The Effect of Methylmalonic Acid Treatment on Human Neuronal Cell Coenzyme Q10 Status and Mitochondrial Function


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
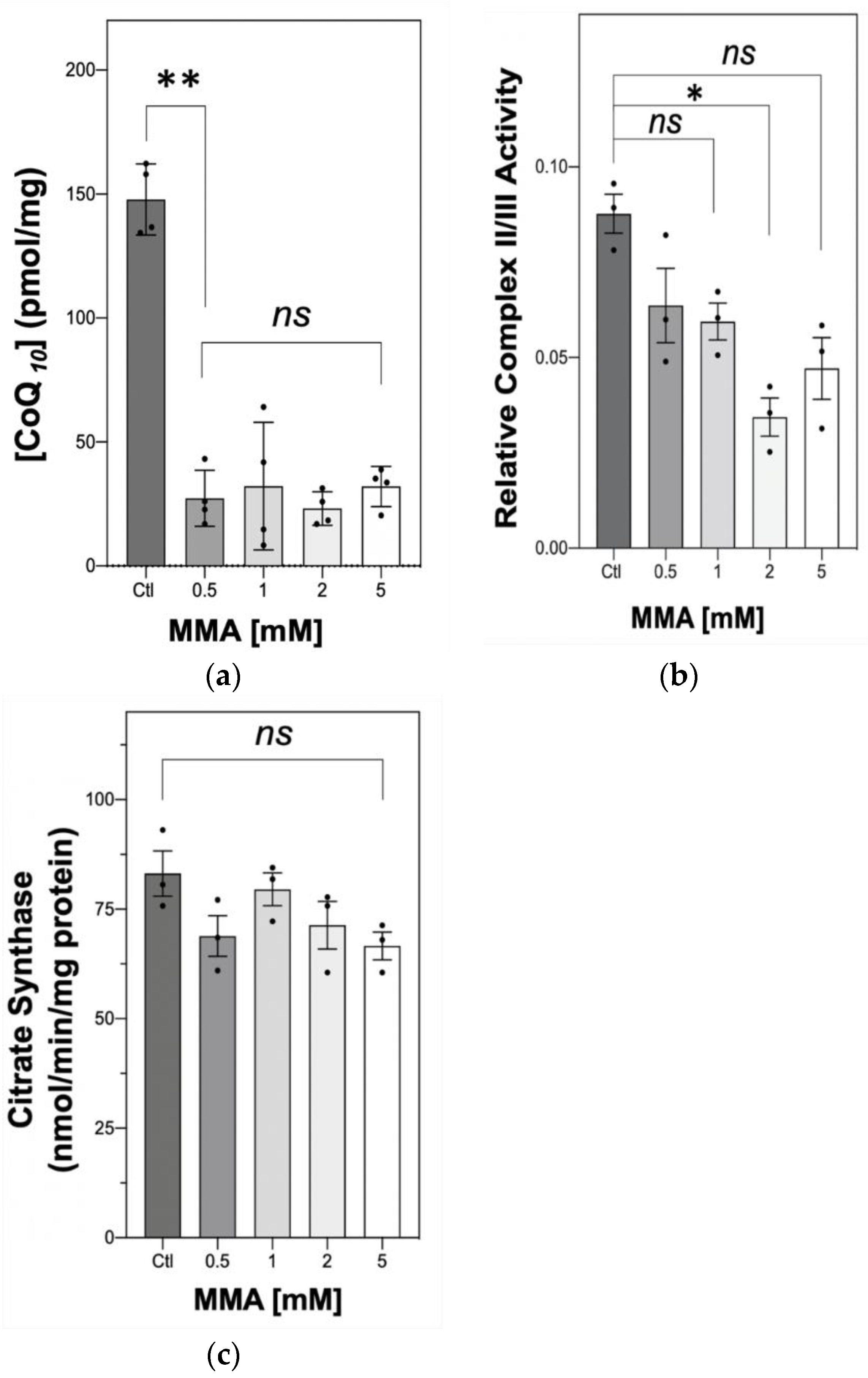
2.1. The Effect of MMA Treatment on Neuronal CoQ10 Status and MRC Complex II–III Activity
2.2. The Effect MMA Treatment on SH-SY5Y Viability and Mitochondrial Membrane Potential
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. High-Performance Liquid Chromatography (HPLC)
4.3. Spectrophotometric Enzyme Assays
4.4. MTT Assay
4.5. Flow Cytometry
4.6. Protein Determination
4.7. Determination of Intracellular MMA Concentrations
4.8. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MMA | Methylmalonic acid |
| mut0 | Complete defect of MCM activity: OMIM 251000 |
| mut- | Partial defect of MCM activity, OMIM 251000 |
| BBB | Blood–brain barrier |
| 2-MCA | 2-Methylcitrate |
| HPLC | High-performance liquid chromatography |
| IMM | Inner mitochondrial membrane |
References
- Haijes, H.A.; Jans, J.J.M.; Tas, S.Y.; Verhoeven-Duif, N.M.; Van Hasselt, P.M. Pathophysiology of propionic and methylmalonic acidemias. Part 1: Complications. Methylmalonic aciduria. An inborn error of metabolism leading to chronic metabolic acidosis. J. Inherit. Metab. Dis. 2019, 42, 730–744. [Google Scholar] [CrossRef] [PubMed]
- Kölker, S.; Schwab, M.; Hörster, F.; Sauer, S.; Hinz, A.; Wolf, N.I.; Mayatepek, E.; Hoffmann, G.F.; Smeitink, J.A.M.; Okun, J.G. Methylmalonic Acid, a Biochemical Hallmark of Methylmalonic Acidurias but No Inhibitor of Mitochondrial Respiratory Chain. J. Biol. Chem. 2003, 278, 47388–47393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenton, W.A.; Gravel, R.A.A.; Rosenblatt, D.S. Disorders of propionate and methylmalonate metabolism. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C.R., Beaudet, A.L., Sky, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2011; pp. 2165–2193. [Google Scholar]
- Baumgartner, M.R.; Hörster, F.; Dionisi-Vici, C.C.; Haliloglu, G.; Karall, D.; Chapman, A.K.; Huemer, M.; Hochuli, M.; Assoun, M.; Ballhausen, D.; et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J. Rare Dis. 2014, 9, 1–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Dick, A.; Montenovo, M.; Horslen, S.; Hansen, R. Cost-effectiveness of liver transplantation in methylmalonic and propionic acidemias. Liver Transplant. 2015, 21, 1208–1218. [Google Scholar] [CrossRef] [Green Version]
- Heidenreich, R.; Natowicz, M.; Hainline, B.E.; Berman, P.; Kelley, R.I.; Hillman, R.E.; Berry, G.T. Acute extrapyramidal syndrome in methylmalonic acidemia: “Metabolic stroke” involving the globus pallidus. J. Pediatr. 1988, 113, 1022–1027. [Google Scholar] [CrossRef]
- Kaplan, P.; Ficicioglu, C.; Mazur, A.T.; Palmieri, M.J.; Berry, G.T. Liver transplantation is not curative for methylmalonic acidopathy caused by methylmalonyl-CoA mutase deficiency. Mol. Genet. Metab. 2006, 88, 322–326. [Google Scholar] [CrossRef]
