MYC as a Multifaceted Regulator of Tumor Microenvironment Leading to Metastasis



Abstract
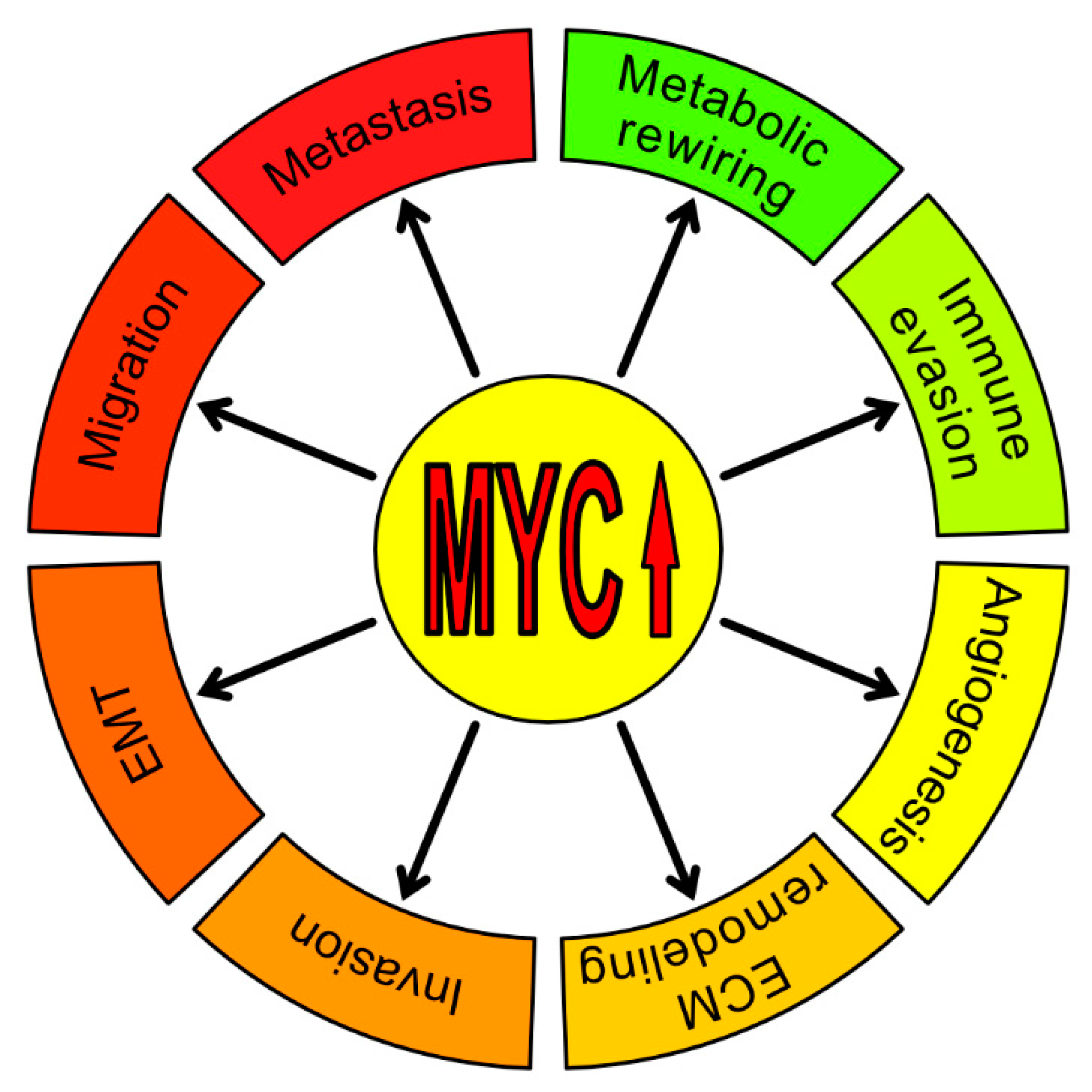
:1. Introduction
2. MYC–A Key Player in the Tumor Microenvironment (TME)
2.1. Tumor-Infiltrating Cells within Tumor Microenvironment: Why MYC Matters?
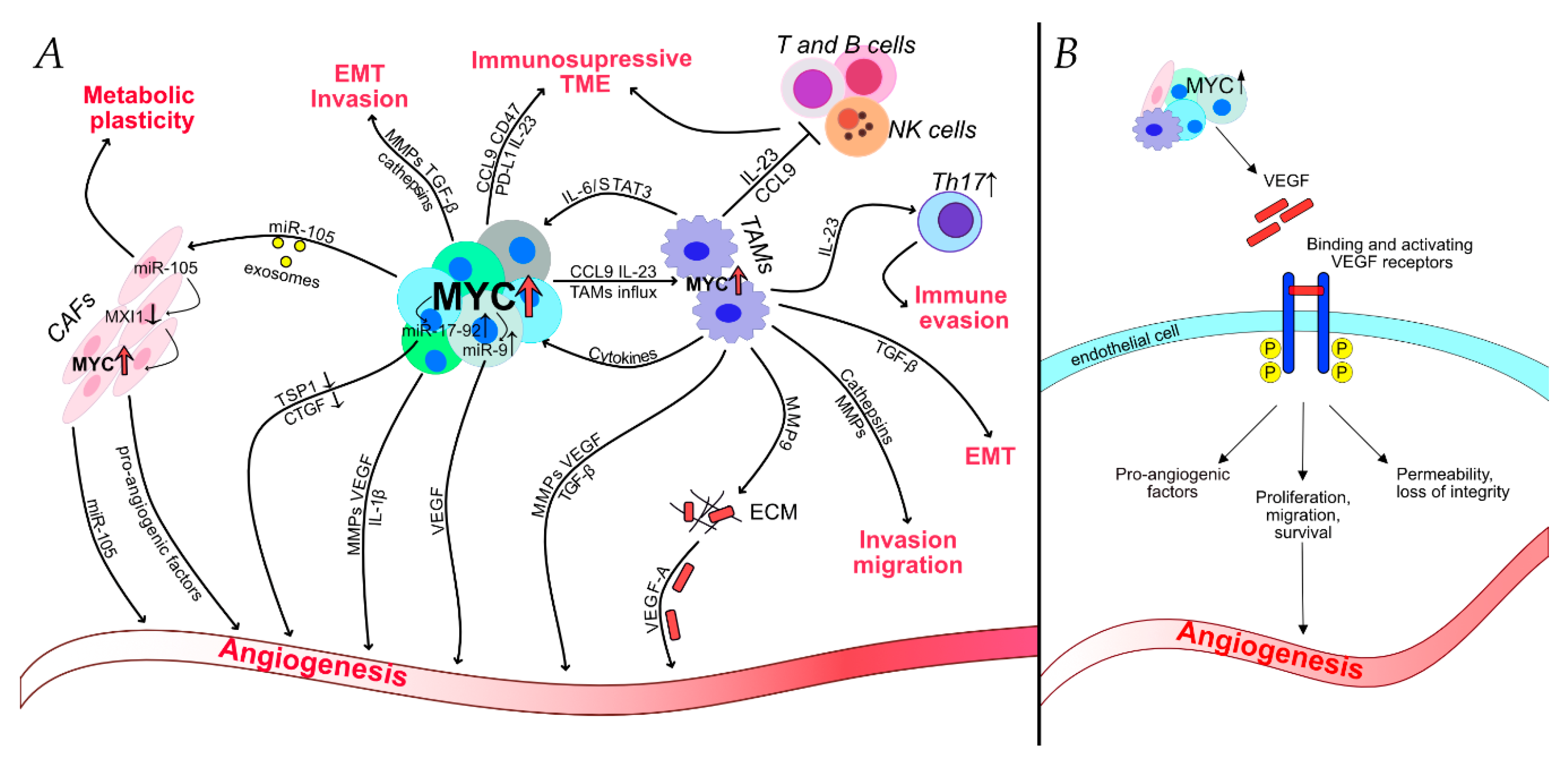
2.2. TAMs–Main Players in MYC-Regulated Tumor Microenvironment
2.3. Angiogenesis–MYC Controls the “Angiogenic Switch”
2.4. Immune Evasion and MYC–The Key Elements of Going under Cover
2.5. MYC in Invasion and Migration–First Steps in Successful Metastasis Formation
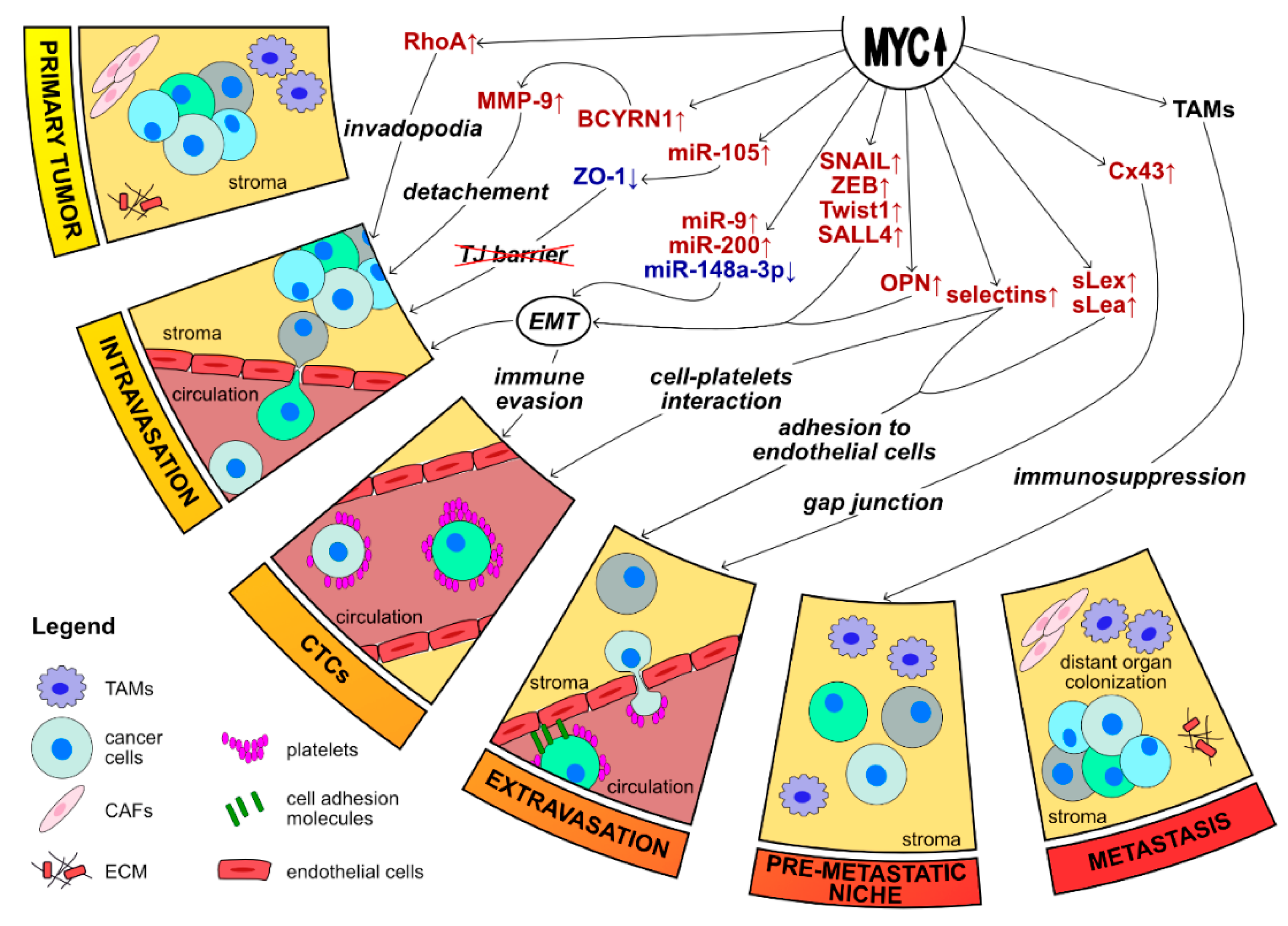
3. MYC–The Regulator of EMT and Metastasis
3.1. MYC Promotes Metastasis via Regulation of EMT
3.2. MYC’s Involvement in the Metastatic Cascade
3.3. Non-Coding RNAs Plays an Integral Role in MYC-Mediated Metastatic Progression
3.4. MYC Recruits TAMs to Promote EMT and Metastasis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EMT | Epithelial-to-Mesenchymal Transition |
| TME | Tumor MicroEnvironment |
| TAMs | Tumor-Associated Macrophages |
| CAFs | Cancer-Associated Fibroblasts |
| ECM | ExtraCellular Matrix |
| NK | Natural Killer |
| miR | microRNA |
| lncRNA | long non-coding RNA |
References
- Dang, C.V.; O’donnell, K.A.; Zeller, K.I.; Nguyen, T.; Osthus, R.C.; Li, F. The C-Myc Target Gene Network. Semin. Cancer Biol. 2006, 16, 253–264. [Google Scholar] [CrossRef]
- Meyer, N.; Penn, L.Z. Reflecting On 25 Years with Myc. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chen, Y.; Olopade, O.I. Myc and Breast Cancer. Genes Cancer 2010, 1, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Kalkat, M.; De Melo, J.; Hickman, K.A.; Lourenco, C.; Redel, C.; Resetca, D.; Tamachi, A.; Tu, W.B.; Penn, L.Z. Myc Deregulation in Primary Human Cancers. Genes 2017, 8, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Liu, H.; Qing, G. Targeting Oncogenic Myc as a Strategy for Cancer Treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albihn, A.; Johnsen, J.I.; Henriksson, M.A. Myc in Oncogenesis and as a Target for Cancer Therapies. Adv. Cancer Res. 2010, 107, 163–224. [Google Scholar] [CrossRef]
- Wolfer, A.; Wittner, B.S.; Irimia, D.; Flavin, R.J.; Lupien, M.; Gunawardane, R.N.; Meyer, C.A.; Lightcap, E.S.; Tamayo, P.; Mesirov, J.P.; et al. Myc Regulation of a “Poor-Prognosis” Metastatic Cancer Cell State. Proc. Natl. Acad. Sci. USA 2010, 107, 3698–3703. [Google Scholar] [CrossRef] [Green Version]
- Casey, S.C.; Baylot, V.; Felsher, D.W. The Myc Oncogene Is a Global Regulator of the Immune Response. Blood 2018, 131, 2007–2015. [Google Scholar] [CrossRef] [Green Version]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gutgemann, I.; Eilers, M.; et al. Myc Regulates the Antitumor Immune Response through Cd47 and Pd-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V. Myc On the Path To Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Dews, M.; Homayouni, A.; Yu, D.; Murphy, D.; Sevignani, C.; Wentzel, E.; Furth, E.E.; Lee, W.M.; Enders, G.H.; Mendell, J.T.; et al. Augmentation of Tumor Angiogenesis by a Myc-Activated Microrna Cluster. Nat. Genet 2006, 38, 1060–1065. [Google Scholar] [CrossRef] [Green Version]
- Whitfield, J.R.; Soucek, L. Tumor Microenvironment: Becoming Sick of Myc. Cell Mol. Life Sci. 2012, 69, 931–934. [Google Scholar] [CrossRef] [Green Version]
- Wolfer, A.; Ramaswamy, S. Myc and Metastasis. Cancer Res. 2011, 71, 2034–2037. [Google Scholar] [CrossRef] [Green Version]
- Venkateswaran, N.; Conacci-Sorrell, M. Myc Leads the Way. Small Gtpases 2020, 11, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The Tumor Microenvironment and Its Role in Promoting Tumor Growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [Green Version]
- Bissell, M.J.; Hines, W.C. Why Don’t We Get More Cancer? a Proposed Role of the Microenvironment in Restraining Cancer Progression. Nat. Med. 2011, 17, 320–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Cai, S.; Wang, G.; Cao, X.; Yang, X.; Luo, X.; Feng, Y.; Hu, J. C-Myc Enhances Colon Cancer Cell-Mediated Angiogenesis through the Regulation of Hif-1alpha. Biochem. Biophys. Res. Commun. 2013, 430, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Kortlever, R.M.; Sodir, N.M.; Wilson, C.H.; Burkhart, D.L.; Pellegrinet, L.; Brown Swigart, L.; Littlewood, T.D.; Evan, G.I. Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 2017, 171, 1301–1315.e14. [Google Scholar] [CrossRef]
- Casacuberta-Serra, S.; Soucek, L. Myc and Ras, the Bonnie and Clyde of Immune Evasion. Transl. Cancer Res. 2018, 7, S457–S459. [Google Scholar] [CrossRef]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Hampton, G.M.; Wahl, G.M. C-Myc Can Induce Dna Damage, Increase Reactive Oxygen Species, and Mitigate P53 Function: A Mechanism for Oncogene-Induced Genetic Instability. Mol. Cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic Instability--An Evolving Hallmark of Cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Sonugur, F.G.; Akbulut, H. The Role of Tumor Microenvironment in Genomic Instability of Malignant Tumors. Front. Genet. 2019, 10, 1063. [Google Scholar] [CrossRef]
- Sodir, N.M.; Swigart, L.B.; Karnezis, A.N.; Hanahan, D.; Evan, G.I.; Soucek, L. Endogenous Myc Maintains the Tumor Microenvironment. Genes Dev. 2011, 25, 907–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pello, O.M.; De Pizzol, M.; Mirolo, M.; Soucek, L.; Zammataro, L.; Amabile, A.; Doni, A.; Nebuloni, M.; Swigart, L.B.; Evan, G.I.; et al. Role of C-Myc in Alternative Activation of Human Macrophages and Tumor-Associated Macrophage Biology. Blood 2012, 119, 411–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhanasekaran, R.; Baylot, V.; Kim, M.; Kuruvilla, S.; Bellovin, D.I.; Adeniji, N.; Rajan Kd, A.; Lai, I.; Gabay, M.; Tong, L.; et al. Myc and Twist1 Cooperate To Drive Metastasis by Eliciting Crosstalk between Cancer and Innate Immunity. Elife 2020, 9. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories To the Crime: Functions of Cells Recruited To the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Dannenmann, S.R.; Thielicke, J.; Stockli, M.; Matter, C.; Von Boehmer, L.; Cecconi, V.; Hermanns, T.; Hefermehl, L.; Schraml, P.; Moch, H.; et al. Tumor-Associated Macrophages Subvert T-Cell Function and Correlate with Reduced Survival in Clear Cell Renal Cell Carcinoma. Oncoimmunology 2013, 2, E23562. [Google Scholar] [CrossRef] [Green Version]
- Tao, L.; Huang, G.; Song, H.; Chen, Y.; Chen, L. Cancer Associated Fibroblasts: An Essential Role in the Tumor Microenvironment. Oncol. Lett. 2017, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Xu, J.; Lan, H. Tumor-Associated Macrophages in Tumor Metastasis: Biological Roles and Clinical Therapeutic Applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef] [PubMed]
- Pello, O.M.; Chevre, R.; Laoui, D.; De Juan, A.; Lolo, F.; Andres-Manzano, M.J.; Serrano, M.; Van Ginderachter, J.A.; Andres, V. In Vivo Inhibition of C-Myc in Myeloid Cells Impairs Tumor-Associated Macrophage Maturation and Pro-Tumoral Activities. PLoS ONE 2012, 7, e45399. [Google Scholar] [CrossRef]
- Yan, W.; Wu, X.; Zhou, W.; Fong, M.Y.; Cao, M.; Liu, J.; Liu, X.; Chen, C.H.; Fadare, O.; Pizzo, D.P.; et al. Cancer-Cell-Secreted Exosomal Mir-105 Promotes Tumour Growth through the Myc-Dependent Metabolic Reprogramming of Stromal Cells. Nat. Cell Biol. 2018, 20, 597–609. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J.; Wynn, T.A. Protective and Pathogenic Functions of Macrophage Subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Bottazzi, B.; Colotta, F.; Sozzani, S.; Ruco, L. The Origin and Function of Tumor-Associated Macrophages. Immunol. Today 1992, 13, 265–270. [Google Scholar] [CrossRef]
