; Vize, P.D. p53 activity is essential for normal development in Xenopus. Curr. Biol. CB 1997, 7, 747–757. [Google Scholar] [CrossRef] [Green Version]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, T.; Nakagawara, A. Role of p53 in Cell Death and Human Cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef] [PubMed]
- Nigro, J.M.; Baker, S.J.; Preisinger, A.C.; Jessup, J.M.; Hostetter, R.; Cleary, K.; Bigner, S.H.; Davidson, N.; Baylin, S.; Devilee, P.; et al. Mutations in the p53 gene occur in diverse human tumour types. Nature 1989, 342, 705–708. [Google Scholar] [CrossRef]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Panuzzo, C.; Signorino, E.; Calabrese, C.; Ali, M.S.; Petiti, J.; Bracco, E.; Cilloni, D. Landscape of Tumor Suppressor Mutations in Acute Myeloid Leukemia. J. Clin. Med. 2020, 9, 802. [Google Scholar] [CrossRef] [Green Version]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A., Jr.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef]
- Rutkowski, R.; Hofmann, K.; Gartner, A. Phylogeny and function of the invertebrate p53 superfamily. Cold Spring Harb. Perspect. Biol. 2010, 2, a001131. [Google Scholar] [CrossRef] [Green Version]
- Nemajerova, A.; Palacios, G.; Nowak, N.J.; Matsui, S.; Petrenko, O. Targeted deletion of p73 in mice reveals its role in T cell development and lymphomagenesis. PLoS ONE 2009, 4, e7784. [Google Scholar] [CrossRef] [Green Version]
- Nemajerova, A.; Petrenko, O.; Trumper, L.; Palacios, G.; Moll, U.M. Loss of p73 promotes dissemination of Myc-induced B cell lymphomas in mice. J. Clin. Investig. 2010, 120, 2070–2080. [Google Scholar] [CrossRef]
- Flores, E.R.; Tsai, K.Y.; Crowley, D.; Sengupta, S.; Yang, A.; McKeon, F.; Jacks, T. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature 2002, 416, 560–564. [Google Scholar] [CrossRef]
- Tophkhane, C.; Yang, S.H.; Jiang, Y.; Ma, Z.; Subramaniam, D.; Anant, S.; Yogosawa, S.; Sakai, T.; Liu, W.G.; Edgerton, S.; et al. p53 inactivation upregulates p73 expression through E2F-1 mediated transcription. PLoS ONE 2012, 7, e43564. [Google Scholar] [CrossRef] [Green Version]
- El-Deiry, W.S.; Harper, J.W.; O’Connor, P.M.; Velculescu, V.E.; Canman, C.E.; Jackman, J.; Pietenpol, J.A.; Burrell, M.; Hill, D.E.; Wang, Y.; et al. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 1994, 54, 1169–1174. [Google Scholar]
- Dotsch, V.; Bernassola, F.; Coutandin, D.; Candi, E.; Melino, G. p63 and p73, the ancestors of p53. Cold Spring Harb. Perspect. Biol. 2010, 2, a004887. [Google Scholar] [CrossRef] [PubMed]
- Melino, G.; Bernassola, F.; Ranalli, M.; Yee, K.; Zong, W.X.; Corazzari, M.; Knight, R.A.; Green, D.R.; Thompson, C.; Vousden, K.H. p73 Induces apoptosis via PUMA transactivation and Bax mitochondrial translocation. J. Biol. Chem. 2004, 279, 8076–8083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moussa, R.S.; Kovacevic, Z.; Richardson, D.R. Differential targeting of the cyclin-dependent kinase inhibitor, p21CIP1/WAF1, by chelators with anti-proliferative activity in a range of tumor cell-types. Oncotarget 2015, 6, 29694–29711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef] [Green Version]