- Hauser, N.S.; Manoli, I.; Graf, J.C.; Sloan, J.; Venditti, C.P. Variable dietary management of methylmalonic acidemia: Metabolic and energetic correlations. Am. J. Clin. Nutr. 2010, 93, 47–56. [Google Scholar] [CrossRef] [Green Version]
- Manoli, I.; Myles, J.G.; Sloan, J.L.; Shchelochkov, O.A.; Venditti, C.P. A critical reappraisal of dietary practices in methylmalonic acidemia raises concerns about the safety of medical foods. Part 1: Isolated methylmalonic acidemias. Genet. Med. 2016, 18, 386–395. [Google Scholar] [CrossRef] [Green Version]
- Han, L.; Wu, S.; Han, F.; Gu, X.-F. Insights into the molecular mechanisms of methylmalonic acidemia using microarray technology. Int. J. Clin. Exp. Med. 2015, 8, 8866–8879. [Google Scholar]
- Fernandes, C.G.; Borges, C.G.; Seminotti, B.; Amaral, A.U.; Knebel, L.A.; Eichler, P.; de Oliveira, A.B.; Leipnitz, G.; Wajner, M. Experimental evidence that metylmalonic acid provokes oxidative damage and compromises antioxidant defences in nerve terminal and striatum of young rats. Cell Mol. Neurobiol. 2011, 31, 775–785. [Google Scholar] [CrossRef]
- Richard, E.; Alvarez-Barrientos, A.; Perez, B.; Desviat, L.; Ugarte, M. Methylmalonic acidaemia leads to increased production of reactive oxygen species and induction of apoptosis through the mitochondrial/caspase pathway. J. Pathol. 2007, 213, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Melo, D.R.; Kowaltowski, A.J.; Wajner, M.; Castilho, R.F. Mitochondrial energy metabolism in neurodegeneration associated with methylmalonic acidemia. J. Bioenerg. Biomembr. 2011, 43, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Shuck, P.F.; Alves, L.; Perrenuzzo, L.F.; Felisberto, F.; Rodrigues, L.B.; Freitas, B.W.; Petronilho, F.; Dal-Pizzol, F.; Streck, E.L.; Ferreira, G.C. Acute renal failure potententiates methylmalonate-induced oxidative stress in brain and kidney of rats. Free Radic. Res. 2013, 45, 661–667. [Google Scholar]
- Jafari, P.; Braissant, O.; Zavadakova, P.; Henry, H.; Bonafé, L.; Ballhausen, D. Brain damage in methylmalonic aciduria: 2-methylcitrate induces cerebral ammonium accumulation and apoptosis in 3D organotypic brain cell cultures. Orphanet J. Rare Dis. 2013, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- De Keyzer, Y.; Valayannopoulos, V.; Benoist, J.-F.; Batteux, F.; Lacaille, F.; Hubert, L.; Chrétien, D.; Chadefeaux-Vekemans, B.; Niaudet, P.; Touati, G.; et al. Multiple OXPHOS Deficiency in the Liver, Kidney, Heart, and Skeletal Muscle of Patients With Methylmalonic Aciduria and Propionic Aciduria. Pediatr. Res. 2009, 66, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Okun, J.G.; Hörster, F.; Farkas, L.M.; Feyh, P.; Hinz, A.; Sauer, S.; Hoffmann, G.F.; Unsicker, K.; Mayatepek, E.; Kölker, S. Neurodegeneration in Methylmalonic Aciduria Involves Inhibition of Complex II and the Tricarboxylic Acid Cycle, and Synergistically Acting Excitotoxicity. J. Biol. Chem. 2002, 277, 14674–14680. [Google Scholar] [CrossRef] [Green Version]