- Balkwill, F.; Mantovani, A. Inflammation and Cancer: Back To Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-Associated Macrophages (Tam) as Major Players of the Cancer-Related Inflammation. J. Leukoc. Biol. 2009, 86, 1065–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S. Alternative Activation of Macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Locati, M. Macrophage Polarization Comes of Age. Immunity 2005, 23, 344–346. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.; Taylor, P.R. Monocyte and Macrophage Heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Martinez, F.O.; Helming, L.; Gordon, S. Alternative Activation of Macrophages: An Immunologic Functional Perspective. Annu. Rev. Immunol. 2009, 27, 451–483. [Google Scholar] [CrossRef] [Green Version]
- Schmieder, A.; Michel, J.; Schonhaar, K.; Goerdt, S.; Schledzewski, K. Differentiation and Gene Expression Profile of Tumor-Associated Macrophages. Semin. Cancer Biol. 2012, 22, 289–297. [Google Scholar] [CrossRef]
- Coussens, L.M.; Tinkle, C.L.; Hanahan, D.; Werb, Z. Mmp-9 Supplied by Bone Marrow-Derived Cells Contributes To Skin Carcinogenesis. Cell 2000, 103, 481–490. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage Polarization: Tumor-Associated Macrophages as a Paradigm for Polarized M2 Mononuclear Phagocytes. Trends. Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Pollard, J.W. Tumour-Educated Macrophages Promote Tumour Progression and Metastasis. Nat. Rev. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Schioppa, T.; Mantovani, A.; Allavena, P. Tumour-Associated Macrophages Are a Distinct M2 Polarised Population Promoting Tumour Progression: Potential Targets of Anti-Cancer Therapy. Eur. J. Cancer 2006, 42, 717–727. [Google Scholar] [CrossRef]
- Talmadge, J.E.; Donkor, M.; Scholar, E. Inflammatory Cell Infiltration of Tumors: Jekyll Or Hyde. Cancer Metastasis Rev. 2007, 26, 373–400. [Google Scholar] [CrossRef]
- Tsutsui, S.; Yasuda, K.; Suzuki, K.; Tahara, K.; Higashi, H.; Era, S. Macrophage Infiltration and Its Prognostic Implications in Breast Cancer: The Relationship with Vegf Expression and Microvessel Density. Oncol. Rep. 2005, 14, 425–431. [Google Scholar] [CrossRef]
- Hansen, B.D.; Schmidt, H.; Von Der Maase, H.; Sjoegren, P.; Agger, R.; Hokland, M. Tumour-Associated Macrophages Are Related To Progression in Patients with Metastatic Melanoma Following Interleukin-2 Based Immunotherapy. Acta Oncol. 2006, 45, 400–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Mulcahy, L.A.; Mohammed, R.A.; Lee, A.H.; Franks, H.A.; Kilpatrick, L.; Yilmazer, A.; Paish, E.C.; Ellis, I.O.; Patel, P.M.; et al. Il-17 Expression by Breast-Cancer-Associated Macrophages: Il-17 Promotes Invasiveness of Breast Cancer Cell Lines. Breast Cancer Res. BCR 2008, 10, R95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steidl, C.; Lee, T.; Shah, S.P.; Farinha, P.; Han, G.; Nayar, T.; Delaney, A.; Jones, S.J.; Iqbal, J.; Weisenburger, D.D.; et al. Tumor-Associated Macrophages and Survival in Classic Hodgkin’s Lymphoma. N. Engl. J. Med. 2010, 362, 875–885. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Yan, Y.; Yang, Y.; Wang, L.; Li, M.; Wang, J.; Liu, X.; Duan, X.; Wang, J. High Infiltration of Tumor-Associated Macrophages Influences Poor Prognosis in Human Gastric Cancer Patients, Associates with the Phenomenon of Emt. Medicine 2016, 95, E2636. [Google Scholar] [CrossRef]
- Zhao, X.; Qu, J.; Sun, Y.; Wang, J.; Liu, X.; Wang, F.; Zhang, H.; Wang, W.; Ma, X.; Gao, X.; et al. Prognostic Significance of Tumor-Associated Macrophages in Breast Cancer: A Meta-Analysis of the Literature. Oncotarget 2017, 8, 30576–30586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Yu, Y.; He, X.; Niu, N.; Li, X.; Zhang, R.; Hu, J.; Ma, J.; Yu, X.; Sun, Y.; et al. Tumor-Associated Macrophages Induce Invasion and Poor Prognosis in Human Gastric Cancer in a Cyclooxygenase-2/Mmp9-Dependent Manner. Am. J. Transl. Res. 2019, 11, 6040–6054. [Google Scholar] [PubMed]
- Tian, K.; Wang, X.; Wu, Y.; Wu, X.; Du, G.; Liu, W.; Wu, R. Relationship of Tumour-Associated Macrophages with Poor Prognosis in Wilms’ Tumour. J. Pediatr. Urol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Shchors, K.; Shchors, E.; Rostker, F.; Lawlor, E.R.; Brown-Swigart, L.; Evan, G.I. The Myc-Dependent Angiogenic Switch in Tumors Is Mediated by Interleukin 1beta. Genes Dev. 2006, 20, 2527–2538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, C.V. Myc, Metabolism, Cell Growth, and Tumorigenesis. Cold Spring Harb. Perspect Med. 2013, 3. [Google Scholar] [CrossRef]
- Ciribilli, Y.; Singh, P.; Spanel, R.; Inga, A.; Borlak, J. Decoding C-Myc Networks of Cell Cycle and Apoptosis Regulated Genes in a Transgenic Mouse Model of Papillary Lung Adenocarcinomas. Oncotarget 2015, 6, 31569–31592. [Google Scholar] [CrossRef] [PubMed]
- Ciribilli, Y.; Singh, P.; Inga, A.; Borlak, J. C-Myc Targeted Regulators of Cell Metabolism in a Transgenic Mouse Model of Papillary Lung Adenocarcinoma. Oncotarget 2016, 7, 65514–65539. [Google Scholar] [CrossRef] [Green Version]
- Pello, O.M. Macrophages and C-Myc Cross Paths. Oncoimmunology 2016, 5, e1151991. [Google Scholar] [CrossRef] [Green Version]
- Jablonski, K.A.; Amici, S.A.; Webb, L.M.; Ruiz-Rosado Jde, D.; Popovich, P.G.; Partida-Sanchez, S.; Guerau-De-Arellano, M. Novel Markers To Delineate Murine M1 and M2 Macrophages. PLoS ONE 2015, 10, e0145342. [Google Scholar] [CrossRef] [Green Version]
- Hadjidaniel, M.D.; Muthugounder, S.; Hung, L.T.; Sheard, M.A.; Shirinbak, S.; Chan, R.Y.; Nakata, R.; Borriello, L.; Malvar, J.; Kennedy, R.J.; et al. Tumor-Associated Macrophages Promote Neuroblastoma Via Stat3 Phosphorylation and Up-Regulation of C-Myc. Oncotarget 2017, 8, 91516–91529. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Jiang, T.; Li, M.Q.; Zheng, X.L.; Zhao, G.J. Transcriptional Regulation of Macrophages Polarization by Micrornas. Front. Immunol. 2018, 9, 1175. [Google Scholar] [CrossRef] [PubMed]
- Solinas, G.; Schiarea, S.; Liguori, M.; Fabbri, M.; Pesce, S.; Zammataro, L.; Pasqualini, F.; Nebuloni, M.; Chiabrando, C.; Mantovani, A.; et al. Tumor-Conditioned Macrophages Secrete Migration-Stimulating Factor: A New Marker for M2-Polarization, Influencing Tumor Cell Motility. J. Immunol. 2010, 185, 642–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benner, B.; Scarberry, L.; Suarez-Kelly, L.P.; Duggan, M.C.; Campbell, A.R.; Smith, E.; Lapurga, G.; Jiang, K.; Butchar, J.P.; Tridandapani, S.; et al. Generation of Monocyte-Derived Tumor-Associated Macrophages Using Tumor-Conditioned Media Provides a Novel Method To Study Tumor-Associated Macrophages In Vitro. J. Immunother. Cancer 2019, 7, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Ye, Y.; Zhu, X. Mmp-9 Secreted by Tumor Associated Macrophages Promoted Gastric Cancer Metastasis through a Pi3k/Akt/Snail Pathway. Biomed. Pharmacother. 2019, 117, 109096. [Google Scholar] [CrossRef]
- Rapp, U.R.; Korn, C.; Ceteci, F.; Karreman, C.; Luetkenhaus, K.; Serafin, V.; Zanucco, E.; Castro, I.; Potapenko, T. Myc Is a Metastasis Gene for Non-Small-Cell Lung Cancer. PLoS ONE 2009, 4, E6029. [Google Scholar] [CrossRef] [Green Version]
- Daurkin, I.; Eruslanov, E.; Stoffs, T.; Perrin, G.Q.; Algood, C.; Gilbert, S.M.; Rosser, C.J.; Su, L.M.; Vieweg, J.; Kusmartsev, S. Tumor-Associated Macrophages Mediate Immunosuppression in the Renal Cancer Microenvironment by Activating the 15-Lipoxygenase-2 Pathway. Cancer Res. 2011, 71, 6400–6409. [Google Scholar] [CrossRef] [Green Version]
- Triner, D.; Shah, Y.M. Hypoxia-Inducible Factors: A Central Link between Inflammation and Cancer. J. Clin. Investig. 2016, 126, 3689–3698. [Google Scholar] [CrossRef]
- Guery, L.; Hugues, S. Th17 Cell Plasticity and Functions in Cancer Immunity. Biomed. Res. Int. 2015, 2015, 314620. [Google Scholar] [CrossRef] [Green Version]
