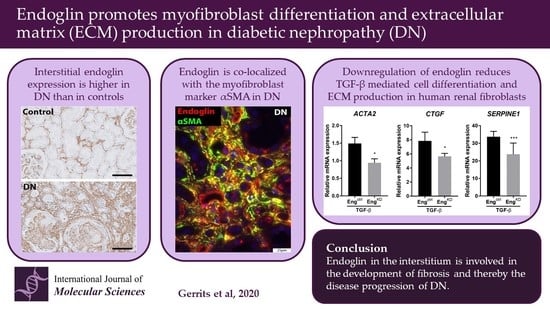
Endoglin Promotes Myofibroblast Differentiation and Extracellular Matrix Production in Diabetic Nephropathy

Abstract
:
1. Introduction
2. Results
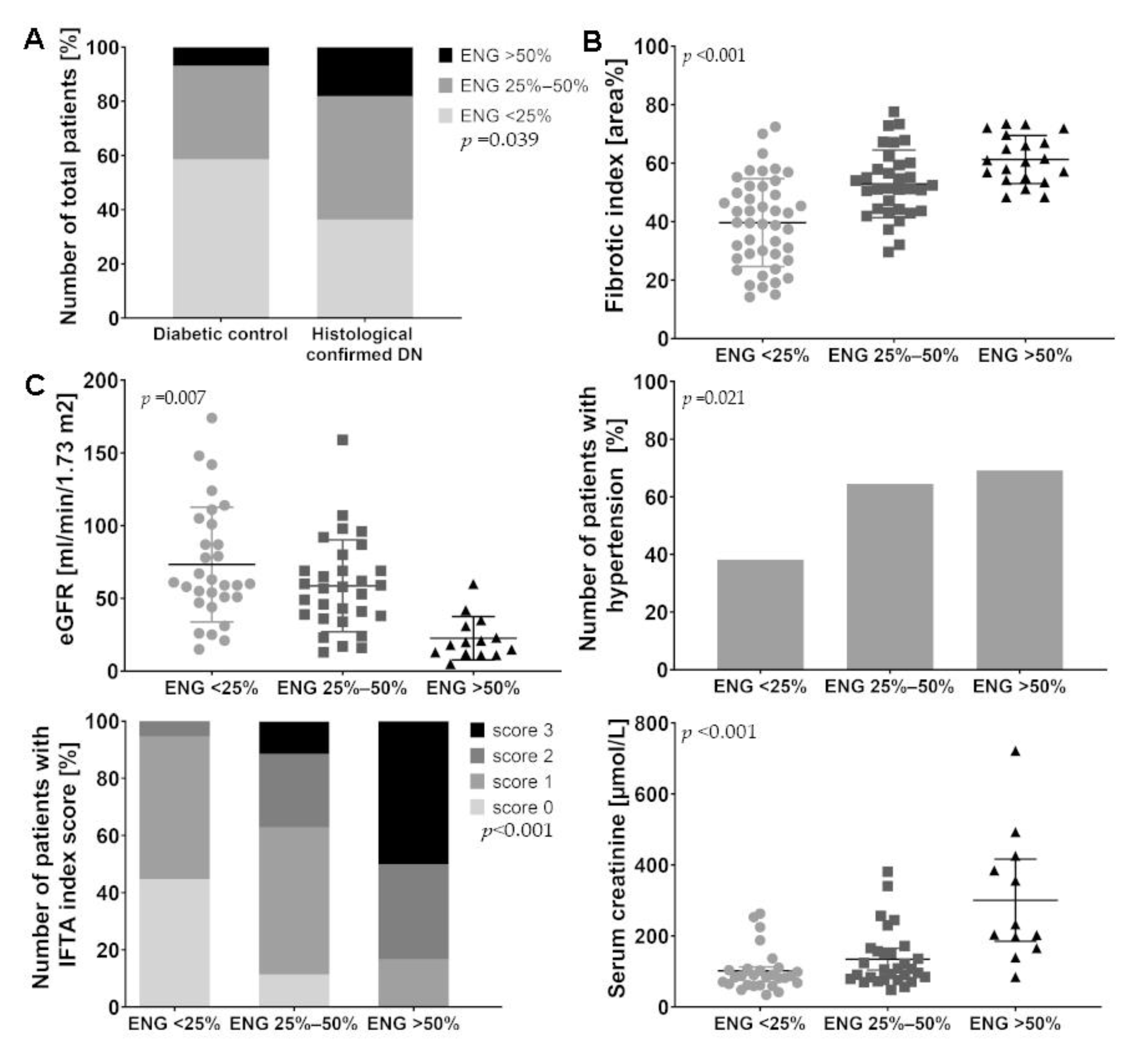