- Caterino, M.; Chandler, R.J.; Sloan, J.L.; Dorko, K.; Cusmano-Ozog, K.; Ingenito, L.; Strom, S.C.; Imperlini, E.; Scolamiero, E.; Venditti, C.P.; et al. The proteome of methylmalonic acidemia (MMA): The elucidation of altered pathways in patient livers. Mol. BioSyst. 2016, 12, 566–574. [Google Scholar] [CrossRef] [Green Version]
- Brusque, A.; Rosa, R.B.; Schuck, P.; Dalcin, K.; Ribeiro, C.; Silva, C.; Wannmacher, C.; Dutra-Filho, C.; Wyse, A.; Briones, P.; et al. Inhibition of the mitochondrial respiratory chain complex activities in rat cerebral cortex by methylmalonic acid. Neurochem. Int. 2002, 40, 593–601. [Google Scholar] [CrossRef]
- Rahman, S.; Hargreaves, I.; Clayton, P.; Heales, S. Neonatal presentation of coenzyme Q10 deficiency. J. Pediatr. 2001, 139, 456–458. [Google Scholar] [CrossRef]
- Hargreaves, I.P. Ubiquinone: Cholesterol’s reclusive cousin. Ann. Clin. Biochem. 2003, 40 Pt 3, 207–218. [Google Scholar] [CrossRef]
- Ernster, L. Ubiquinol: An endogenous antioxidant in aerobic organisms. J. Mol. Med. 1993, 71, S60–S65. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hekimi, S. Understanding Ubiquinone. Trends Cell Biol. 2016, 26, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Haas, D.; Niklowitz, P.; Hörster, F.; Baumgartner, E.R.; Prasad, C.; Rodenburg, R.J.T.; Hoffmann, G.F.; Menke, T.; Okun, J.G. Coenzyme Q10 is decreased in fibroblasts of patients with methylmalonic aciduria but not in mevalonic aciduria. J. Inherit. Metab. Dis. 2009, 32, 570–575. [Google Scholar] [CrossRef]
- Manoli, I.; Sysol, J.R.; Li, L.; Houillier, P.; Garone, C.; Wang, C.; Zerfas, P.M.; Cusmano-Ozog, K.; Young, S.; Trivedi, N.S.; et al. Targeting proximal tubule mitochondrial dysfunction attenuates the renal disease of methylmalonic acidemia. Proc. Natl. Acad. Sci. USA 2013, 110, 13552–13557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, G.F.; Meier-Augenstein, W.; Stöckler, S.; Surtees, R.; Rating, D.; Nyhan, W.L. Physiology and pathophysiology of organic acids in cerebrospinal fluid. J. Inherit. Metab. Dis. 1993, 16, 648–669. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.D.; Kanabus, M.; Anderson, G.; Hargreaves, I.P.; Rutherford, T.; Donnell, M.O.; Cross, J.H.; Rahman, S.; Eaton, S.; Heales, S. The ketogenic diet component decanoic acid increases mitochondrial citrate synthase and complex I activity in neuronal cells. J. Neurochem. 2014, 129, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Ericsson, J.; Dallner, G. Distribution, Biosynthesis, and Function of Mevalonate Pathway Lipids. Subcell. Biochem. 1993, 21, 229–272. [Google Scholar]