- Vidak, E.; Javorsek, U.; Vizovisek, M.; Turk, B. Cysteine Cathepsins and Their Extracellular Roles: Shaping the Microenvironment. Cells 2019, 8, 264. [Google Scholar] [CrossRef] [Green Version]
- Rudzinska, M.; Parodi, A.; Soond, S.M.; Vinarov, A.Z.; Korolev, D.O.; Morozov, A.O.; Daglioglu, C.; Tutar, Y.; Zamyatnin, A.A., Jr. The Role of Cysteine Cathepsins in Cancer Progression and Drug Resistance. Int. J. Mol. Sci. 2019, 20, 3602. [Google Scholar] [CrossRef]
- Yu, T.; Gan, S.; Zhu, Q.; Dai, D.; Li, N.; Wang, H.; Chen, X.; Hou, D.; Wang, Y.; Pan, Q.; et al. Modulation of M2 Macrophage Polarization by the Crosstalk between Stat6 and Trim24. Nat. Commun. 2019, 10, 4353. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, J.S.; Fuentes-Duculan, J.; Suarez-Farinas, M.; Pierson, K.C.; Pitts-Kiefer, A.; Fan, L.; Belkin, D.A.; Wang, C.Q.; Bhuvanendran, S.; Johnson-Huang, L.M.; et al. Tumor-Associated Macrophages in the Cutaneous Scc Microenvironment Are Heterogeneously Activated. J. Invest. Dermatol. 2011, 131, 1322–1330. [Google Scholar] [CrossRef] [Green Version]
- Scodeller, P.; Simon-Gracia, L.; Kopanchuk, S.; Tobi, A.; Kilk, K.; Saalik, P.; Kurm, K.; Squadrito, M.L.; Kotamraju, V.R.; Rinken, A.; et al. Precision Targeting of Tumor Macrophages with a Cd206 Binding Peptide. Sci. Rep. 2017, 7, 14655. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Buranych, A.; Sarkar, D.; Fisher, P.B.; Wang, X.Y. The Role of Tumor-Associated Macrophages in Tumor Vascularization. Vasc Cell 2013, 5, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Folkman, J. Patterns and Emerging Mechanisms of the Angiogenic Switch During Tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.Z.; Pollard, J.W. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, S.R.; Schmid, M.C. Macrophages as Key Drivers of Cancer Progression and Metastasis. Mediat. Inflamm. 2017, 2017, 9624760. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.; Muthana, M.; Coffelt, S.B.; Lewis, C.E. The Role of Myeloid Cells in the Promotion of Tumour Angiogenesis. Nat. Rev. Cancer 2008, 8, 618–631. [Google Scholar] [CrossRef]
- Baeriswyl, V.; Christofori, G. The Angiogenic Switch in Carcinogenesis. Semin. Cancer Biol. 2009, 19, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Mezquita, P.; Parghi, S.S.; Brandvold, K.A.; Ruddell, A. Myc Regulates Vegf Production in B Cells by Stimulating Initiation of Vegf Mrna Translation. Oncogene 2005, 24, 889–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R.; Zeisberg, M. Fibroblasts in Cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef]
- Mundim, F.G.; Pasini, F.S.; Brentani, M.M.; Soares, F.A.; Nonogaki, S.; Waitzberg, A.F. Myc Is Expressed in the Stromal and Epithelial Cells of Primary Breast Carcinoma and Paired Nodal Metastases. Mol. Clin. Oncol. 2015, 3, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.E.; Hughes, R. Inflammation and Breast Cancer. Microenvironmental Factors Regulating Macrophage Function in Breast Tumours: Hypoxia and Angiopoietin-2. Breast Cancer Res. BCR 2007, 9, 209. [Google Scholar] [CrossRef] [Green Version]
- Grimshaw, M.J. Endothelins and Hypoxia-Inducible Factor in Cancer. Endocr. Relat. Cancer 2007, 14, 233–244. [Google Scholar] [CrossRef]
- Riabov, V.; Gudima, A.; Wang, N.; Mickley, A.; Orekhov, A.; Kzhyshkowska, J. Role of Tumor Associated Macrophages in Tumor Angiogenesis and Lymphangiogenesis. Front. Physiol. 2014, 5, 75. [Google Scholar] [CrossRef] [Green Version]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-Associated Macrophages as Major Players in the Tumor Microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef] [Green Version]
- Tabernero, J. The Role of Vegf and Egfr Inhibition: Implications for Combining Anti-Vegf and Anti-Egfr Agents. Mol. Cancer Res. 2007, 5, 203–220. [Google Scholar] [CrossRef] [Green Version]
- Goel, H.L.; Mercurio, A.M. Vegf Targets the Tumour Cell. Nat. Rev. Cancer 2013, 13, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Linde, N.; Lederle, W.; Depner, S.; Van Rooijen, N.; Gutschalk, C.M.; Mueller, M.M. Vascular Endothelial Growth Factor-Induced Skin Carcinogenesis Depends On Recruitment and Alternative Activation of Macrophages. J. Pathol. 2012, 227, 17–28. [Google Scholar] [CrossRef]
- Wheeler, K.C.; Jena, M.K.; Pradhan, B.S.; Nayak, N.; Das, S.; Hsu, C.D.; Wheeler, D.S.; Chen, K.; Nayak, N.R. Vegf May Contribute To Macrophage Recruitment and M2 Polarization in the Decidua. PLoS ONE 2018, 13, e0191040. [Google Scholar] [CrossRef]
- Yang, C.M.; Chien, C.S.; Ma, Y.H.; Hsiao, L.D.; Lin, C.H.; Wu, C. Bradykinin B2 Receptor-Mediated Proliferation Via Activation of the Ras/Raf/Mek/Mapk Pathway in Rat Vascular Smooth Muscle Cells. J. Biomed. Sci. 2003, 10, 208–218. [Google Scholar] [CrossRef]
- Ciribilli, Y.; Borlak, J. Oncogenomics of C-Myc Transgenic Mice Reveal Novel Regulators of Extracellular Signaling, Angiogenesis and Invasion with Clinical Significance for Human Lung Adenocarcinoma. Oncotarget 2017, 8, 101808–101831. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Ou, X.; Song, T.; Zhang, W.; Cong, F.; Zhang, S.; Xiong, Y. Adenosine A2b Receptor Stimulates Angiogenesis by Inducing Vegf and Enos in Human Microvascular Endothelial Cells. Exp. Biol. Med. 2015, 240, 1472–1479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kessler, O.; Shraga-Heled, N.; Lange, T.; Gutmann-Raviv, N.; Sabo, E.; Baruch, L.; Machluf, M.; Neufeld, G. Semaphorin-3f Is an Inhibitor of Tumor Angiogenesis. Cancer Res. 2004, 64, 1008–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neufeld, G.; Sabag, A.D.; Rabinovicz, N.; Kessler, O. Semaphorins in Angiogenesis and Tumor Progression. Cold Spring Harb. Perspect Med. 2012, 2, A006718. [Google Scholar] [CrossRef]
- Nakayama, H.; Kusumoto, C.; Nakahara, M.; Fujiwara, A.; Higashiyama, S. Semaphorin 3f and Netrin-1: The Novel Function as a Regulator of Tumor Microenvironment. Front. Physiol. 2018, 9, 1662. [Google Scholar] [CrossRef]
- Brandvold, K.A.; Neiman, P.; Ruddell, A. Angiogenesis Is an Early Event in the Generation of Myc-Induced Lymphomas. Oncogene 2000, 19, 2780–2785. [Google Scholar] [CrossRef] [Green Version]
- Baudino, T.A.; Mckay, C.; Pendeville-Samain, H.; Nilsson, J.A.; Maclean, K.H.; White, E.L.; Davis, A.C.; Ihle, J.N.; Cleveland, J.L. C-Myc Is Essential for Vasculogenesis and Angiogenesis During Development and Tumor Progression. Genes Dev. 2002, 16, 2530–2543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magid, R.; Murphy, T.J.; Galis, Z.S. Expression of Matrix Metalloproteinase-9 in Endothelial Cells Is Differentially Regulated by Shear Stress. Role of C-Myc. J. Biol. Chem. 2003, 278, 32994–32999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shchors, K.; Nozawa, H.; Xu, J.; Rostker, F.; Swigart-Brown, L.; Evan, G.; Hanahan, D. Increased Invasiveness of Mmp-9-Deficient Tumors in Two Mouse Models of Neuroendocrine Tumorigenesis. Oncogene 2013, 32, 502–513. [Google Scholar] [CrossRef] [Green Version]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Yang, F.; Zhang, T.; Wang, W.; Xi, W.; Li, Y.; Zhang, D.; Huo, Y.; Zhang, J.; Yang, A.; et al. Mir-9 Promotes Tumorigenesis and Angiogenesis and Is Activated by Myc and Oct4 in Human Glioma. J. Exp. Clin. Cancer Res. 2019, 38, 99. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Fong, M.Y.; Min, Y.; Somlo, G.; Liu, L.; Palomares, M.R.; Yu, Y.; Chow, A.; O’connor, S.T.; Chin, A.R.; et al. Cancer-Secreted Mir-105 Destroys Vascular Endothelial Barriers To Promote Metastasis. Cancer Cell 2014, 25, 501–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bui, T.V.; Mendell, J.T. Myc: Maestro of Micrornas. Genes Cancer 2010, 1, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Ernst, A.; Campos, B.; Meier, J.; Devens, F.; Liesenberg, F.; Wolter, M.; Reifenberger, G.; Herold-Mende, C.; Lichter, P.; Radlwimmer, B. De-Repression of Ctgf Via the Mir-17-92 Cluster Upon Differentiation of Human Glioblastoma Spheroid Cultures. Oncogene 2010, 29, 3411–3422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, J.L.; Dews, M.; Minn, A.J.; Thomas-Tikhonenko, A. Targeting of Tgfbeta Signature and Its Essential Component Ctgf by Mir-18 Correlates with Improved Survival in Glioblastoma. RNA 2013, 19, 177–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Psathas, J.N.; Thomas-Tikhonenko, A. Myc and the Art of Microrna Maintenance. Cold Spring Harb. Perspect Med. 2014, 4. [Google Scholar] [CrossRef] [Green Version]