- Balint, E.; Bates, S.; Vousden, K.H. Mdm2 binds p73 alpha without targeting degradation. Oncogene 1999, 18, 3923–3929. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Chen, L.; Jost, C.A.; Maya, R.; Keller, D.; Wang, X.; Kaelin, W.G., Jr.; Oren, M.; Chen, J.; Lu, H. MDM2 suppresses p73 function without promoting p73 degradation. Mol. Cell. Biol. 1999, 19, 3257–3266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehman, J.; Zhang, H.J.; Toth, P.T.; Zhang, Y.; Marsboom, G.; Hong, Z.; Salgia, R.; Husain, A.N.; Wietholt, C.; Archer, S.L. Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2012, 26, 2175–2186. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.T.; Liu, X.; Ye, C.Y.; Tao, L.X.; Zhou, H.; Zhang, H.Y. Iron-induced energy supply deficiency and mitochondrial fragmentation in neurons. J. Neurochem. 2018, 147, 816–830. [Google Scholar] [CrossRef] [Green Version]
- Hage, S.; Stanga, S.; Marinangeli, C.; Octave, J.N.; Dewachter, I.; Quetin-Leclercq, J.; Kienlen-Campard, P. Characterization of Pterocarpus erinaceus kino extract and its gamma-secretase inhibitory properties. J. Ethnopharmacol. 2015, 163, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Panuzzo, C.; Volpe, G.; Cibrario Rocchietti, E.; Casnici, C.; Crotta, K.; Crivellaro, S.; Carra, G.; Lorenzatti, R.; Peracino, B.; Torti, D.; et al. New alternative splicing BCR/ABL-OOF shows an oncogenic role by lack of inhibition of BCR GTPase activity and an increased of persistence of Rac activation in chronic myeloid leukemia. Oncoscience 2015, 2, 880–891. [Google Scholar] [CrossRef]
- Carturan, S.; Petiti, J.; Rosso, V.; Calabrese, C.; Signorino, E.; Bot-Sartor, G.; Nicoli, P.; Gallo, D.; Bracco, E.; Morotti, A.; et al. Variable but consistent pattern of Meningioma 1 gene (MN1) expression in different genetic subsets of acute myelogenous leukaemia and its potential use as a marker for minimal residual disease detection. Oncotarget 2016, 7, 74082–74096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valente, A.J.; Maddalena, L.A.; Robb, E.L.; Moradi, F.; Stuart, J.A. A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochem. 2017, 119, 315–326. [Google Scholar] [CrossRef]
- Morotti, A.; Panuzzo, C.; Crivellaro, S.; Carra, G.; Guerrasio, A.; Saglio, G. HAUSP compartmentalization in chronic myeloid leukemia. Eur. J. Haematol. 2015, 94, 318–321. [Google Scholar] [CrossRef] [PubMed]
- Stanga, S.; Brambilla, L.; Tasiaux, B.; Dang, A.H.; Ivanoiu, A.; Octave, J.N.; Rossi, D.; van Pesch, V.; Kienlen-Campard, P. A Role for GDNF and Soluble APP as Biomarkers of Amyotrophic Lateral Sclerosis Pathophysiology. Front. Neurol. 2018, 9, 384. [Google Scholar] [CrossRef]
- Stanga, S.; Vrancx, C.; Tasiaux, B.; Marinangeli, C.; Karlstrom, H.; Kienlen-Campard, P. Specificity of presenilin-1- and presenilin-2-dependent gamma-secretases towards substrate processing. J. Cell. Mol. Med. 2018, 22, 823–833. [Google Scholar] [CrossRef] [Green Version]
- Hage, S.; Marinangeli, C.; Stanga, S.; Octave, J.N.; Quetin-Leclercq, J.; Kienlen-Campard, P. Gamma-secretase inhibitor activity of a Pterocarpus erinaceus extract. Neuro Degener. Dis. 2014, 14, 39–51. [Google Scholar] [CrossRef]
- Davis, S.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, G.K.; Michaud, J.; Scott, H.S. Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics 2005, 21, 2067–2075. [Google Scholar] [CrossRef]
- Kinsella, R.J.; Kahari, A.; Haider, S.; Zamora, J.; Proctor, G.; Spudich, G.; Almeida-King, J.; Staines, D.; Derwent, P.; Kerhornou, A.; et al. Ensembl BioMarts: A hub for data retrieval across taxonomic space. Database J. Biol. Databases Curation 2011, 2011, bar030. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, C.F.; Anthony, K.; Krupa, S.; Buchoff, J.; Day, M.; Hannay, T.; Buetow, K.H. PID: The Pathway Interaction Database. Nucleic Acids Res. 2009, 37, D674–D679. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, N.F.; Gundersen, G.W.; Rahman, A.; Grimes, M.L.; Rikova, K.; Hornbeck, P.; Ma’ayan, A. Clustergrammer, a web-based heatmap visualization and analysis tool for high-dimensional biological data. Sci. Data 2017, 4, 170151. [Google Scholar] [CrossRef] [PubMed]