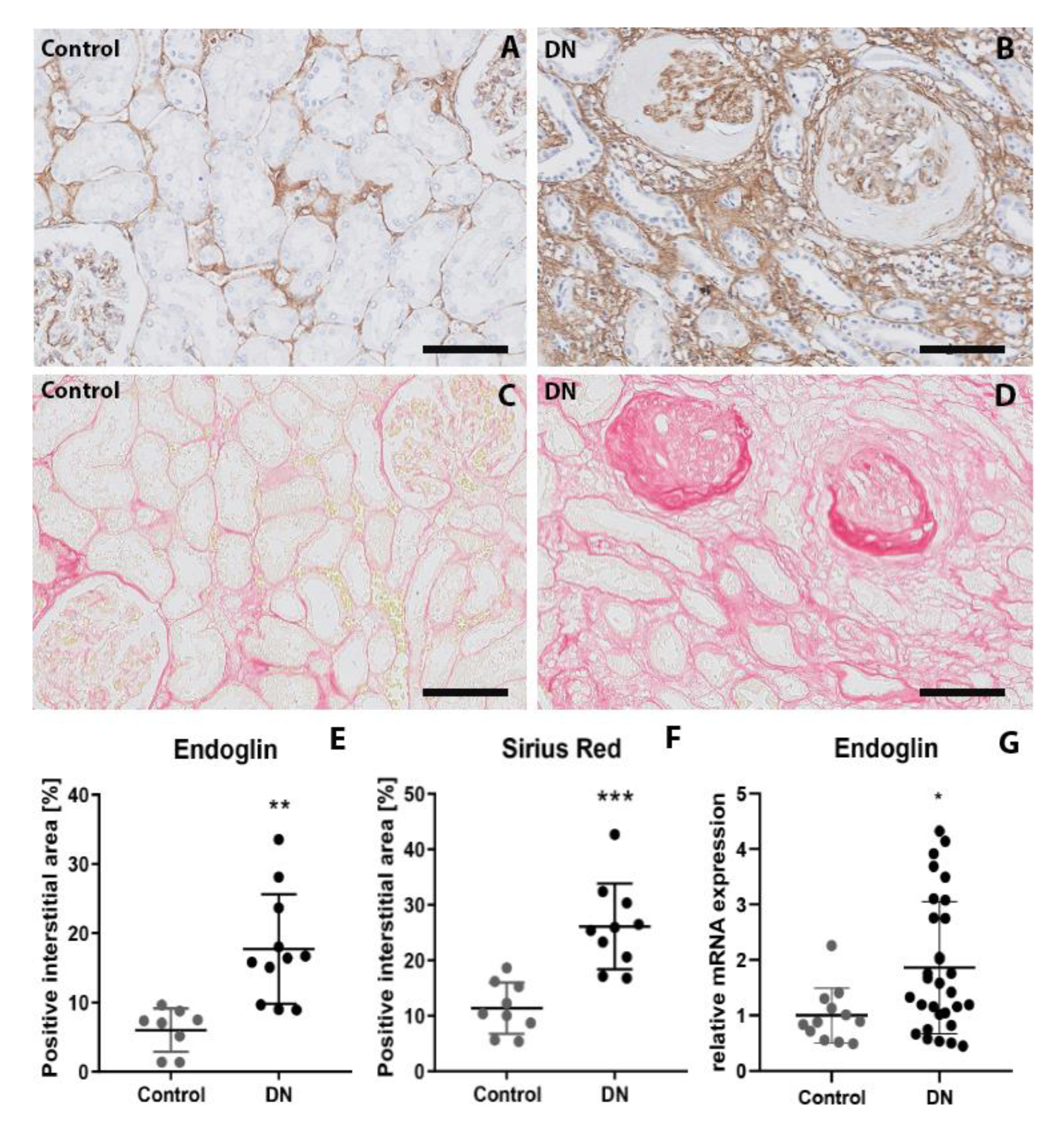
2.1. Patients with Diabetic Nephropathy Have Increased Endoglin Expression that is Correlated with Various Clinical Parameters
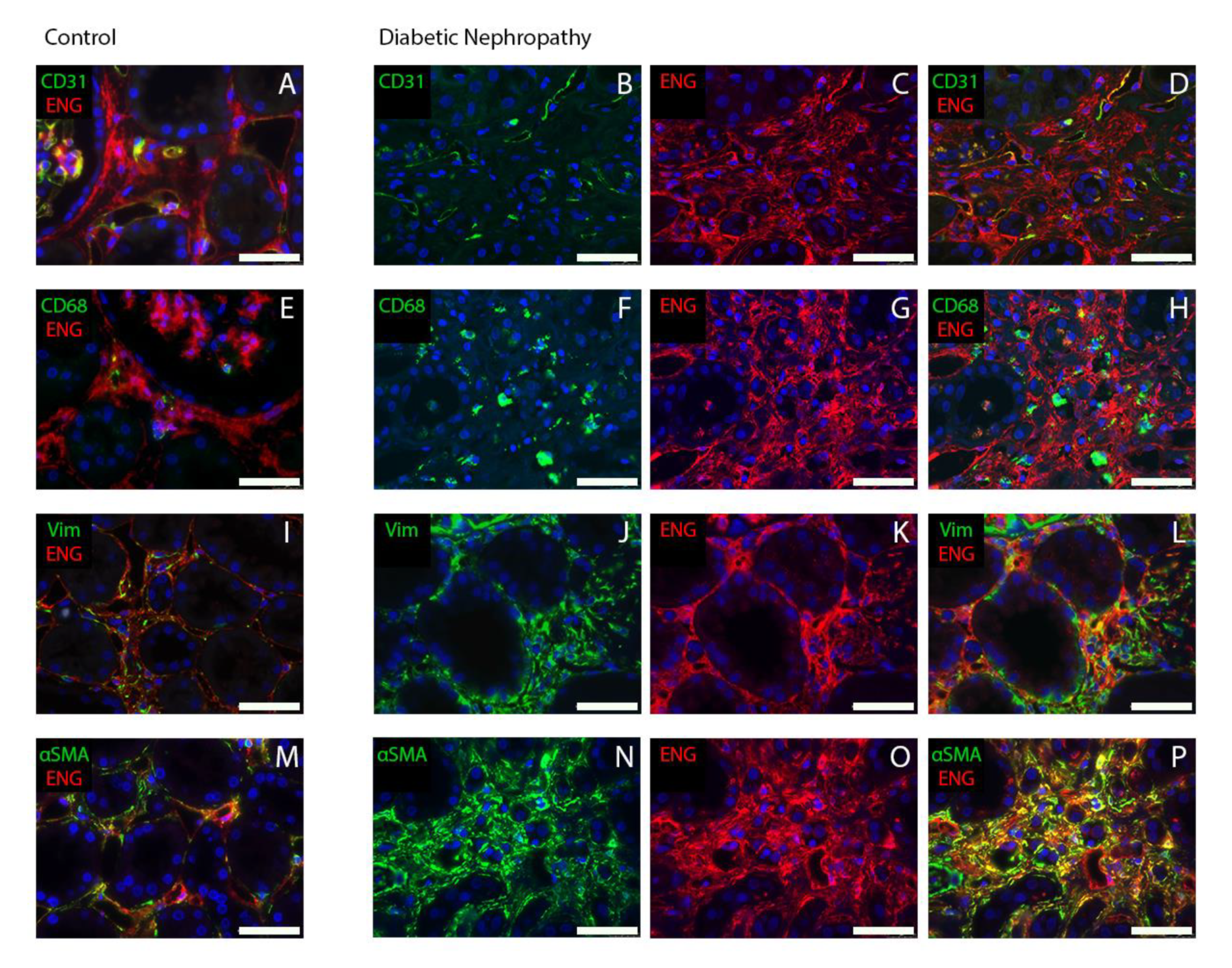