- Dews, M.; Fox, J.L.; Hultine, S.; Sundaram, P.; Wang, W.; Liu, Y.Y.; Furth, E.; Enders, G.H.; El-Deiry, W.; Schelter, J.M.; et al. The Myc-Mir-17~92 Axis Blunts Tgf{Beta} Signaling and Production of Multiple Tgf{Beta}-Dependent Antiangiogenic Factors. Cancer Res. 2010, 70, 8233–8246. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V.; Le, A.; Gao, P. Myc-Induced Cancer Cell Energy Metabolism and Therapeutic Opportunities. Clin. Cancer Res. 2009, 15, 6479–6483. [Google Scholar] [CrossRef] [Green Version]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The Reverse Warburg Effect: Aerobic Glycolysis in Cancer Associated Fibroblasts and the Tumor Stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [Green Version]
- Pello, O.M.; Andres, V. Role of C-Myc in Tumor-Associated Macrophages and Cancer Progression. Oncoimmunology 2013, 2, e22984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atsaves, V.; Tsesmetzis, N.; Chioureas, D.; Kis, L.; Leventaki, V.; Drakos, E.; Panaretakis, T.; Grander, D.; Medeiros, L.J.; Young, K.H.; et al. Pd-L1 Is Commonly Expressed and Transcriptionally Regulated by Stat3 and Myc in Alk-Negative Anaplastic Large-Cell Lymphoma. Leukemia 2017, 31, 1633–1637. [Google Scholar] [CrossRef]
- Kim, E.Y.; Kim, A.; Kim, S.K.; Chang, Y.S. Myc Expression Correlates with Pd-L1 Expression in Non-Small Cell Lung Cancer. Lung Cancer 2017, 110, 63–67. [Google Scholar] [CrossRef]
- Lian, S.; Xie, R.; Ye, Y.; Xie, X.; Li, S.; Lu, Y.; Li, B.; Cheng, Y.; Katanaev, V.L.; Jia, L. Simultaneous Blocking of Cd47 and Pd-L1 Increases Innate and Adaptive Cancer Immune Responses and Cytokine Release. Ebiomedicine 2019, 42, 281–295. [Google Scholar] [CrossRef] [Green Version]
- Topper, M.J.; Vaz, M.; Chiappinelli, K.B.; Destefano Shields, C.E.; Niknafs, N.; Yen, R.C.; Wenzel, A.; Hicks, J.; Ballew, M.; Stone, M.; et al. Epigenetic Therapy Ties Myc Depletion To Reversing Immune Evasion and Treating Lung Cancer. Cell 2017, 171, 1284–1300.e21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaynes, J.M.; Sable, R.; Ronzetti, M.; Bautista, W.; Knotts, Z.; Abisoye-Ogunniyan, A.; Li, D.; Calvo, R.; Dashnyam, M.; Singh, A.; et al. Mannose Receptor (Cd206) Activation in Tumor-Associated Macrophages Enhances Adaptive and Innate Antitumor Immune Responses. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Morvan, M.G.; Lanier, L.L. Nk Cells and Cancer: You Can Teach Innate Cells New Tricks. Nat. Rev. Cancer 2016, 16, 7–19. [Google Scholar] [CrossRef]
- Zlotnik, A.; Yoshie, O. Chemokines: A New Classification System and Their Role in Immunity. Immunity 2000, 12, 121–127. [Google Scholar] [CrossRef] [Green Version]
- Langowski, J.L.; Kastelein, R.A.; Oft, M. Swords Into Plowshares: Il-23 Repurposes Tumor Immune Surveillance. Trends. Immunol. 2007, 28, 207–212. [Google Scholar] [CrossRef]
- Kortylewski, M.; Xin, H.; Kujawski, M.; Lee, H.; Liu, Y.; Harris, T.; Drake, C.; Pardoll, D.; Yu, H. Regulation of the Il-23 and Il-12 Balance by Stat3 Signaling in the Tumor Microenvironment. Cancer Cell 2009, 15, 114–123. [Google Scholar] [CrossRef] [Green Version]
- Teng, M.W.; Andrews, D.M.; Mclaughlin, N.; Von Scheidt, B.; Ngiow, S.F.; Moller, A.; Hill, G.R.; Iwakura, Y.; Oft, M.; Smyth, M.J. Il-23 Suppresses Innate Immune Response Independently of Il-17a During Carcinogenesis and Metastasis. Proc. Natl. Acad. Sci. USA 2010, 107, 8328–8333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.W.; Joyce, J.A. Alternative Activation of Tumor-Associated Macrophages by Il-4: Priming for Protumoral Functions. Cell Cycle 2010, 9, 4824–4835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Yi, T.; Zhang, W.; Pardoll, D.M.; Yu, H. Il-17 Enhances Tumor Development in Carcinogen-Induced Skin Cancer. Cancer Res. 2010, 70, 10112–10120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, D.; Li, H.; Yusuf, N.; Elmets, C.A.; Athar, M.; Katiyar, S.K.; Xu, H. Il-17 Mediated Inflammation Promotes Tumor Growth and Progression in the Skin. PLoS ONE 2012, 7, e32126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuen, D.S.; Kim, B.S.; Chung, Y. Il-17-Producing Cells in Tumor Immunity: Friends Or Foes? Immune. Netw. 2020, 20, e6. [Google Scholar] [CrossRef]
- Bailey, S.R.; Nelson, M.H.; Himes, R.A.; Li, Z.; Mehrotra, S.; Paulos, C.M. Th17 Cells in Cancer: The Ultimate Identity Crisis. Front. Immunol. 2014, 5, 276. [Google Scholar] [CrossRef] [Green Version]
- Thibaudin, M.; Chaix, M.; Boidot, R.; Vegran, F.; Derangere, V.; Limagne, E.; Berger, H.; Ladoire, S.; Apetoh, L.; Ghiringhelli, F. Human Ectonucleotidase-Expressing Cd25(High) Th17 Cells Accumulate in Breast Cancer Tumors and Exert Immunosuppressive Functions. Oncoimmunology 2016, 5, e1055444. [Google Scholar] [CrossRef] [Green Version]
- Weaver, C.T.; Harrington, L.E.; Mangan, P.R.; Gavrieli, M.; Murphy, K.M. Th17: An Effector Cd4 T Cell Lineage with Regulatory T Cell Ties. Immunity 2006, 24, 677–688. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Rodriguez, J.; Wohlfert, E.A.; Handon, R.; Meylan, F.; Wu, J.Z.; Anderson, S.M.; Kirby, M.R.; Belkaid, Y.; Schwartzberg, P.L. Itk-Mediated Integration of T Cell Receptor and Cytokine Signaling Regulates the Balance between Th17 and Regulatory T Cells. J. Exp. Med. 2014, 211, 529–543. [Google Scholar] [CrossRef]
- Chalmin, F.; Mignot, G.; Bruchard, M.; Chevriaux, A.; Vegran, F.; Hichami, A.; Ladoire, S.; Derangere, V.; Vincent, J.; Masson, D.; et al. Stat3 and Gfi-1 Transcription Factors Control Th17 Cell Immunosuppressive Activity Via the Regulation of Ectonucleotidase Expression. Immunity 2012, 36, 362–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konjevic, G.M.; Vuletic, A.M.; Mirjacic Martinovic, K.M.; Larsen, A.K.; Jurisic, V.B. The Role of Cytokines in the Regulation of Nk Cells in the Tumor Environment. Cytokine 2019, 117, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.; Mojares, E.; Del Rio Hernandez, A. Role of Extracellular Matrix in Development and Cancer Progression. Int. J. Mol. Sci. 2018, 19, 3028. [Google Scholar] [CrossRef] [Green Version]
- Gocheva, V.; Wang, H.W.; Gadea, B.B.; Shree, T.; Hunter, K.E.; Garfall, A.L.; Berman, T.; Joyce, J.A. Il-4 Induces Cathepsin Protease Activity in Tumor-Associated Macrophages To Promote Cancer Growth and Invasion. Genes Dev. 2010, 24, 241–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merdad, A.; Karim, S.; Schulten, H.J.; Dallol, A.; Buhmeida, A.; Al-Thubaity, F.; Gari, M.A.; Chaudhary, A.G.; Abuzenadah, A.M.; Al-Qahtani, M.H. Expression of Matrix Metalloproteinases (Mmps) in Primary Human Breast Cancer: Mmp-9 as a Potential Biomarker for Cancer Invasion and Metastasis. Anticancer Res. 2014, 34, 1355–1366. [Google Scholar] [PubMed]
- Yan, H.; Li, J.; Ying, Y.; Xie, H.; Chen, H.; Xu, X.; Zheng, X. Mir-300 in the Imprinted Dlk1-Dio3 Domain Suppresses the Migration of Bladder Cancer by Regulating the Sp1/Mmp9 Pathway. Cell Cycle 2018, 17, 2790–2801. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Yang, L.; Xu, X.; Lu, M.; Guo, R.; Li, D.; Huang, Q.; Liu, Y.; Deng, G.; Xu, Y. Mir-34a Inhibits Esophageal Squamous Cell Carcinoma Progression Via Regulation of Foxm1. Oncol. Lett. 2019, 17, 706–712. [Google Scholar] [CrossRef]