- Cilloni, D.; Ravera, S.; Calabrese, C.; Gaidano, V.; Niscola, P.; Balleari, E.; Gallo, D.; Petiti, J.; Signorino, E.; Rosso, V.; et al. Iron overload alters the energy metabolism in patients with myelodysplastic syndromes: Results from the multicenter FISM BIOFER study. Sci. Rep. 2020, 10, 9156. [Google Scholar] [CrossRef]
- Hinkle, P.C. P/O ratios of mitochondrial oxidative phosphorylation. Biochim. Biophys. Acta 2005, 1706, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Vaseva, A.V.; Moll, U.M. The mitochondrial p53 pathway. Biochim. Biophys. Acta 2009, 1787, 414–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, L.; Cao, H.; Wang, G.; Lyu, Y.; Sun, X.; An, J.; Wu, Z.; Huang, Q.; Liu, B.; Xing, J. Drp1-mediated mitochondrial fission promotes cell proliferation through crosstalk of p53 and NF-kappaB pathways in hepatocellular carcinoma. Oncotarget 2016, 7, 65001–65011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Leng, R.P. MDM2 mediates p73 ubiquitination: A new molecular mechanism for suppression of p73 function. Oncotarget 2015, 6, 21479–21492. [Google Scholar] [CrossRef] [Green Version]
- Tomasini, R.; Seux, M.; Nowak, J.; Bontemps, C.; Carrier, A.; Dagorn, J.C.; Pebusque, M.J.; Iovanna, J.L.; Dusetti, N.J. TP53INP1 is a novel p73 target gene that induces cell cycle arrest and cell death by modulating p73 transcriptional activity. Oncogene 2005, 24, 8093–8104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zacharski, L.R.; Chow, B.K.; Howes, P.S.; Shamayeva, G.; Baron, J.A.; Dalman, R.L.; Malenka, D.J.; Ozaki, C.K.; Lavori, P.W. Decreased cancer risk after iron reduction in patients with peripheral arterial disease: Results from a randomized trial. J. Natl. Cancer Inst. 2008, 100, 996–1002. [Google Scholar] [CrossRef] [Green Version]
- Pullarkat, V. Iron overload in patients undergoing hematopoietic stem cell transplantation. Adv. Hematol. 2010, 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilken, A.; Langebrake, C.; Wolschke, C.; Kersten, J.F.; Rohde, H.; Nielsen, P.; Kroger, N. Impact of non-transferrin-bound iron (NTBI) in comparison to serum ferritin on outcome after allogeneic stem cell transplantation (ASCT). Ann. Hematol. 2017, 96, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- Lohrum, M.A.; Vousden, K.H. Regulation and activation of p53 and its family members. Cell Death Differ. 1999, 6, 1162–1168. [Google Scholar] [CrossRef] [Green Version]
- Moussa, R.S.; Park, K.C.; Kovacevic, Z.; Richardson, D.R. Ironing out the role of the cyclin-dependent kinase inhibitor, p21 in cancer: Novel iron chelating agents to target p21 expression and activity. Free Radic. Biol. Med. 2019, 133, 276–294. [Google Scholar] [CrossRef]
- Kruse, J.P.; Gu, W. SnapShot: p53 posttranslational modifications. Cell 2008, 133, 930.e1. [Google Scholar] [CrossRef] [Green Version]
- Sermeus, A.; Michiels, C. Reciprocal influence of the p53 and the hypoxic pathways. Cell Death Dis. 2011, 2, e164. [Google Scholar] [CrossRef] [Green Version]