2.2. Endoglin Is Expressed by Interstitial Myofibroblasts
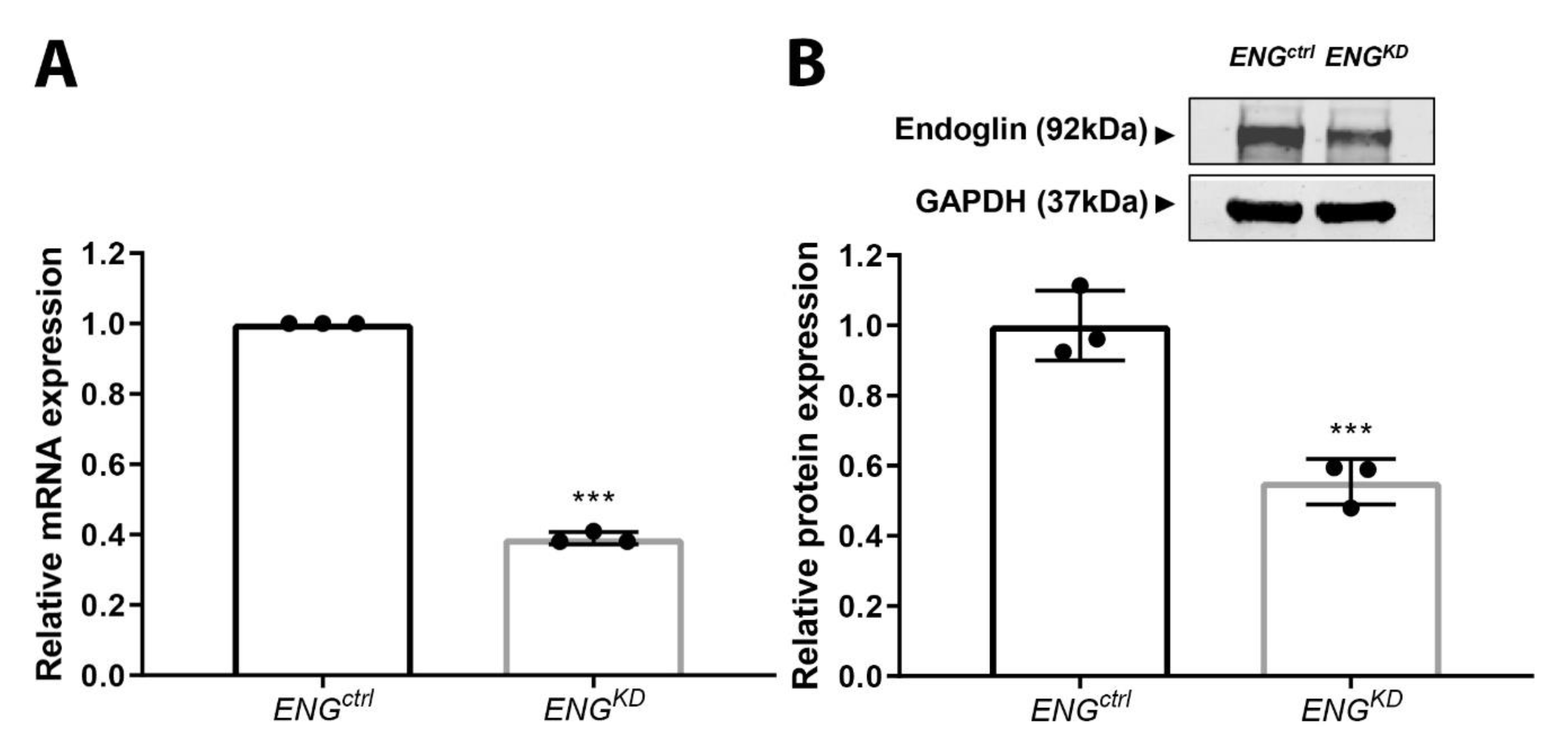
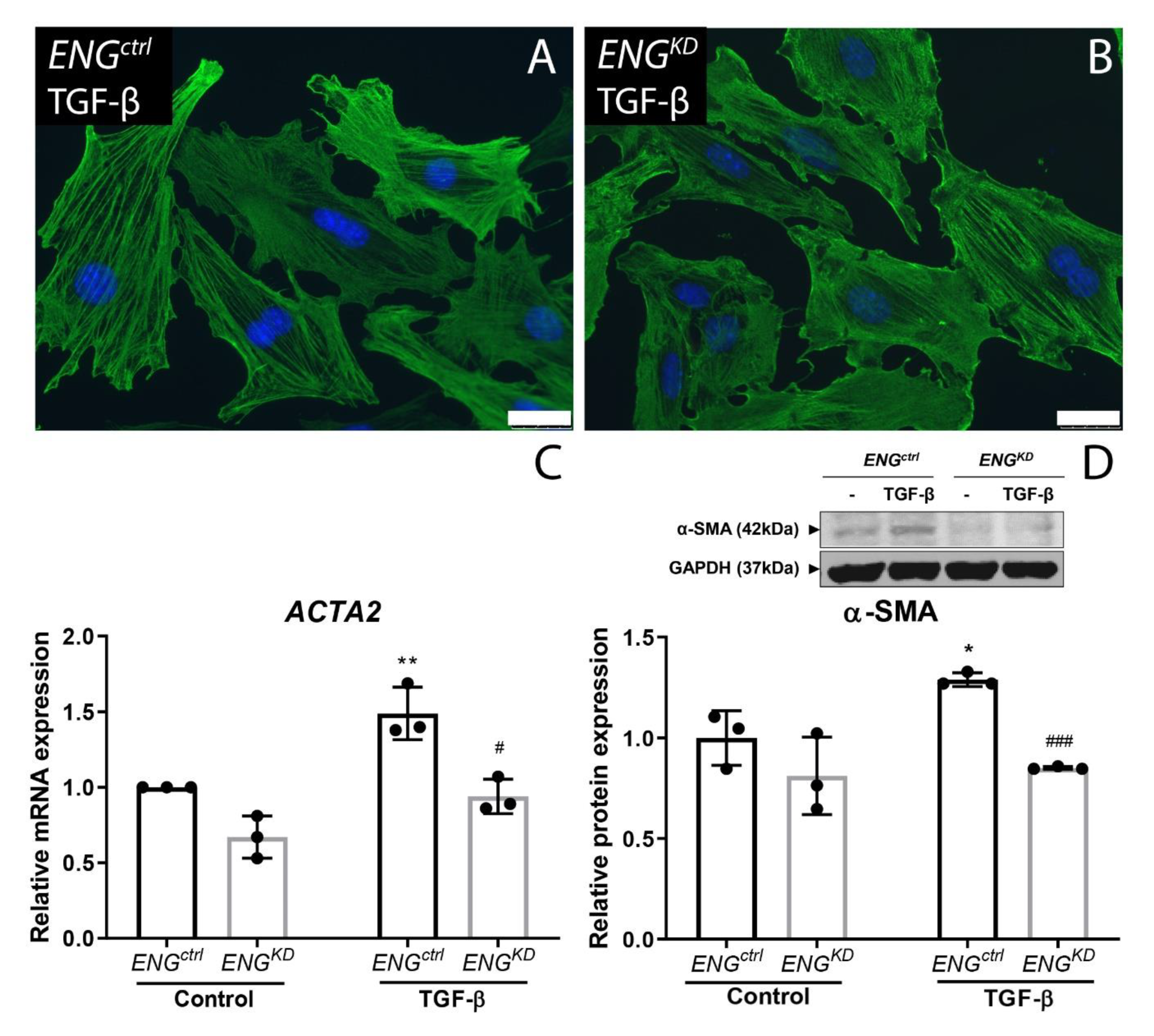
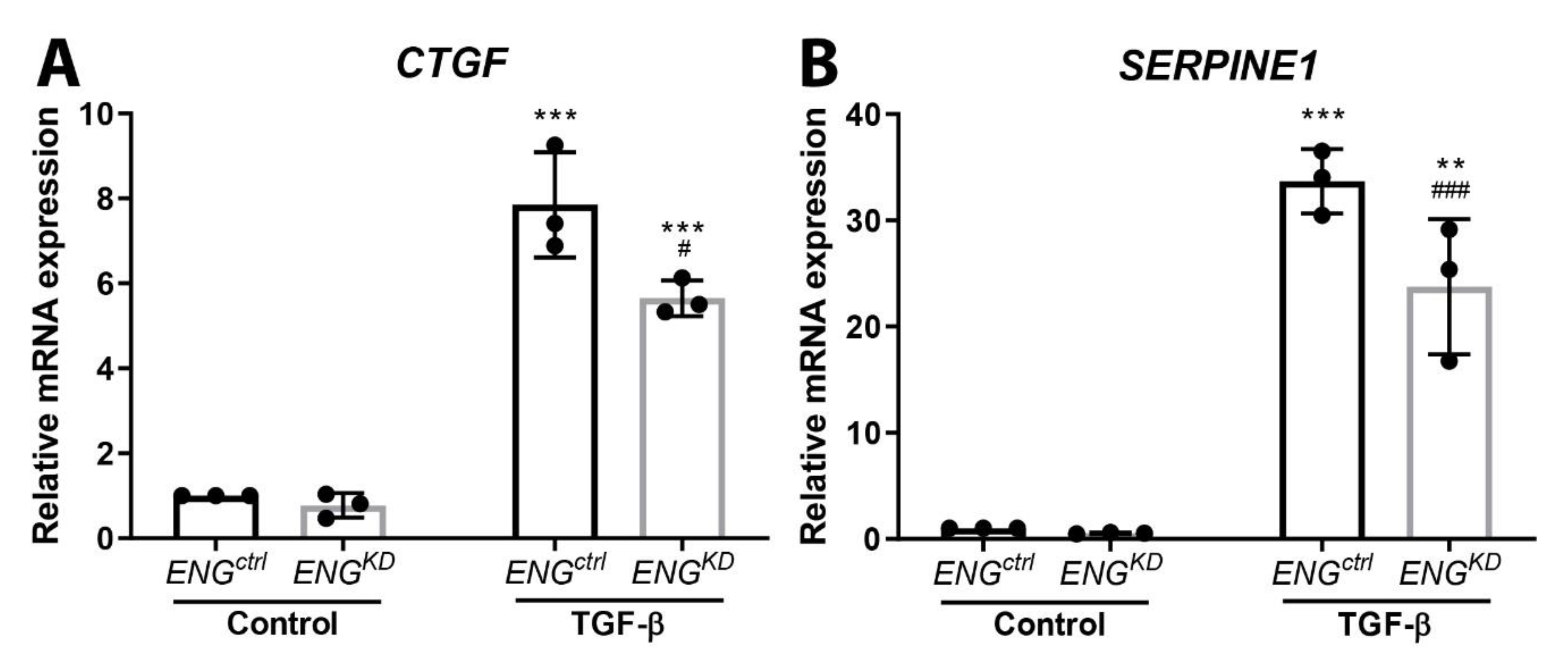
2.3. Endoglin Knockdown Reduces the Expression of TGF-β Downstream Targets and