- Dai, L.; Hu, W.; Yang, Z.; Chen, D.; He, B.; Chen, Y.; Zhou, L.; Xie, H.; Wu, J.; Zheng, S. Upregulated Expression of Hoxb7 in Intrahepatic Cholangiocarcinoma Is Associated with Tumor Cell Metastasis and Poor Prognosis. Lab. Investig. J. Tech. Methods Pathol. 2019, 99, 736–748. [Google Scholar] [CrossRef]
- Liu, G.; Yin, L.; Ouyang, X.; Zeng, K.; Xiao, Y.; Li, Y. M2 Macrophages Promote Hcc Cells Invasion and Migration Via Mir-149-5p/Mmp9 Signaling. J. Cancer 2020, 11, 1277–1287. [Google Scholar] [CrossRef] [Green Version]
- Bjorklund, M.; Koivunen, E. Gelatinase-Mediated Migration and Invasion of Cancer Cells. Biochim. Biophys. Acta 2005, 1755, 37–69. [Google Scholar] [CrossRef] [Green Version]
- Cauwe, B.; Van Den Steen, P.E.; Opdenakker, G. The Biochemical, Biological, and Pathological Kaleidoscope of Cell Surface Substrates Processed by Matrix Metalloproteinases. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 113–185. [Google Scholar] [CrossRef] [Green Version]
- Simian, M.; Hirai, Y.; Navre, M.; Werb, Z.; Lochter, A.; Bissell, M.J. The Interplay of Matrix Metalloproteinases, Morphogens and Growth Factors Is Necessary for Branching of Mammary Epithelial Cells. Development 2001, 128, 3117–3131. [Google Scholar] [PubMed]
- Olson, O.C.; Joyce, J.A. Cysteine Cathepsin Proteases: Regulators of Cancer Progression and Therapeutic Response. Nat. Rev. Cancer 2015, 15, 712–729. [Google Scholar] [CrossRef] [PubMed]
- Brindle, N.R.; Joyce, J.A.; Rostker, F.; Lawlor, E.R.; Swigart-Brown, L.; Evan, G.; Hanahan, D.; Shchors, K. Deficiency for the Cysteine Protease Cathepsin L Impairs Myc-Induced Tumorigenesis in a Mouse Model of Pancreatic Neuroendocrine Cancer. PLoS ONE 2015, 10, e0120348. [Google Scholar] [CrossRef]
- Vizovisek, M.; Fonovic, M.; Turk, B. Cysteine Cathepsins in Extracellular Matrix Remodeling: Extracellular Matrix Degradation and Beyond. Matrix Biol. 2019, 75–76, 141–159. [Google Scholar] [CrossRef]
- Sobotic, B.; Vizovisek, M.; Vidmar, R.; Van Damme, P.; Gocheva, V.; Joyce, J.A.; Gevaert, K.; Turk, V.; Turk, B.; Fonovic, M. Proteomic Identification of Cysteine Cathepsin Substrates Shed from the Surface of Cancer Cells. Mol. Cell. Proteom. MCP 2015, 14, 2213–2228. [Google Scholar] [CrossRef] [Green Version]
- Gocheva, V.; Zeng, W.; Ke, D.; Klimstra, D.; Reinheckel, T.; Peters, C.; Hanahan, D.; Joyce, J.A. Distinct Roles for Cysteine Cathepsin Genes in Multistage Tumorigenesis. Genes Dev. 2006, 20, 543–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohashi, K.; Naruto, M.; Nakaki, T.; Sano, E. Identification of Interleukin-8 Converting Enzyme as Cathepsin L. Biochim. Biophys. Acta 2003, 1649, 30–39. [Google Scholar] [CrossRef]
- Repnik, U.; Starr, A.E.; Overall, C.M.; Turk, B. Cysteine Cathepsins Activate Elr Chemokines and Inactivate Non-Elr Chemokines. J. Biol. Chem. 2015, 290, 13800–13811. [Google Scholar] [CrossRef] [Green Version]
- Breznik, B.; Motaln, H.; Lah Turnsek, T. Proteases and Cytokines as Mediators of Interactions between Cancer and Stromal Cells in Tumours. Biol. Chem. 2017, 398, 709–719. [Google Scholar] [CrossRef] [Green Version]
- Kurozumi, S.; Alsaeed, S.; Orah, N.; Miligy, I.M.; Joseph, C.; Aljohani, A.; Toss, M.S.; Fujii, T.; Shirabe, K.; Green, A.R.; et al. Clinicopathological Significance of Lipocalin 2 Nuclear Expression in Invasive Breast Cancer. Breast Cancer Res. Treat. 2020, 179, 557–564. [Google Scholar] [CrossRef]
- Takenaka, K.; Ishikawa, S.; Kawano, Y.; Yanagihara, K.; Miyahara, R.; Otake, Y.; Morioka, Y.; Takahashi, C.; Noda, M.; Wada, H.; et al. Expression of a Novel Matrix Metalloproteinase Regulator, Reck, and Its Clinical Significance in Resected Non-Small Cell Lung Cancer. Eur. J. Cancer 2004, 40, 1617–1623. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Li, W.; Zhu, J.; Deng, S.; Tao, X. Low Expression of Reck in Oral Squamous Cell Carcinoma Patients Induces a Shorter Survival Rate through an Imbalance of Reck/Mmps. Int. J. Clin. Exp. Pathol. 2020, 13, 501–508. [Google Scholar] [PubMed]
- Busser, B.; Sancey, L.; Brambilla, E.; Coll, J.L.; Hurbin, A. The Multiple Roles of Amphiregulin in Human Cancer. Biochim. Biophys. Acta 2011, 1816, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Kometani, K.; Hashida, H.; Matsunaga, A.; Miyoshi, H.; Hosogi, H.; Aoki, M.; Oshima, M.; Hattori, M.; Takabayashi, A.; et al. Smad4-Deficient Intestinal Tumors Recruit Ccr1+ Myeloid Cells That Promote Invasion. Nat. Genet 2007, 39, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Mi, J.; Zhou, B.P. Metabolic Rewiring in Cancer-Associated Fibroblasts Provides a Niche for Oncogenesis and Metastatic Dissemination. Mol. Cell Oncol. 2016, 3, e1056331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ippolito, L.; Morandi, A.; Taddei, M.L.; Parri, M.; Comito, G.; Iscaro, A.; Raspollini, M.R.; Magherini, F.; Rapizzi, E.; Masquelier, J.; et al. Cancer-Associated Fibroblasts Promote Prostate Cancer Malignancy Via Metabolic Rewiring and Mitochondrial Transfer. Oncogene 2019, 38, 5339–5355. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.E.; Lisanti, M.P.; Sotgia, F. Catabolic Cancer-Associated Fibroblasts Transfer Energy and Biomass To Anabolic Cancer Cells, Fueling Tumor Growth. Semin. Cancer Biol. 2014, 25, 47–60. [Google Scholar] [CrossRef]
- Coller, H.A. Myc Sets a Tumour-Stroma Metabolic Loop. Nat. Cell Biol. 2018, 20, 506–507. [Google Scholar] [CrossRef]
- Labelle, M.; Hynes, R.O. The Initial Hours of Metastasis: The Importance of Cooperative Host-Tumor Cell Interactions During Hematogenous Dissemination. Cancer Discov. 2012, 2, 1091–1099. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Shi, J.; Chai, K.; Ying, X.; Zhou, B.P. The Role of Snail in Emt and Tumorigenesis. Curr. Cancer Drug Targets 2013, 13, 963–972. [Google Scholar] [CrossRef]
- Mittal, V. Epithelial Mesenchymal Transition in Tumor Metastasis. Annu. Rev. Pathol. 2018, 13, 395–412. [Google Scholar] [CrossRef]
- Cho, H.; Herzka, T.; Zheng, W.; Qi, J.; Wilkinson, J.E.; Bradner, J.E.; Robinson, B.D.; Castillo-Martin, M.; Cordon-Cardo, C.; Trotman, L.C. Rapidcap, a Novel Gem Model for Metastatic Prostate Cancer Analysis and Therapy, Reveals Myc as a Driver of Pten-Mutant Metastasis. Cancer Discov. 2014, 4, 318–333. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Sleeman, J.P. Complex Networks Orchestrate Epithelial-Mesenchymal Transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; De Herreros, A.G. Epithelial-Mesenchymal Transition in Cancer. Mol. Oncol. 2017, 11, 715–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular Mechanisms of Epithelial-Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisberg, M.; Neilson, E.G. Biomarkers for Epithelial-Mesenchymal Transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Nelson, C.M. New Insights Into the Regulation of Epithelial-Mesenchymal Transition and Tissue Fibrosis. Int. Rev. Cell Mol. Biol. 2012, 294, 171–221. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New Insights Into the Mechanisms of Epithelial-Mesenchymal Transition and Implications for Cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Cowling, V.H.; Cole, M.D. E-Cadherin Repression Contributes To C-Myc-Induced Epithelial Cell Transformation. Oncogene 2007, 26, 3582–3586. [Google Scholar] [CrossRef] [Green Version]