- Davey, G.P.; Peuchen, S.; Clark, J.B. Energy thresholds in brain mitochondria. Potential involvement in neurodegeneration. J. Biol. Chem. 1998, 273, 12753–12777. [Google Scholar] [CrossRef] [Green Version]
- Yubero, D.; Montero, R.; Martín, M.A.; Montoya, J.; Ribes, A.; Grazina, M.; Trevisson, E.; Rodriguez-Aguilera, J.C.; Hargreaves, I.; Salviati, L.; et al. Secondary coenzyme Q 10 deficiencies in oxidative phosphorylation (OXPHOS) and non-OXPHOS disorders. Mitochondrion 2016, 30, 51–58. [Google Scholar] [CrossRef]
- Stepien, K.M.; Heaton, R.A.; Rankin, S.; Murphy, A.; Bentley, J.; Sexton, D.W.; Hargreaves, I. Evidence of Oxidative Stress and Secondary Mitochondrial Dysfunction in Metabolic and Non-Metabolic Disorders. J. Clin. Med. 2017, 6, 71. [Google Scholar] [CrossRef] [Green Version]
- Neergheen, V.; Hargreaves, I.P. Secondary coenzyme Q10 deficiency: Causes and consequence. In Coenzyme Q10: Uses, Health Effects and Use in Disease; Grigoryeva, S., Ed.; Nova Science Publishers: New York, NY, USA, 2018; pp. 89–110. [Google Scholar]
- Yubero, D.; O’Callaghan, M.; Montero, R.; Ormazábal, A.; Armstrong, J.; Espinós, C.; Rodríguez, A.M.; Jou, C.; Castejón-Ponce, E.; Aracil, A.M.; et al. Association between coenzyme Q10 and glucose transporter (GLUT1) deficiency. BMC Pediatr. 2014, 14, 284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockert, J.C.; Horobin, R.W.; Colombo, L.L.; Blázquez-Castro, A. Tetrazolium salts and formazan products in Cell Biology: Viability assessment, fluorescence imaging, and labeling perspectives. Acta Histochem. 2018, 120, 159–167. [Google Scholar] [CrossRef] [Green Version]
- Shepherd, J.A.; Garland, P.B. Citrate synthase activity from rat liver. Methods Enzymol. 1969, 13, 11–19. [Google Scholar]
- Xie, H.-R.; Hu, L.; Li, G.-Y. SH-SY5Y human neuroblastoma cell line: In vitro cell model of dopaminergic neurons in Parkinson’s disease. Chin. Med. J. 2010, 123, 1086–1092. [Google Scholar]
- Schneider, L.; Giordano, S.; Zelickson, B.R.; Johnson, Z.M.; Benavides, G.A.; Ouyang, X.; Fineberg, N.; Darley-Usmar, V.M.; Zhang, J. Differentiation of SH-SY5Y cells to a neuronal phenotype changes cellular bioenergetics and the response to oxidative stress. Free Radic. Biol. Med. 2011, 51, 2007–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinar-Sueiro, S.; Martínez-Fernández, R.; Lage, S.; Aldámiz-Echevarría, L.; Vecino, E. Optic neuropathy in methylmalonic acidemia: The role of neuroprotection. J. Inherit. Metab. Dis. 2010, 33, 199–203. [Google Scholar] [CrossRef]
- Biedler, J.L.; Roffler-Tarlov, S.; Schachner, M.; Freedman, L.S. Multiple neurotransmitter synthesis by human neuroblastoma cel lines and clones. Cancer Res. 1978, 7S, S41–S50. [Google Scholar]
- Bhagavan, H.N.; Chopra, R.K. Coenzyme Q10: Absorption, tissue uptake, metabolism and pharmacokinetics. Free Radic. Res. 2006, 40, 445–453. [Google Scholar] [CrossRef]