Inhibits Fibroblast Differentiation
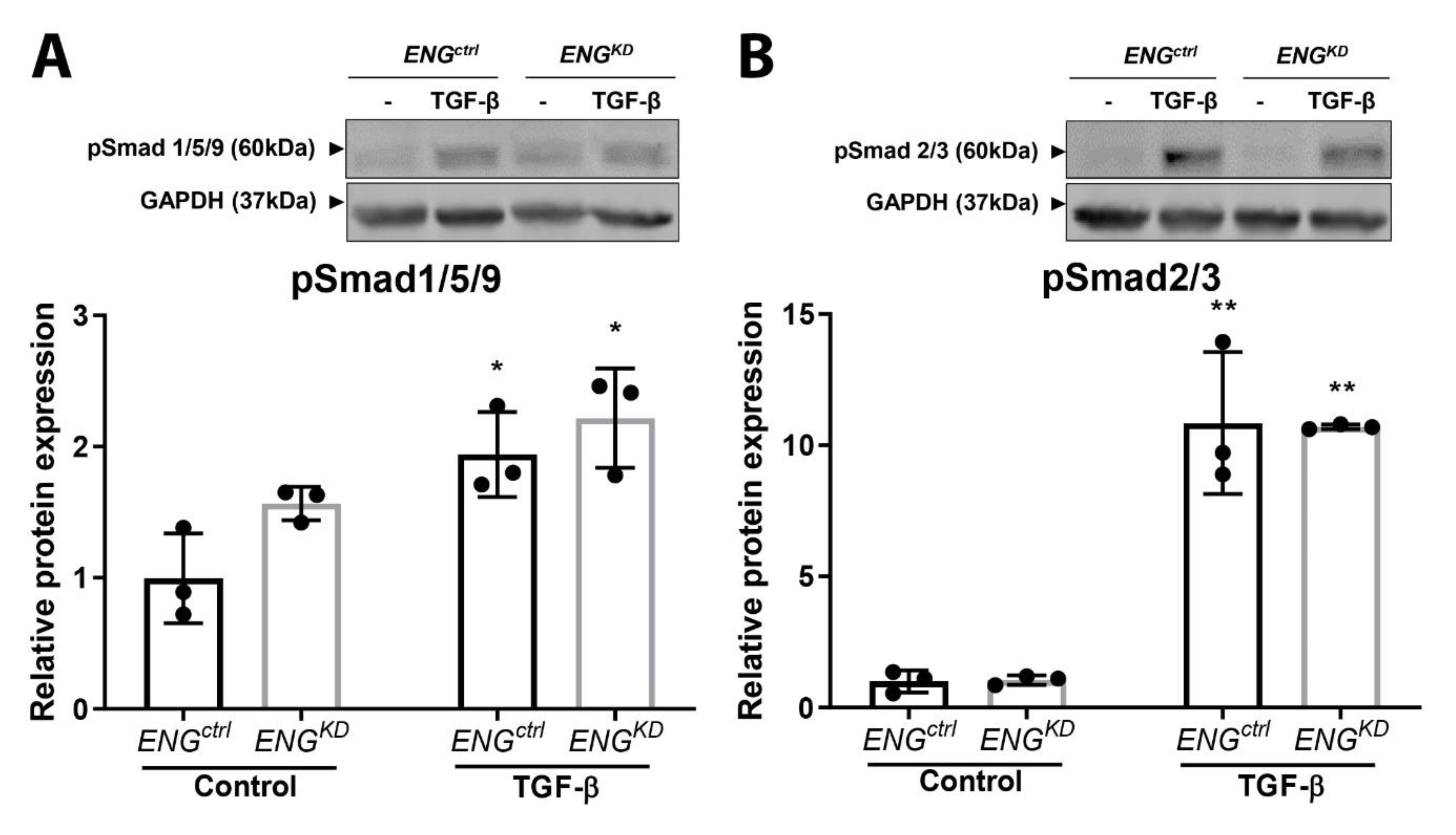
2.4. The Effect of Endoglin Knockdown on Myofibroblast Differentiation and ECM Production Cannot be Explained by Differences in Canonical TGF-β Signaling.