- Cho, K.B.; Cho, M.K.; Lee, W.Y.; Kang, K.W. Overexpression of C-Myc Induces Epithelial Mesenchymal Transition in Mammary Epithelial Cells. Cancer Lett. 2010, 293, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.P.; Verrecchia, A.; Faga, G.; Doni, M.; Perna, D.; Martinato, F.; Guccione, E.; Amati, B. A Positive Role for Myc in Tgfbeta-Induced Snail Transcription and Epithelial-To-Mesenchymal Transition. Oncogene 2009, 28, 422–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, D.; Cui, C.; Xie, L.; Cai, L.; Yu, J. Sterol Regulatory Element-Binding Protein 1 Cooperates with C-Myc To Promote Epithelial-Mesenchymal Transition in Colorectal Cancer. Oncol. Lett. 2018, 15, 5959–5965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruyere, F.; Namdarian, B.; Corcoran, N.M.; Pedersen, J.; Ockrim, J.; Voelzke, B.B.; Mete, U.; Costello, A.J.; Hovens, C.M. Snail Expression Is an Independent Predictor of Tumor Recurrence in Superficial Bladder Cancers. Urol. Oncol. 2010, 28, 591–596. [Google Scholar] [CrossRef]
- Jin, H.; Yu, Y.; Zhang, T.; Zhou, X.; Zhou, J.; Jia, L.; Wu, Y.; Zhou, B.P.; Feng, Y. Snail Is Critical for Tumor Growth and Metastasis of Ovarian Carcinoma. Int. J. Cancer 2010, 126, 2102–2111. [Google Scholar] [CrossRef]
- Park, S.M.; Park, S.H.; Ryu, K.J.; Kim, I.K.; Han, H.; Kim, H.J.; Kim, S.H.; Hong, K.S.; Kim, H.; Kim, M.; et al. Downregulation of Chip Promotes Ovarian Cancer Metastasis by Inducing Snail-Mediated Epithelial-Mesenchymal Transition. Mol. Oncol. 2019, 13, 1280–1295. [Google Scholar] [CrossRef] [Green Version]
- Kudo-Saito, C.; Shirako, H.; Takeuchi, T.; Kawakami, Y. Cancer Metastasis Is Accelerated through Immunosuppression During Snail-Induced Emt of Cancer Cells. Cancer Cell 2009, 15, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Liu, L.; Mao, J.; Zhang, Z.; Wang, Q.; Li, Q. Pim1 Mediates Epithelial-Mesenchymal Transition by Targeting Smads and C-Myc in the Nucleus and Potentiates Clear-Cell Renal-Cell Carcinoma Oncogenesis. Cell Death Dis. 2018, 9, 307. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Liu, X.; Lu, Y.; Wang, Y.; Cao, W.; Liu, X.; Hu, H.; Wang, H. Pim1 Is Responsible for Il-6-Induced Breast Cancer Cell Emt and Stemness Via C-Myc Activation. Breast Cancer 2019, 26, 663–671. [Google Scholar] [CrossRef] [Green Version]
- Guan, F.; Handa, K.; Hakomori, S.I. Specific Glycosphingolipids Mediate Epithelial-To-Mesenchymal Transition of Human and Mouse Epithelial Cell Lines. Proc. Natl. Acad. Sci. USA 2009, 106, 7461–7466. [Google Scholar] [CrossRef] [Green Version]
- Laubli, H.; Borsig, L. Selectins Promote Tumor Metastasis. Semin. Cancer Biol. 2010, 20, 169–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.S.; Ip, C.K.M.; Tang, M.Y.H.; Tang, M.K.S.; Tong, Y.; Zhang, J.; Hassan, A.A.; Mak, A.S.C.; Yung, S.; Chan, T.M.; et al. Sialyl Lewis(X)-P-Selectin Cascade Mediates Tumor-Mesothelial Adhesion in Ascitic Fluid Shear Flow. Nat. Commun. 2019, 10, 2406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakuma, K.; Aoki, M.; Kannagi, R. Transcription Factors C-Myc and Cdx2 Mediate E-Selectin Ligand Expression in Colon Cancer Cells Undergoing Egf/Bfgf-Induced Epithelial-Mesenchymal Transition. Proc. Natl. Acad. Sci. USA 2012, 109, 7776–7781. [Google Scholar] [CrossRef] [Green Version]
- Julien, S.; Ivetic, A.; Grigoriadis, A.; Qize, D.; Burford, B.; Sproviero, D.; Picco, G.; Gillett, C.; Papp, S.L.; Schaffer, L.; et al. Selectin Ligand Sialyl-Lewis X Antigen Drives Metastasis of Hormone-Dependent Breast Cancers. Cancer Res. 2011, 71, 7683–7693. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.X.; Liang, Y.; Gao, W. Clinicopathological and Prognostic Significance of Sialyl Lewis X Overexpression in Patients with Cancer: A Meta-Analysis. Oncotargets Ther. 2016, 9, 3113–3125. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Ma, F.; Liu, C.; Zhu, K.; Li, W.; Xu, Y.; Li, G.; Niu, Z.; Liu, J.; Chen, D.; et al. Ube2o Promotes the Proliferation, Emt and Stemness Properties of Breast Cancer Cells through the Ube2o/Ampkalpha2/Mtorc1-Myc Positive Feedback Loop. Cell Death Dis. 2020, 11, 10. [Google Scholar] [CrossRef]
- Vila, I.K.; Yao, Y.; Kim, G.; Xia, W.; Kim, H.; Kim, S.J.; Park, M.K.; Hwang, J.P.; Gonzalez-Billalabeitia, E.; Hung, M.C.; et al. A Ube2o-Ampkalpha2 Axis That Promotes Tumor Initiation and Progression Offers Opportunities for Therapy. Cancer Cell 2017, 31, 208–224. [Google Scholar] [CrossRef] [Green Version]
- Forghanifard, M.M.; Moghbeli, M.; Raeisossadati, R.; Tavassoli, A.; Mallak, A.J.; Boroumand-Noughabi, S.; Abbaszadegan, M.R. Role of Sall4 in the Progression and Metastasis of Colorectal Cancer. J. Biomed. Sci. 2013, 20, 6. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Xu, Z.; Xu, X.; Zhang, B.; Wu, H.; Wang, M.; Zhang, X.; Yang, T.; Cai, J.; Yan, Y.; et al. Sall4, a Novel Marker for Human Gastric Carcinogenesis and Metastasis. Oncogene 2014, 33, 5491–5500. [Google Scholar] [CrossRef] [Green Version]
- Itou, J.; Matsumoto, Y.; Yoshikawa, K.; Toi, M. Sal-Like 4 (Sall4) Suppresses Cdh1 Expression and Maintains Cell Dispersion in Basal-Like Breast Cancer. FEBS Lett. 2013, 587, 3115–3121. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Zhang, J.; Yang, X.; Fang, C.; Xu, H.; Xi, X. Sall4 as an Epithelial-Mesenchymal Transition and Drug Resistance Inducer through the Regulation of C-Myc in Endometrial Cancer. PLoS ONE 2015, 10, e0138515. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Zhou, C.; Lou, X.; Xiao, Z.; Zhu, H.; Wang, Q.; Wang, Y.; Lu, N.; He, S.; Zhan, Q.; et al. Pttg Overexpression Promotes Lymph Node Metastasis in Human Esophageal Squamous Cell Carcinoma. Cancer Res. 2009, 69, 3283–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, Y.L.; Wu, C.Y.; Hung, J.Y.; Lin, Y.S.; Huang, M.S.; Kuo, P.L. Galectin-1 Promotes Lung Cancer Tumor Metastasis by Potentiating Integrin Alpha6beta4 and Notch1/Jagged2 Signaling Pathway. Carcinogenesis 2013, 34, 1370–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, M.H.; Park, J.H.; Choi, H.J.; Park, M.K.; Won, H.Y.; Park, Y.J.; Lee, C.H.; Oh, S.H.; Song, Y.S.; Kim, H.S.; et al. Dot1l Cooperates with the C-Myc-P300 Complex To Epigenetically Derepress Cdh1 Transcription Factors in Breast Cancer Progression. Nat. Commun. 2015, 6, 7821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Kong, G. Roles and Epigenetic Regulation of Epithelial-Mesenchymal Transition and Its Transcription Factors in Cancer Initiation and Progression. Cell Mol. Life Sci. 2016, 73, 4643–4660. [Google Scholar] [CrossRef]
- Kerkhoff, E.; Houben, R.; Loffler, S.; Troppmair, J.; Lee, J.E.; Rapp, U.R. Regulation of C-Myc Expression by Ras/Raf Signalling. Oncogene 1998, 16, 211–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soucek, L.; Whitfield, J.R.; Sodir, N.M.; Masso-Valles, D.; Serrano, E.; Karnezis, A.N.; Swigart, L.B.; Evan, G.I. Inhibition of Myc Family Proteins Eradicates Kras-Driven Lung Cancer in Mice. Genes Dev. 2013, 27, 504–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.Y.; Cha, J.; Kim, S.K.; Park, J.H.; Song, K.H.; Kim, P.; Kim, M.Y. C-Myc Drives Breast Cancer Metastasis To the Brain, But Promotes Synthetic Lethality with Trail. Mol. Cancer Res. 2019, 17, 544–554. [Google Scholar] [CrossRef] [Green Version]
- Kazan, J.M.; El-Saghir, J.; Saliba, J.; Shaito, A.; Jalaleddine, N.; El-Hajjar, L.; Al-Ghadban, S.; Yehia, L.; Zibara, K.; El-Sabban, M. Cx43 Expression Correlates with Breast Cancer Metastasis in Mda-Mb-231 Cells in Vitro, in a Mouse Xenograft Model and in Human Breast Cancer Tissues. Cancers 2019, 11, 460. [Google Scholar] [CrossRef] [Green Version]
- Lamiche, C.; Clarhaut, J.; Strale, P.O.; Crespin, S.; Pedretti, N.; Bernard, F.X.; Naus, C.C.; Chen, V.C.; Foster, L.J.; Defamie, N.; et al. The Gap Junction Protein Cx43 Is Involved in the Bone-Targeted Metastatic Behaviour of Human Prostate Cancer Cells. Clin. Exp. Metastasis 2012, 29, 111–122. [Google Scholar] [CrossRef]