- Dongiovanni, P.; Fracanzani, A.L.; Cairo, G.; Megazzini, C.P.; Gatti, S.; Rametta, R.; Fargion, S.; Valenti, L. Iron-dependent regulation of MDM2 influences p53 activity and hepatic carcinogenesis. Am. J. Pathol. 2010, 176, 1006–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khurana, A.; Shafer, D.A. MDM2 antagonists as a novel treatment option for acute myeloid leukemia: Perspectives on the therapeutic potential of idasanutlin (RG7388). OncoTargets Ther. 2019, 12, 2903–2910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiech, M.; Olszewski, M.B.; Tracz-Gaszewska, Z.; Wawrzynow, B.; Zylicz, M.; Zylicz, A. Molecular mechanism of mutant p53 stabilization: The role of HSP70 and MDM2. PLoS ONE 2012, 7, e51426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stindt, M.H.; Muller, P.A.; Ludwig, R.L.; Kehrloesser, S.; Dotsch, V.; Vousden, K.H. Functional interplay between MDM2, p63/p73 and mutant p53. Oncogene 2015, 34, 4300–4310. [Google Scholar] [CrossRef] [PubMed]
- Kravchenko, J.E.; Ilyinskaya, G.V.; Komarov, P.G.; Agapova, L.S.; Kochetkov, D.V.; Strom, E.; Frolova, E.I.; Kovriga, I.; Gudkov, A.V.; Feinstein, E.; et al. Small-molecule RETRA suppresses mutant p53-bearing cancer cells through a p73-dependent salvage pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 6302–6307. [Google Scholar] [CrossRef] [Green Version]
- Stanga, S.; Caretto, A.; Boido, M.; Vercelli, A. Mitochondrial Dysfunctions: A Red Thread across Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 3719. [Google Scholar] [CrossRef]
- Panuzzo, C.; Jovanovski, A.; Pergolizzi, B.; Pironi, L.; Stanga, S.; Fava, C.; Cilloni, D. Mitochondria: A Galaxy in the Hematopoietic and Leukemic Stem Cell Universe. Int. J. Mol. Sci. 2020, 21, 3928. [Google Scholar] [CrossRef]
- Horowitz, M.P.; Greenamyre, J.T. Mitochondrial iron metabolism and its role in neurodegeneration. J. Alzheimer’s Dis. JAD 2010, 20 (Suppl. 2), S551–S568. [Google Scholar] [CrossRef] [Green Version]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Kim, Y.Y.; Um, J.H.; Yoon, J.H.; Lee, D.Y.; Lee, Y.J.; Kim, D.H.; Park, J.I.; Yun, J. p53 regulates mitochondrial dynamics by inhibiting Drp1 translocation into mitochondria during cellular senescence. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 2451–2464. [Google Scholar] [CrossRef] [Green Version]
- Yoon, Y.S.; Yoon, D.S.; Lim, I.K.; Yoon, S.H.; Chung, H.Y.; Rojo, M.; Malka, F.; Jou, M.J.; Martinou, J.C.; Yoon, G. Formation of elongated giant mitochondria in DFO-induced cellular senescence: Involvement of enhanced fusion process through modulation of Fis1. J. Cell. Physiol. 2006, 209, 468–480. [Google Scholar] [CrossRef] [PubMed]
- Gardner, P.R. Aconitase is a sensitive and critical target of oxygen poisoning in cultured mammalian cells and in rat lungs. Proc. Natl. Acad. Sci. USA 1994, 91, 12248–12252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta 2012, 1817, 1833–1838. [Google Scholar] [CrossRef] [Green Version]
- Ravera, S.; Podesta, M.; Sabatini, F.; Fresia, C.; Columbaro, M.; Bruno, S.; Fulcheri, E.; Ramenghi, L.A.; Frassoni, F. Mesenchymal stem cells from preterm to term newborns undergo a significant switch from anaerobic glycolysis to the oxidative phosphorylation. Cell. Mol. Life Sci. CMLS 2018, 75, 889–903. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HL60 | NB4 | MOLM-13 | ||||
|---|---|---|---|---|---|---|
| NT | DFX | NT | DFX | NT | DFX | |
| OCR_P/M (nmol O/min/106 cells) | 33.86 ± 1.25 | 19.51 ± 1.84 **** | 30.98 ± 1.74 | 14.03 ± 1.23 **** | 18.55 ± 1.19 | 9.96 ± 0.89 *** |
| ATPsynth_P/M (nmol ATP/min/106 cells) | 82.89 ± 0.91 | 37.46 ± 1.86 **** | 75.98 ± 3.11 | 16.58 ± 1.84 **** | 44.96 ± 1.77 | 11.24 ± 0.93 *** |
| P/O_P/M | 2.45 ± 0.08 | 1.62 ± 0.03 **** | 2.45 ± 0.12 | 1.18 ± 0.04 **** | 2.43 ± 0.08 | 1.13 ± 0.04 *** |