3. Discussion
4. Materials and Methods
4.1. Autopsy Cohort
4.2. Biopsy Cohort
4.3. Clinical Data
4.4. Immunohistochemistry
4.5. Scoring of Sections and Analysis of Digital Images
4.6. Immunofluorescence Staining of Biopsy Material
4.7. Cell Culture
4.8. Analysis of Endoglin Expression
4.9. Immunofluorescence Staining of Cell Culture
4.10. Analysis of TGF-β-Induced Changes in Gene and Protein Expression
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DN | Diabetic nephropathy |
| ECM | Extra cellular matrix |
| TGF-β | Transforming growth factor-beta |
| CTGF | Connective tissue growth factor |
| SERPINE-1 | Plasminogen activator inhibitor-1 |
| αSMA | Alpha smooth muscle actin |
| UUO | Unilateral ureter obstruction |
| ENG | Endoglin |
| eGFR | Estimated glomerular filtration rate |
| IFTA | Interstitial fibrosis and tubular atrophy |
| IgG | Immunoglobulin G |
| shRNA | Small hairpin RNA |
| LUMC | Leiden University Medical Centre |
| qPCR | Quantitative real-time PCR |
| Ct | Cycle threshold |
| PBS | Phosphate-buffered saline |
| TBS | Tris-buffered saline |
| SDS | Sodium dodecyl sulfate |
| BSA | Bovine serum albumin |
References
- Gross, J.L.; De Azevedo, M.J.; Silveiro, S.P.; Canani, L.H.; Caramori, M.L.; Zelmanovitz, T. Diabetic nephropathy: Diagnosis, prevention, and treatment. Diabetes Care 2004, 28, 164–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrows, N.R.; Li, Y.; Geiss, L.S. Incidence of treatment for end-stage renal disease among individuals with diabetes in the U.S. continues to decline. Diabetes Care 2010, 33, 73–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rheinberger, M.; Boger, C.A. Diabetic nephropathy: New insights into diagnosis, prevention and treatment. Dtsch. Med. Wochenschr. 2014, 139, 704–706. [Google Scholar] [PubMed]
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A Common Pathway to Organ Injury and Failure. N. Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [CrossRef]
- Menn-Josephy, H.; Lee, C.S.; Nolin, A.; Christov, M.; Rybin, D.V.; Weinberg, J.M.; Henderson, J.; Bonegio, R.; Havasi, A. Renal Interstitial Fibrosis: An Imperfect Predictor of Kidney Disease Progression in Some Patient Cohorts. Am. J. Nephrol. 2016, 44, 289–299. [Google Scholar] [CrossRef] [Green Version]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-beta signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.K.; Sheppard, D.; Chapman, H.A. TGF-beta1 Signaling and Tissue Fibrosis. Cold Spring Harb. Perspect. Biol. 2018, 10, a022293. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Samarakoon, R.; Overstreet, J.M.; Higgins, P.J. TGF-beta signaling in tissue fibrosis: Redox controls, target genes and therapeutic opportunities. Cell Signal 2013, 25, 264–268. [Google Scholar] [CrossRef] [Green Version]
- Hinz, B.; Celetta, G.; Tomasek, J.J.; Gabbiani, G.; Chaponnier, C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol. Biol. Cell 2001, 12, 2730–2741. [Google Scholar] [CrossRef] [Green Version]
- Desmouliere, A.; Geinoz, A.; Gabbiani, F.; Gabbiani, G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 1993, 122, 103–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ignotz, R.A.; Massagué, J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J. Biol. Chem. 1986, 261, 4337–4345. [Google Scholar] [PubMed]
- Zhang, K.; Rekhter, M.D.; Gordon, D.; Phan, S.H. Myofibroblasts and their role in lung collagen gene expression during pulmonary fibrosis. A combined immunohistochemical and in situ hybridization study. Am. J. Pathol. 1994, 145, 114–125. [Google Scholar]
- Gougos, A.; Letarte, M. Identification of a human endothelial cell antigen with monoclonal antibody 44G4 produced against a pre-B leukemic cell line. J. Immunol. 1988, 141, 1925–1933. [Google Scholar] [PubMed]
- Lastres, P.; Bellón, T.; Cabañas, C.; Sánchez-Madrid, F.; Acevedo, A.; Gougos, A.; Letarte, M.; Bernabéu, C. Regulated expression on human macrophages of endoglin, an Arg-Gly-Asp-containing surface antigen. Eur. J. Immunol. 1992, 22, 393–397. [Google Scholar] [CrossRef]
- Adam, P.J.; Clesham, G.J.; Weissberg, P.L. Expression of endoglin mRNA and protein in human vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 1998, 247, 33–37. [Google Scholar] [CrossRef]
- St-Jacques, S.; Cymerman, U.; Pece, N.; Letarte, M. Molecular characterization and in situ localization of murine endoglin reveal that it is a transforming growth factor-beta binding protein of endothelial and stromal cells. Endocrinology 1994, 134, 2645–2657. [Google Scholar] [CrossRef]
- López-Novoa, J.M.; Bernabeu, C. The physiological role of endoglin in the cardiovascular system. Am. J. Physiol. Circ. Physiol. 2010, 299, H959–H974. [Google Scholar] [CrossRef] [Green Version]
- Clemente, M.; Núñez, O.; Lorente, R.; Rincón, D.; Matilla, A.; Salcedo, M.; Catalina, M.V.; Ripoll, C.; Iacono, O.L.; Bañares, R.; et al. Increased intrahepatic and circulating levels of endoglin, a TGF-beta1 co-receptor, in patients with chronic hepatitis C virus infection: Relationship to histological and serum markers of hepatic fibrosis. J. Viral Hepat. 2006, 13, 625–632. [Google Scholar] [CrossRef]
- Burke, J.P.; Watson, R.W.; Mulsow, J.J.; Docherty, N.G.; Coffey, J.C.; O’Connell, P.R. Endoglin negatively regulates transforming growth factor beta1-induced profibrotic responses in intestinal fibroblasts. Br. J. Surg. 2010, 97, 892–901. [Google Scholar] [CrossRef]