- Martinez, C.; Churchman, M.; Freeman, T.; Ilyas, M. Osteopontin Provides Early Proliferative Drive and May Be Dependent Upon Aberrant C-Myc Signalling in Murine Intestinal Tumours. Exp. Mol. Pathol. 2010, 88, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Mcallister, S.S.; Gifford, A.M.; Greiner, A.L.; Kelleher, S.P.; Saelzler, M.P.; Ince, T.A.; Reinhardt, F.; Harris, L.N.; Hylander, B.L.; Repasky, E.A.; et al. Systemic Endocrine Instigation of Indolent Tumor Growth Requires Osteopontin. Cell 2008, 133, 994–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allan, A.L.; George, R.; Vantyghem, S.A.; Lee, M.W.; Hodgson, N.C.; Engel, C.J.; Holliday, R.L.; Girvan, D.P.; Scott, L.A.; Postenka, C.O.; et al. Role of the Integrin-Binding Protein Osteopontin in Lymphatic Metastasis of Breast Cancer. Am. J. Pathol. 2006, 169, 233–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anborgh, P.H.; Mutrie, J.C.; Tuck, A.B.; Chambers, A.F. Role of the Metastasis-Promoting Protein Osteopontin in the Tumour Microenvironment. J. Cell Mol. Med. 2010, 14, 2037–2044. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Chen, Q.; Alam, A.; Cui, J.; Suen, K.C.; Soo, A.P.; Eguchi, S.; Gu, J.; Ma, D. The Role of Osteopontin in the Progression of Solid Organ Tumour. Cell Death Dis. 2018, 9, 356. [Google Scholar] [CrossRef] [PubMed]
- Li, N.Y.; Weber, C.E.; Mi, Z.; Wai, P.Y.; Cuevas, B.D.; Kuo, P.C. Osteopontin Up-Regulates Critical Epithelial-Mesenchymal Transition Transcription Factors To Induce an Aggressive Breast Cancer Phenotype. J. Am. Coll Surg. 2013, 217, 17–26, Discussion 26. [Google Scholar] [CrossRef]
- Kothari, A.N.; Arffa, M.L.; Chang, V.; Blackwell, R.H.; Syn, W.K.; Zhang, J.; Mi, Z.; Kuo, P.C. Osteopontin-A Master Regulator of Epithelial-Mesenchymal Transition. J. Clin. Med. 2016, 5, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro-Giner, F.; Aceto, N. Tracking Cancer Progression: From Circulating Tumor Cells To Metastasis. Genome Med. 2020, 12, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.H.; Lee, S.W.; Li, C.F.; Wang, J.; Yang, W.L.; Wu, C.Y.; Wu, J.; Nakayama, K.I.; Kang, H.Y.; Huang, H.Y.; et al. Deciphering the Transcriptional Complex Critical for Rhoa Gene Expression and Cancer Metastasis. Nat. Cell Biol. 2010, 12, 457–467. [Google Scholar] [CrossRef]
- Weaver, A.M. Invadopodia: Specialized Cell Structures for Cancer Invasion. Clin. Exp. Metastasis 2006, 23, 97–105. [Google Scholar] [CrossRef]
- Struckhoff, A.P.; Rana, M.K.; Worthylake, R.A. Rhoa Can Lead the Way in Tumor Cell Invasion and Metastasis. Front. Biosci. (Landmark Ed.) 2011, 16, 1915–1926. [Google Scholar] [CrossRef]
- Lasorella, A.; Noseda, M.; Beyna, M.; Yokota, Y.; Iavarone, A. Id2 Is a Retinoblastoma Protein Target and Mediates Signalling by Myc Oncoproteins. Nature 2000, 407, 592–598. [Google Scholar] [CrossRef]
- Cotta, C.V.; Leventaki, V.; Atsaves, V.; Vidaki, A.; Schlette, E.; Jones, D.; Medeiros, L.J.; Rassidakis, G.Z. The Helix-Loop-Helix Protein Id2 Is Expressed Differentially and Induced by Myc in T-Cell Lymphomas. Cancer 2008, 112, 552–561. [Google Scholar] [CrossRef]
- Coma, S.; Amin, D.N.; Shimizu, A.; Lasorella, A.; Iavarone, A.; Klagsbrun, M. Id2 Promotes Tumor Cell Migration and Invasion through Transcriptional Repression of Semaphorin 3f. Cancer Res. 2010, 70, 3823–3832. [Google Scholar] [CrossRef] [Green Version]
- Iaccarino, I. Lncrnas and Myc: An Intricate Relationship. Int. J. Mol. Sci. 2017, 18, 1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.; Jeon, Y.J.; Cui, R.; Lee, J.H.; Peng, Y.; Kim, S.H.; Tili, E.; Alder, H.; Croce, C.M. Role of Myc-Regulated Long Noncoding Rnas in Cell Cycle Regulation and Tumorigenesis. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Young, J.; Prabhala, H.; Pan, E.; Mestdagh, P.; Muth, D.; Teruya-Feldstein, J.; Reinhardt, F.; Onder, T.T.; Valastyan, S.; et al. Mir-9, a Myc/Mycn-Activated Microrna, Regulates E-Cadherin and Cancer Metastasis. Nat. Cell Biol. 2010, 12, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Zhu, H.; Yang, S.; Wang, Z.; Bai, J.; Xu, N. C-Myc Suppressed E-Cadherin through Mir-9 At the Post-Transcriptional Level. Cell Biol. Int. 2013, 37, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Khew-Goodall, Y.; Goodall, G.J. Myc-Modulated Mir-9 Makes More Metastases. Nat. Cell Biol. 2010, 12, 209–211. [Google Scholar] [CrossRef]
- Chang, T.C.; Yu, D.; Lee, Y.S.; Wentzel, E.A.; Arking, D.E.; West, K.M.; Dang, C.V.; Thomas-Tikhonenko, A.; Mendell, J.T. Widespread Microrna Repression by Myc Contributes To Tumorigenesis. Nat. Genet 2008, 40, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Boyerinas, B.; Park, S.M.; Shomron, N.; Hedegaard, M.M.; Vinther, J.; Andersen, J.S.; Feig, C.; Xu, J.; Burge, C.B.; Peter, M.E. Identification of Let-7-Regulated Oncofetal Genes. Cancer Res. 2008, 68, 2587–2591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dangi-Garimella, S.; Yun, J.; Eves, E.M.; Newman, M.; Erkeland, S.J.; Hammond, S.M.; Minn, A.J.; Rosner, M.R. Raf Kinase Inhibitory Protein Suppresses a Metastasis Signalling Cascade Involving Lin28 and Let-7. Embo J. 2009, 28, 347–358. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.C.; Zeitels, L.R.; Hwang, H.W.; Chivukula, R.R.; Wentzel, E.A.; Dews, M.; Jung, J.; Gao, P.; Dang, C.V.; Beer, M.A.; et al. Lin-28b Transactivation Is Necessary for Myc-Mediated Let-7 Repression and Proliferation. Proc. Natl. Acad. Sci. USA 2009, 106, 3384–3389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viswanathan, S.R.; Powers, J.T.; Einhorn, W.; Hoshida, Y.; Ng, T.L.; Toffanin, S.; O’sullivan, M.; Lu, J.; Phillips, L.A.; Lockhart, V.L.; et al. Lin28 Promotes Transformation and Is Associated with Advanced Human Malignancies. Nat. Genet 2009, 41, 843–848. [Google Scholar] [CrossRef] [Green Version]
- King, C.E.; Cuatrecasas, M.; Castells, A.; Sepulveda, A.R.; Lee, J.S.; Rustgi, A.K. Lin28b Promotes Colon Cancer Progression and Metastasis. Cancer Res. 2011, 71, 4260–4268. [Google Scholar] [CrossRef] [Green Version]
- Helland, A.; Anglesio, M.S.; George, J.; Cowin, P.A.; Johnstone, C.N.; House, C.M.; Sheppard, K.E.; Etemadmoghadam, D.; Melnyk, N.; Rustgi, A.K.; et al. Deregulation of Mycn, Lin28b and Let7 in a Molecular Subtype of Aggressive High-Grade Serous Ovarian Cancers. PLoS ONE 2011, 6, e18064. [Google Scholar] [CrossRef] [Green Version]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The Mir-200 Family and Mir-205 Regulate Epithelial To Mesenchymal Transition by Targeting Zeb1 and Sip1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The Mir-200 Family Determines the Epithelial Phenotype of Cancer Cells by Targeting the E-Cadherin Repressors Zeb1 and Zeb2. Genes Dev. 2008, 22, 894–907. [Google Scholar] [CrossRef] [Green Version]
- Korpal, M.; Lee, E.S.; Hu, G.; Kang, Y. The Mir-200 Family Inhibits Epithelial-Mesenchymal Transition and Cancer Cell Migration by Direct Targeting of E-Cadherin Transcriptional Repressors Zeb1 and Zeb2. J. Biol. Chem. 2008, 283, 14910–14914. [Google Scholar] [CrossRef] [Green Version]
- Title, A.C.; Hong, S.J.; Pires, N.D.; Hasenohrl, L.; Godbersen, S.; Stokar-Regenscheit, N.; Bartel, D.P.; Stoffel, M. Genetic Dissection of the Mir-200-Zeb1 Axis Reveals Its Importance in Tumor Differentiation and Invasion. Nat. Commun. 2018, 9, 4671. [Google Scholar] [CrossRef]
- Yang, J.; Wu, S.P.; Wang, W.J.; Jin, Z.R.; Miao, X.B.; Wu, Y.; Gou, D.M.; Liu, Q.Z.; Yao, K.T. A Novel Mir-200c/C-Myc Negative Regulatory Feedback Loop Is Essential To the Emt Process, Csc Biology and Drug Sensitivity in Nasopharyngeal Cancer. Exp. Cell Res. 2020, 391, 111817. [Google Scholar] [CrossRef]
- Zhang, J.P.; Zeng, C.; Xu, L.; Gong, J.; Fang, J.H.; Zhuang, S.M. Microrna-148a Suppresses the Epithelial-Mesenchymal Transition and Metastasis of Hepatoma Cells by Targeting Met/Snail Signaling. Oncogene 2014, 33, 4069–4076. [Google Scholar] [CrossRef]