| OCR_Succ (nmol O/min/106 cells) | 22.50 ± 1.08 | 12.87 ± 0.73 **** | 20.52 ± 1.02 | 9.26 ± 0.85 **** | 12.58 ± 0.54 | 6.78 ± 0.47 **** |
| ATPsynth_Succ (nmol ATP/min/106 cells) | 34.96 ± 2.85 | 14.95 ± 0.58 **** | 32.05 ± 0.68 | 6.99 ± 0.31 **** | 18.97 ± 1.45 | 4.73 ±0.66 **** |
| P/O_Succ | 1.56 ± 0.09 | 1.00 ± 0.05 **** | 1.57 ± 0.07 | 0.76 ± 0.04 **** | 1.55 ± 0.09 | 0.72 ± 0.04 **** |
| HL60 | NB4 | MOLM-13 | ||||
|---|---|---|---|---|---|---|
| NT | DFX | NT | DFX | NT | DFX | |
| ATP (mM/mg) | 2.39 ± 0.09 | 1.58 ± 0.22 *** | 2.24 ± 0.10 | 1.29 ± 0.04 *** | 2.17 ± 0.06 | 1.23 ± 0.04 *** |
| AMP (mM/mg) | 0.84 ± 0.03 | 1.21 ± 0.04 *** | 0.83 ± 0.04 | 1.40 ± 0.06 *** | 0.95 ± 0.05 | 1.47 ± 0.02 *** |
| ATP/AMP | 2.86 ± 0.07 | 1.45 ± 0.18 **** | 2.71 ± 0.21 | 0.92 ± 0.06 **** | 2.28 ± 0.16 | 0.84 ± 0.03 **** |
| Age, Years Median 78 (Range 56–82) | Diagnosis | Karyotype | Bm Blast (%) | Ferritin (ng/mL) |
|---|---|---|---|---|
| 81 | MDS-SLD | Normal | 2.0 | N/A |
| 58 | Isolated del (5q) | 46, XX, 5q- | 1.5 | 1191 |
| 67 | AML | Normal | 25.0 | 2250 |
| 78 | MDS-MLD | 46, XY, del 9 (q22:q32) | 2.2 | 2292 |
| 70 | MDS-RS-SLD | 47, XY, +8 | 2.0 | 2587 |
| 82 | MDS-EB-II | Normal | 5.0 | 975 |
| 69 | MDS-EB-II | N/A | 15.0 | 700 |
| 56 | MDS-MLD | N/A | 3.0 | 4706 |
| 78 | MDS-EB-I | Normal | 7.0 | 1207 |
| 69 | MDS-SLD | N/A | N/A | 2643 |
| 64 | MDS-MLD | Normal | 4.0 | 1314 |
| 71 | MDS-SLD | Normal | 3.0 | N/A |
| 79 | Isolated del (5q) | 46, XY, 5q- | 2.5 | 746 |
| 82 | MDS-EB-I | Normal | N/A | N/A. |
| 81 | AML | N/A | 20.0 | 2272 |
| 63 | Healthy donor 1 | Normal | - | 200 |
| 52 | Healthy donor 2 | Normal | - | 13 |
| 56 | Healthy donor 3 | Normal | - | 80 |
| 57 | Healthy donor 4 | Normal | - | 83 |
| 53 | Healthy donor 5 | Normal | - | 49 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calabrese, C.; Panuzzo, C.; Stanga, S.; Andreani, G.; Ravera, S.; Maglione, A.; Pironi, L.; Petiti, J.; Shahzad Ali, M.; Scaravaglio, P.; et al. Deferasirox-Dependent Iron Chelation Enhances Mitochondrial Dysfunction and Restores p53 Signaling by Stabilization of p53 Family Members in Leukemic Cells. Int. J. Mol. Sci. 2020, 21, 7674. https://doi.org/10.3390/ijms21207674
Calabrese C, Panuzzo C, Stanga S, Andreani G, Ravera S, Maglione A, Pironi L, Petiti J, Shahzad Ali M, Scaravaglio P, et al. Deferasirox-Dependent Iron Chelation Enhances Mitochondrial Dysfunction and Restores p53 Signaling by Stabilization of p53 Family Members in Leukemic Cells. International Journal of Molecular Sciences. 2020; 21(20):7674. https://doi.org/10.3390/ijms21207674
Chicago/Turabian StyleCalabrese, Chiara, Cristina Panuzzo, Serena Stanga, Giacomo Andreani, Silvia Ravera, Alessandro Maglione, Lucrezia Pironi, Jessica Petiti, Muhammad Shahzad Ali, Patrizia Scaravaglio, and et al. 2020. "Deferasirox-Dependent Iron Chelation Enhances Mitochondrial Dysfunction and Restores p53 Signaling by Stabilization of p53 Family Members in Leukemic Cells" International Journal of Molecular Sciences 21, no. 20: 7674. https://doi.org/10.3390/ijms21207674
APA StyleCalabrese, C., Panuzzo, C., Stanga, S., Andreani, G., Ravera, S., Maglione, A., Pironi, L., Petiti, J., Shahzad Ali, M., Scaravaglio, P., Napoli, F., Fava, C., De Gobbi, M., Frassoni, F., Saglio, G., Bracco, E., Pergolizzi, B., & Cilloni, D. (2020). Deferasirox-Dependent Iron Chelation Enhances Mitochondrial Dysfunction and Restores p53 Signaling by Stabilization of p53 Family Members in Leukemic Cells. International Journal of Molecular Sciences, 21(20), 7674. https://doi.org/10.3390/ijms21207674