- Chen, K.; Mehta, J.L.; Li, D.; Joseph, L.; Joseph, J. Transforming growth factor beta receptor endoglin is expressed in cardiac fibroblasts and modulates profibrogenic actions of angiotensin II. Circ. Res. 2004, 95, 1167–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shyu, K.G. The Role of Endoglin in Myocardial Fibrosis. Acta Cardiol. Sin. 2017, 33, 461–467. [Google Scholar] [PubMed]
- Docherty, N.G.; Lopez-Novoa, J.M.; Arevalo, M.; Düwel, A.; Rodriguez-Peña, A.; Pérez-Barriocanal, F.; Bernabéu, C.; Eleno, N. Endoglin regulates renal ischaemia-reperfusion injury. Nephrol. Dial. Transplant. 2006, 21, 2106–2119. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Peña, A.; Eleno, N.; Düwell, A.; Arévalo, M.; Pérez-Barriocanal, F.; Flores, O.; Docherty, N.; Bernabeu, C.; Letarte, M.; López-Novoa, J.M. Endoglin upregulation during experimental renal interstitial fibrosis in mice. Hypertension 2002, 40, 713–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieto, M.; Rodríguez-Peña, A.B.; Düwel, A.; Rivas, J.V.; Docherty, N.G.; Pérez-Barriocanal, F.; Arévalo, M.; Vary, C.P.H.; Bernabéu, C.; Lopez-Novoa, J.M.; et al. Temporal changes in renal endoglin and TGF-beta1 expression following ureteral obstruction in rats. J. Physiol. Biochem. 2005, 61, 457–467. [Google Scholar] [CrossRef]
- Oujo, B.; Muñoz-Félix, J.M.; Arevalo, M.; Nuñez-Gomez, E.; Perez-Roque, L.; Pericacho, M.; Gonzalez-Nunez, M.; Langa, C.; Martinez-Salgado, C.; Pérez-Barriocanal, F.; et al. L-Endoglin overexpression increases renal fibrosis after unilateral ureteral obstruction. PLoS ONE 2014, 9, e110365. [Google Scholar] [CrossRef]
- Kapur, N.K.; Wilson, S.; Yunis, A.A.; Qiao, X.; Mackey, E.; Paruchuri, V.; Baker, C.; Aronovitz, M.J.; Karumanchi, S.A.; Letarte, M.; et al. Reduced endoglin activity limits cardiac fibrosis and improves survival in heart failure. Circulation 2012, 125, 2728–2738. [Google Scholar] [CrossRef] [Green Version]
- Scharpfenecker, M.; Floot, B.; Russell, N.; Coppes, R.P.; Stewart, F.A.; Coppes, R.P. Endoglin haploinsufficiency attenuates radiation-induced deterioration of kidney function in mice. Radiother. Oncol. 2013, 108, 464–468. [Google Scholar] [CrossRef]
- Paauwe, M.; Schoonderwoerd, M.J.A.; Helderman, R.; Harryvan, T.J.; Groenewoud, A.; van Pelt, G.W.; Bor, R.; Hemmer, D.M.; Versteeg, H.H.; Snaar-Jagalska, B.E.; et al. Endoglin Expression on Cancer-Associated Fibroblasts Regulates Invasion and Stimulates Colorectal Cancer Metastasis. Clin. Cancer Res. 2018, 24, 6331–6344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, A.M.; Ponticos, M.; Shi-Wen, X.; Denton, C.P.; Abraham, D.J. Elevated CCN2 expression in scleroderma: A putative role for the TGFbeta accessory receptors TGFbetaRIII and endoglin. J. Cell Commun. Signal. 2011, 5, 173–177. [Google Scholar] [CrossRef] [Green Version]
- Roy-Chaudhury, P.; Simpson, J.G.; Power, D.A. Endoglin, a transforming growth factor-beta-binding protein, is upregulated in chronic progressive renal disease. Exp. Nephrol. 1997, 5, 55–60. [Google Scholar] [PubMed]
- Bader, R.; Bader, H.; Grund, K.; Mackensen-Haen, S.; Christ, H.; Bohle, A. Structure and function of the kidney in diabetic glomerulosclerosis. Correlations between morphological and functional parameters. Pathol. Res. Pr. 1980, 167, 204–216. [Google Scholar] [CrossRef]
- White, K.E.; Bilous, R.W. Type 2 diabetic patients with nephropathy show structural-functional relationships that are similar to type 1 disease. J. Am. Soc. Nephrol. 2000, 11, 1667–1673. [Google Scholar]
- Scharpfenecker, M.; Floot, B.; Russell, N.S.; Ten Dijke, P.; Stewart, F.A. Endoglin haploinsufficiency reduces radiation-induced fibrosis and telangiectasia formation in mouse kidneys. Radiother. Oncol. 2009, 92, 484–491. [Google Scholar] [CrossRef]
- Scharpfenecker, M.; Floot, B.; Russell, N.S.; Stewart, F.A. The TGF-beta co-receptor endoglin regulates macrophage infiltration and cytokine production in the irradiated mouse kidney. Radiother. Oncol. 2012, 105, 313–320. [Google Scholar] [CrossRef]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Galli, A.; Bochaton-Piallat, M.L.; Gabbiani, G. The myofibroblast: One function, multiple origins. Am. J. Pathol. 2007, 170, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Keane, T.J.; Horejs, C.-M.; Stevens, M.M. Scarring vs. functional healing: Matrix-based strategies to regulate tissue repair. Adv. Drug Deliv. Rev. 2018, 129, 407–419. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef]
- Asada, N.; Takase, M.; Nakamura, J.; Oguchi, A.; Asada, M.; Suzuki, N.; Yamamura, K.-I.; Nagoshi, N.; Shibata, S.; Rao, T.N.; et al. Dysfunction of fibroblasts of extrarenal origin underlies renal fibrosis and renal anemia in mice. J. Clin. Investig. 2011, 121, 3981–3990. [Google Scholar] [CrossRef] [Green Version]
- Ina, K.; Kitamura, H.; Tatsukawa, S.; Takayama, T.; Fujikura, Y.; Shimada, T. Transformation of interstitial fibroblasts and tubulointerstitial fibrosis in diabetic nephropathy. Med. Mol. Morphol. 2002, 35, 87–95. [Google Scholar] [CrossRef]
- Coimbra, T.M.; Janssen, U.; Gröne, H.J.; Ostendorf, T.; Kunter, U.; Schmidt, H.; Brabant, G.; Floege, J. Early events leading to renal injury in obese Zucker (fatty) rats with type II diabetes. Kidney Int. 2000, 57, 167–182. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Barbero, A.; Obreo, J.; Eleno, N.; Rodríguez-Peña, A.; Düwel, A.; Jerkić, M.; Sánchez-Rodríguez, A.; Bernabéu, C.; Lopez-Novoa, J.M. Endoglin expression in human and rat mesangial cells and its upregulation by TGF-beta1. Biochem. Biophys. Res. Commun. 2001, 282, 142–147. [Google Scholar] [CrossRef]