- Wang, X.; Liang, Z.; Xu, X.; Li, J.; Zhu, Y.; Meng, S.; Li, S.; Wang, S.; Xie, B.; Ji, A.; et al. Mir-148a-3p Represses Proliferation and Emt by Establishing Regulatory Circuits between Erbb3/Akt2/C-Myc and Dnmt1 in Bladder Cancer. Cell Death Dis. 2016, 7, e2503. [Google Scholar] [CrossRef] [Green Version]
- Fong, M.Y.; Zhou, W.; Liu, L.; Alontaga, A.Y.; Chandra, M.; Ashby, J.; Chow, A.; O’connor, S.T.; Li, S.; Chin, A.R.; et al. Breast-Cancer-Secreted Mir-122 Reprograms Glucose Metabolism in Premetastatic Niche To Promote Metastasis. Nat. Cell Biol. 2015, 17, 183–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhang, Z.; Chen, F.; Hu, T.; Peng, W.; Gu, Q.; Sun, Y. The Diverse Oncogenic and Tumor Suppressor Roles of Microrna-105 in Cancer. Front. Oncol. 2019, 9, 518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Z.; Zhou, R.; Liu, C.; Wang, Y.; Zhan, W.; Shao, Z.; Liu, J.; Zhang, F.; Xu, L.; Zhou, X.; et al. Microrna-105 Is Involved in Tnf-Alpha-Related Tumor Microenvironment Enhanced Colorectal Cancer Progression. Cell Death Dis. 2017, 8, 3213. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Wang, Z.; Liu, D.; Xie, P. Hoxc13-As-Mir-122-5p-Satb1-C-Myc Feedback Loop Promotes Migration, Invasion and Emt Process in Glioma. Oncotargets Ther. 2019, 12, 7165–7173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Wu, W.; Ding, J.; Yang, J. Linc01433/Mir-2116-3p/Myc Feedback Loop Promotes Cell Proliferation, Migration, and the Epithelial-Mesenchymal Transition in Breast Cancer. Cancer Biother Radiopharm 2019, 34, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Yu, T.; Hu, H.; He, K. Knockdown of the Long Non-Coding Rna Hottip Inhibits Colorectal Cancer Cell Proliferation and Migration and Induces Apoptosis by Targeting Sgk1. Biomed. Pharmacother. 2018, 98, 286–296. [Google Scholar] [CrossRef]
- Tang, Y.; Ji, F. Lncrna Hottip Facilitates Osteosarcoma Cell Migration, Invasion and Epithelial-Mesenchymal Transition by Forming a Positive Feedback Loop with C-Myc. Oncol. Lett. 2019, 18, 1649–1656. [Google Scholar] [CrossRef] [Green Version]
- Rui, Y.; Hu, M.; Wang, P.; Zhang, C.; Xu, H.; Li, Y.; Zhang, Y.; Gu, J.; Wang, Q. Lncrna Hottip Mediated Dkk1 Downregulation Confers Metastasis and Invasion in Colorectal Cancer Cells. Histol. Histopathol. 2019, 34, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Lu, Y.R. Bcyrn1, a C-Myc-Activated Long Non-Coding Rna, Regulates Cell Metastasis of Non-Small-Cell Lung Cancer. Cancer Cell Int. 2015, 15, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quail, D.F.; Joyce, J.A. Microenvironmental Regulation of Tumor Progression and Metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Kawata, M.; Koinuma, D.; Ogami, T.; Umezawa, K.; Iwata, C.; Watabe, T.; Miyazono, K. Tgf-Beta-Induced Epithelial-Mesenchymal Transition of A549 Lung Adenocarcinoma Cells Is Enhanced by Pro-Inflammatory Cytokines Derived from Raw 264.7 Macrophage Cells. J. Biochem. 2012, 151, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-Associated Macrophages: From Mechanisms To Therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.; Brabletz, T.; Kang, Y.; Longmore, G.D.; Nieto, M.A.; Stanger, B.Z.; Yang, J.; Weinberg, R.A. Upholding a Role for Emt in Breast Cancer Metastasis. Nature 2017, 547, E1–E3. [Google Scholar] [CrossRef]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; Macdonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. Vegfr1-Positive Haematopoietic Bone Marrow Progenitors Initiate the Pre-Metastatic Niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef]
- Chen, X.W.; Yu, T.J.; Zhang, J.; Li, Y.; Chen, H.L.; Yang, G.F.; Yu, W.; Liu, Y.Z.; Liu, X.X.; Duan, C.F.; et al. Cyp4a in Tumor-Associated Macrophages Promotes Pre-Metastatic Niche Formation and Metastasis. Oncogene 2017, 36, 5045–5057. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, L.R.; Borriello, L.; Entenberg, D.; Condeelis, J.S.; Oktay, M.H.; Karagiannis, G.S. The Emerging Roles of Macrophages in Cancer Metastasis and Response To Chemotherapy. J. Leukoc. Biol. 2019, 106, 259–274. [Google Scholar] [CrossRef]
- Bouvard, C.; Lim, S.M.; Ludka, J.; Yazdani, N.; Woods, A.K.; Chatterjee, A.K.; Schultz, P.G.; Zhu, S. Small Molecule Selectively Suppresses Myc Transcription in Cancer Cells. Proc. Natl. Acad. Sci. USA 2017, 114, 3497–3502. [Google Scholar] [CrossRef] [Green Version]
- Allen-Petersen, B.L.; Sears, R.C. Mission Possible: Advances in Myc Therapeutic Targeting in Cancer. Biodrugs 2019, 33, 539–553. [Google Scholar] [CrossRef] [Green Version]
- Esser, A.K.; Ross, M.H.; Fontana, F.; Su, X.; Gabay, A.; Fox, G.C.; Xu, Y.; Xiang, J.; Schmieder, A.H.; Yang, X.; et al. Nanotherapy Delivery of C-Myc Inhibitor Targets Protumor Macrophages and Preserves Antitumor Macrophages in Breast Cancer. Theranostics 2020, 10, 7510–7526. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Processes | Mechanisms | References | |
|---|---|---|---|
| MMP-9 | Invasion, migration Angiogenesis EMT | Remodeling and degradation of ECM Release VEGF from ECM PI3K/AKT signaling pathway | [24,64] |
| VEGF | Angiogenesis | Blood vessel permeability, loss of integrity | [17,24,65] |
| TGF- β | EMT | via SMAD/SNAIL | [24,64] |
| ALOX15 | Alternative macrophage activation | M2/TAMs marker | [24,66] |
| Immunosuppression | via IL-10/CCL-2 | ||
| HIF-1α | Angiogenesis | HIF-1α/VEGF axis | [24,67] |
| Immunosuppression | Macrophage recruitment | ||
| CCL9 | Immune evasion | Macrophage influx, T cell loss | [18] |
| Angiogenesis | via VEFG | ||
| IL-23 | Immune evasion | Expulsion of T, B and NK cells, Th17 influx | [18,68] |
| CTCS | Invasion | Remodeling and degradation of ECM | [24,69,70] |
| STAT6 | Alternative macrophage activation | Downregulation of TRIM24 | [24,71] |
| CD209 | Alternative macrophage activation | M2/TAMs marker | [24,72] |
| MRC1 | Alternative macrophage activation | M2/TAMs marker | [24,73] |
| PPARγ | Alternative macrophage activation | M2/TAMs marker | [24] |
| SCARB1 | Alternative macrophage activation | M2/TAMs marker | [24] |
| Processes | Target/Signaling Pathways | References | |
|---|---|---|---|
| microRNAs activated by Myc | |||
| miR-17-92 cluster: | |||
| miR-18a miR-19 | Angiogenesis | CTGF ↓ TSP-1 ↓ | [11,106,107,109] |
| miR-105 | Metabolic rewiring Angiogenesis Metastasis, EMT | MIX1 ↓ ZO-1 ↓ ZO-1 ↓ RAP2C ↑ | [104,136,137,235,236] |
| miR-9 | Angiogenesis Metastasis, EMT | HIF-1α/VEGF axis E-Cadherin ↓ | [103,218] |
| microRNAs suppressed by Myc | |||
| miR-148a-3p | EMT | ERBB3/AKT2/MYC axis | [233] |
| miR-200 | EMT | Zeb-1 ↑ Zeb-2 ↑ | [108,231] |
| long non-coding RNAs activated by Myc | |||
| HOXC13-AS | Migration, invasion, EMT | MYC ↑ | [237] |
| LIN28B | EMT | let-7 ↓ HMHA2 ↓ Snail ↑ | [172,221,222,223,224] |
| HOTTIP | Migration, invasion, EMT Metastasis | MYC ↑ β-catenin ↑ Vimetin ↑ DKK1 ↓ | [239,240,241] |
| LINC01433 | Migration, EMT | Myc ↑ (via sponging miR-2116-3p) | [238] |
| BCYRN1 | Metastasis | MMP-9 ↑ MMP-13 ↑ | [242] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meškytė, E.M.; Keskas, S.; Ciribilli, Y. MYC as a Multifaceted Regulator of Tumor Microenvironment Leading to Metastasis. Int. J. Mol. Sci. 2020, 21, 7710. https://doi.org/10.3390/ijms21207710
Meškytė EM, Keskas S, Ciribilli Y. MYC as a Multifaceted Regulator of Tumor Microenvironment Leading to Metastasis. International Journal of Molecular Sciences. 2020; 21(20):7710. https://doi.org/10.3390/ijms21207710
Chicago/Turabian StyleMeškytė, Erna Marija, Sabiha Keskas, and Yari Ciribilli. 2020. "MYC as a Multifaceted Regulator of Tumor Microenvironment Leading to Metastasis" International Journal of Molecular Sciences 21, no. 20: 7710. https://doi.org/10.3390/ijms21207710
APA StyleMeškytė, E. M., Keskas, S., & Ciribilli, Y. (2020). MYC as a Multifaceted Regulator of Tumor Microenvironment Leading to Metastasis. International Journal of Molecular Sciences, 21(20), 7710. https://doi.org/10.3390/ijms21207710