- Duncan, A.J.; Heales, S.J.; Mills, K.; Eaton, S.; Land, J.M.; Hargreaves, I.P. Determination of Coenzyme Q10 Status in Blood Mononuclear Cells, Skeletal Muscle, and Plasma by HPLC with Di-Propoxy-Coenzyme Q10 as an Internal Standard. Clin. Chem. 2005, 51, 2380–2382. [Google Scholar] [CrossRef] [Green Version]
- Boitier, E.; Degoul, F.; Desguerre, I.; Charpentier, C.; François, M.; Ponsot, G.; Diry, M.; Rustin, P.; Marsac, C. A case of mitochondrial encephalomyopathy associated with a muscle coenzyme Q10 deficiency. J. Neurol. Sci. 1998, 156, 41–46. [Google Scholar] [CrossRef]
- King, T.S. Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1967; Volume 10, pp. 217–235. [Google Scholar]
- Selak, M.A.; de Chadarevian, J.P.; Melvin, J.J.; Grover, W.D.; Salganicoff, L.; Kaye, E.M. Mitochondrial activity in pompe’s disease. Pediatr. Neurol. 2000, 23, 54–57. [Google Scholar] [CrossRef]
- Wu, C.-W.; Ping, Y.-H.; Yen, J.-C.; Chang, C.-Y.; Wang, S.-F.; Yeh, C.-L.; Chi, C.-W.; Lee, H.-C. Enhanced oxidative stress and aberrant mitochondrial biogenesis in human neuroblastoma SH-SY5Y cells during methamphetamine induced apoptosis. Toxicol. Appl. Pharmacol. 2007, 220, 243–251. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
- Blom, H.J.; van Rooij, A.; Hogeveen, M. A simple high-throughput method for the assessment of plasma methylmalonic acid by liquid chromatohraphy-tan mass spectrometry. Clin. Chem. Lab. Med. 2007, 45, 645–650. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Proctor, E.C.; Turton, N.; Boan, E.J.; Bennett, E.; Philips, S.; Heaton, R.A.; Hargreaves, I.P. The Effect of Methylmalonic Acid Treatment on Human Neuronal Cell Coenzyme Q10 Status and Mitochondrial Function. Int. J. Mol. Sci. 2020, 21, 9137. https://doi.org/10.3390/ijms21239137
Proctor EC, Turton N, Boan EJ, Bennett E, Philips S, Heaton RA, Hargreaves IP. The Effect of Methylmalonic Acid Treatment on Human Neuronal Cell Coenzyme Q10 Status and Mitochondrial Function. International Journal of Molecular Sciences. 2020; 21(23):9137. https://doi.org/10.3390/ijms21239137
Chicago/Turabian StyleProctor, Emma C., Nadia Turton, Elle Jo Boan, Emily Bennett, Suzannah Philips, Robert A. Heaton, and Iain P. Hargreaves. 2020. "The Effect of Methylmalonic Acid Treatment on Human Neuronal Cell Coenzyme Q10 Status and Mitochondrial Function" International Journal of Molecular Sciences 21, no. 23: 9137. https://doi.org/10.3390/ijms21239137
APA StyleProctor, E. C., Turton, N., Boan, E. J., Bennett, E., Philips, S., Heaton, R. A., & Hargreaves, I. P. (2020). The Effect of Methylmalonic Acid Treatment on Human Neuronal Cell Coenzyme Q10 Status and Mitochondrial Function. International Journal of Molecular Sciences, 21(23), 9137. https://doi.org/10.3390/ijms21239137