- Diez-Marques, L.; Ortega, R.; Langa, C.; Rodríguez-Barbero, A.; Lopez-Novoa, J.M.; Lamas, S.; Bernabéu, C. Expression of endoglin in human mesangial cells: Modulation of extracellular matrix synthesis. Biochim. Biophys. Acta 2002, 1587, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Desmoulière, A.; Darby, I.A.; Gabbiani, G. Normal and pathologic soft tissue remodeling: Role of the myofibroblast, with special emphasis on liver and kidney fibrosis. Lab. Investig. 2003, 83, 1689–1707. [Google Scholar] [CrossRef]
- Evans, R.A.; Tian, Y.C.; Steadman, R.; Phillips, A.O. TGF-beta1-mediated fibroblast-myofibroblast terminal differentiation-the role of Smad proteins. Exp. Cell Res. 2003, 282, 90–100. [Google Scholar] [CrossRef]
- Meurer, S.K.; Alsamman, M.; Sahin, H.; Wasmuth, H.E.; Kisseleva, T.; Brenner, D.A.; Trautwein, C.; Weiskirchen, R.; Scholten, D. Overexpression of endoglin modulates TGF-beta1-signalling pathways in a novel immortalized mouse hepatic stellate cell line. PLoS ONE 2013, 8, e56116. [Google Scholar] [CrossRef] [Green Version]
- Velasco, S.; Alvarez-Munoz, P.; Pericacho, M.; Dijke, P.T.; Bernabeu, C.; Lopez-Novoa, J.M.; Rodriguez-Barbero, A. L- and S-endoglin differentially modulate TGFbeta1 signaling mediated by ALK1 and ALK5 in L6E9 myoblasts. J. Cell Sci. 2008, 121, 913–919. [Google Scholar] [CrossRef] [Green Version]
- Morris, E.; Chrobak, I.; Bujor, A.; Hant, F.; Mummery, C.; Ten Dijke, P.; Trojanowska, M. Endoglin promotes TGF-beta/Smad1 signaling in scleroderma fibroblasts. J. Cell. Physiol. 2011, 226, 3340–3348. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Félix, J.M.; Oujo, B.; Lopez-Novoa, J.M. The role of endoglin in kidney fibrosis. Expert Rev. Mol. Med. 2014, 16, e18. [Google Scholar] [CrossRef]
- Finnson, K.W.; Philip, A. Endoglin in liver fibrosis. J. Cell Commun. Signal. 2011, 6, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, X.M.; Tang, P.M.; Li, J.; Lan, H.Y. TGF-beta/Smad signaling in renal fibrosis. Front. Physiol. 2015, 6, 82. [Google Scholar] [CrossRef] [Green Version]
- Wilkes, M.C.; Mitchell, H.; Penheiter, S.G.; Dore, J.J.; Suzuki, K.; Edens, M.; Sharma, D.K.; Pagano, R.E.; Leof, E.B. Transforming growth factor-beta activation of phosphatidylinositol 3-kinase is independent of Smad2 and Smad3 and regulates fibroblast responses via p21-activated kinase-2. Cancer Res. 2005, 65, 10431–10440. [Google Scholar] [CrossRef] [Green Version]
- Daniels, C.E.; Wilkes, M.C.; Edens, M.; Kottom, T.J.; Murphy, S.J.; Limper, A.H.; Leof, E.B. Imatinib mesylate inhibits the profibrogenic activity of TGF-beta and prevents bleomycin-mediated lung fibrosis. J. Clin. Investig. 2004, 114, 1308–1316. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, H.; Meyer, C.; Li, J.; Nadalin, S.; Konigsrainer, A.; Weng, H.; Dooley, S.; Ten Dijke, P. Transforming growth factor-beta (TGF-beta)-mediated connective tissue growth factor (CTGF) expression in hepatic stellate cells requires Stat3 signaling activation. J. Biol. Chem. 2013, 288, 30708–30719. [Google Scholar] [CrossRef] [Green Version]
- Dees, C.; Tomcik, M.; Palumbo-Zerr, K.; Distler, A.; Beyer, C.; Lang, V.; Horn, A.; Zerr, P.; Zwerina, J.; Gelse, K.; et al. JAK-2 as a novel mediator of the profibrotic effects of transforming growth factor beta in systemic sclerosis. Arthritis Rheum. 2012, 64, 3006–3015. [Google Scholar] [CrossRef]
- Bus, P.; Gerrits, T.; Heemskerk, S.A.; Zandbergen, M.; Wolterbeek, R.; Bruijn, J.A.; Baelde, H.J.; Scharpfenecker, M. Endoglin Mediates Vascular Endothelial Growth Factor-A-Induced Endothelial Cell Activation by Regulating Akt Signaling. Am. J. Pathol. 2018, 188, 2924–2935. [Google Scholar] [CrossRef] [Green Version]
- Lindenmeyer, M.T.; Kretzler, M.; Boucherot, A.; Berra, S.; Yasuda, Y.; Henger, A.; Eichinger, F.; Gaiser, S.; Schmid, H.; Rastaldi, M.P.; et al. Interstitial vascular rarefaction and reduced VEGF-A expression in human diabetic nephropathy. J. Am. Soc. Nephrol. 2007, 18, 1765–1776. [Google Scholar] [CrossRef] [Green Version]
- Baelde, H.-J.; Eikmans, M.; Lappin, D.; Doran, P.; Hohenadel, D.; Brinkkoetter, P.-T.; Van Der Woude, F.-J.; Waldherr, R.; Rabelink, T.-J.; De Heer, E.; et al. Reduction of VEGF-A and CTGF expression in diabetic nephropathy is associated with podocyte loss. Kidney Int. 2007, 71, 637–645. [Google Scholar] [CrossRef] [Green Version]
- Apolo, A.B.; Karzai, F.H.; Trepel, J.B.; Alarcon, S.; Lee, S.; Lee, M.J.; Tomita, Y.; Cao, L.; Yu, Y.; Merino, M.J.; et al. A Phase II Clinical Trial of TRC105 (Anti-Endoglin Antibody) in Adults with Advanced/Metastatic Urothelial Carcinoma. Clin. Genitourin. Cancer 2017, 15, 77–85. [Google Scholar] [CrossRef]
- Duffy, A.; Ulahannan, S.V.; Cao, L.; Rahma, O.; Makarova-Rusher, O.; Kleiner, D.; Fioravanti, S.; Walker, M.; Carey, S.; Yu, Y.; et al. A phase II study of TRC105 in patients with hepatocellular carcinoma who have progressed on sorafenib. United Eur. Gastroenterol. J. 2015, 3, 453–461. [Google Scholar] [CrossRef]
- TRACON Pharmaceuticals I. [Internet] Pipeline; TRC205. [Updated 2017]. Available online: http://traconpharma.com/trc205.php (accessed on 9 November 2018).
- Klessens, C.Q.; Woutman, T.D.; Veraar, K.A.; Zandbergen, M.; Valk, E.J.; Rotmans, J.; Wolterbeek, R.; Bruijn, J.A.; Bajema, I.M. An autopsy study suggests that diabetic nephropathy is underdiagnosed. Kidney Int. 2016, 90, 149–156. [Google Scholar] [CrossRef]
- Tervaert, T.W.; Mooyaart, A.L.; Amann, K.; Cohen, A.H.; Cook, H.T.; Drachenberg, C.B.; Ferrario, F.; Fogo, A.B.; Haas, M.; de Heer, E.; et al. Pathologic classification of diabetic nephropathy. J. Am. Soc. Nephrol. 2010, 21, 556–563. [Google Scholar] [CrossRef] [Green Version]
- Müller, G.A.; Frank, J.; Rodemann, H.P.; Engler-Blum, G. Human renal fibroblast cell lines (tFKIF and tNKF) are new tools to investigate pathophysiologic mechanisms of renal interstitial fibrosis. Exp. Nephrol. 1995, 3, 127–133. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Histological Confirmed DN (n = 55) | Diabetic Controls (n = 25) | p | ||
|---|---|---|---|---|---|
| Male sex | n (%) | 34 (61.8) | 17 (68.0) | 0.594 * | |
| Age years | Mean ± SD | 67.4 ± 13.8 | 67.4 ± 12.7 | 0.996 † | |
| Type 1 Diabetes | n (%) | 5 (10.4) | 3 (13.0) | 0.541 * | |
| eGFR (ml/min/1.73 m2) | Mean ± SD | 51.7 ± 34.9 | 72.4 ± 38.7 | 0.035 † | |
| Serum creatinine (µmol/L) | Mean ± SD | 167.3 ± 116.6 | 103.7 ± 51. 5 | 0.024 † | |
| Systolic blood pressure (mmHg) | Mean ± SD | 140.8 ± 32.2 | 134.3 ± 28.2 | 0.509 † | |
| Diastolic blood pressure (mmHg) | Mean ± SD | 76.4 ± 11.6 | 78.2 ± 18.5 | 0.682 † | |
| HbA1c (%) | Mean ± SD | 8.2 ± 2.2 | 7.1 ± 1.1 | 0.205 † | |
| Hypertension present | n (%) | 26 (59.1) | 11 (52.4) | 0.609 * | |
| IFTA | Index score 0 | n (%) | 9 (16.4) | 12 (48.0) | |
| Index score 1 | n (%) | 28(50.9) | 10 (40.0) | ||
| Index score 2 | n (%) | 11 (20.0) | 3 (12.0) | ||
| Index score 3 | n (%) | 7 (12.7) | 0 (0.0) | 0.013 * | |
| Gene | Forward | Reverse |
|---|---|---|
| ACTA2 | 5’-TTCAATGTCCCAGCCATGTA-3’ | 5’-GAAGGAATAGCCACGCTCAG-3’ |
| CTGF | 5’-CCTGGTCCAGACCACAGAGT-3’ | 5’-TGGAGATTTTGGGAGTACGG-3’ |
| ENG | 5’-CACTAGCCAGGTCTCGAAGG-3’ | 5’-CTGAGGACCAGAAGCACCTC-3’ |
| HPRT | 5’-AGATGGTCAAGGTCGCAAGC-3’ | 5’-TCAAGGGCATATCCTACAACAAAC-3’ |
| SERPINE1 | 5’-ACTGGAAAGGCAACATGACC-3’ | 5’-TGACAGCTGTGGATGAGGAG-3’ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerrits, T.; Zandbergen, M.; Wolterbeek, R.; Bruijn, J.A.; Baelde, H.J.; Scharpfenecker, M. Endoglin Promotes Myofibroblast Differentiation and Extracellular Matrix Production in Diabetic Nephropathy. Int. J. Mol. Sci. 2020, 21, 7713. https://doi.org/10.3390/ijms21207713
Gerrits T, Zandbergen M, Wolterbeek R, Bruijn JA, Baelde HJ, Scharpfenecker M. Endoglin Promotes Myofibroblast Differentiation and Extracellular Matrix Production in Diabetic Nephropathy. International Journal of Molecular Sciences. 2020; 21(20):7713. https://doi.org/10.3390/ijms21207713
Chicago/Turabian StyleGerrits, Tessa, Malu Zandbergen, Ron Wolterbeek, Jan A. Bruijn, Hans J. Baelde, and Marion Scharpfenecker. 2020. "Endoglin Promotes Myofibroblast Differentiation and Extracellular Matrix Production in Diabetic Nephropathy" International Journal of Molecular Sciences 21, no. 20: 7713. https://doi.org/10.3390/ijms21207713
APA StyleGerrits, T., Zandbergen, M., Wolterbeek, R., Bruijn, J. A., Baelde, H. J., & Scharpfenecker, M. (2020). Endoglin Promotes Myofibroblast Differentiation and Extracellular Matrix Production in Diabetic Nephropathy. International Journal of Molecular Sciences, 21(20), 7713. https://doi.org/10.3390/ijms21207713