MYC in Brain Development and Cancer


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
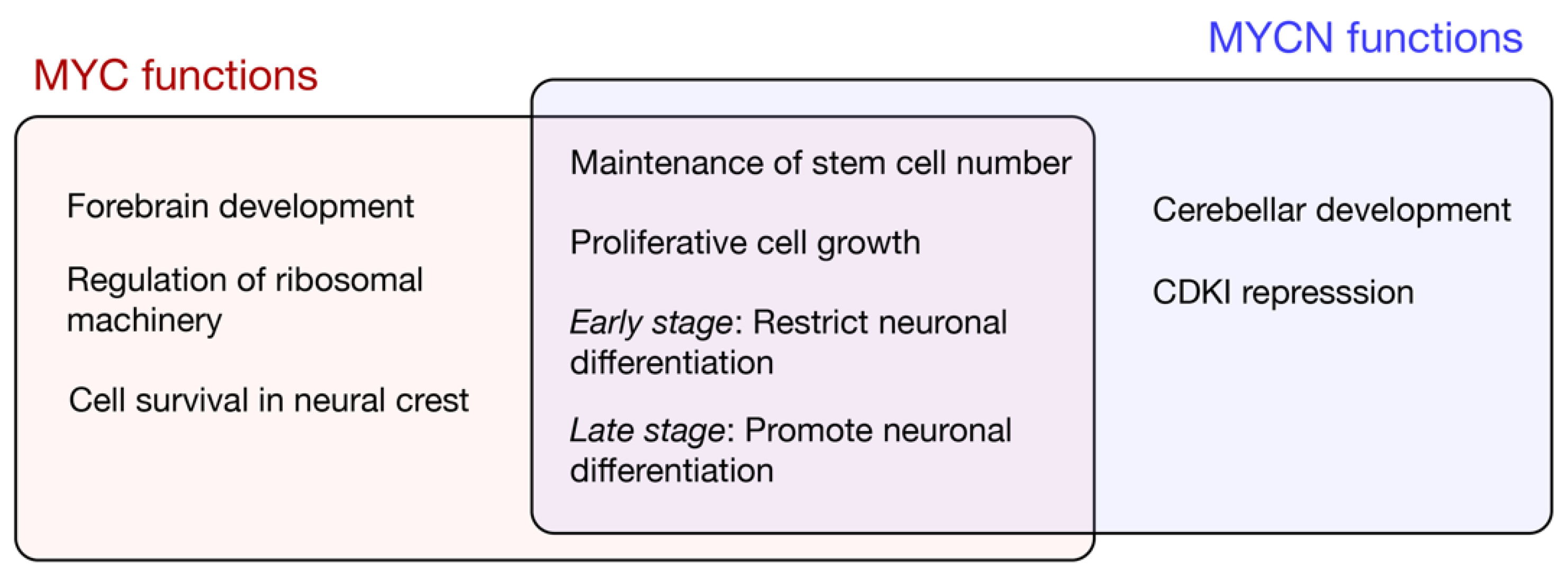
2. Distinct Expression of the MYC Family during Mammalian Brain Development
3. MYC and MYCN Regulate Brain Development in Mammals
4. Neural Development in Nonmammalian Vertebrates Is Controlled by MYC
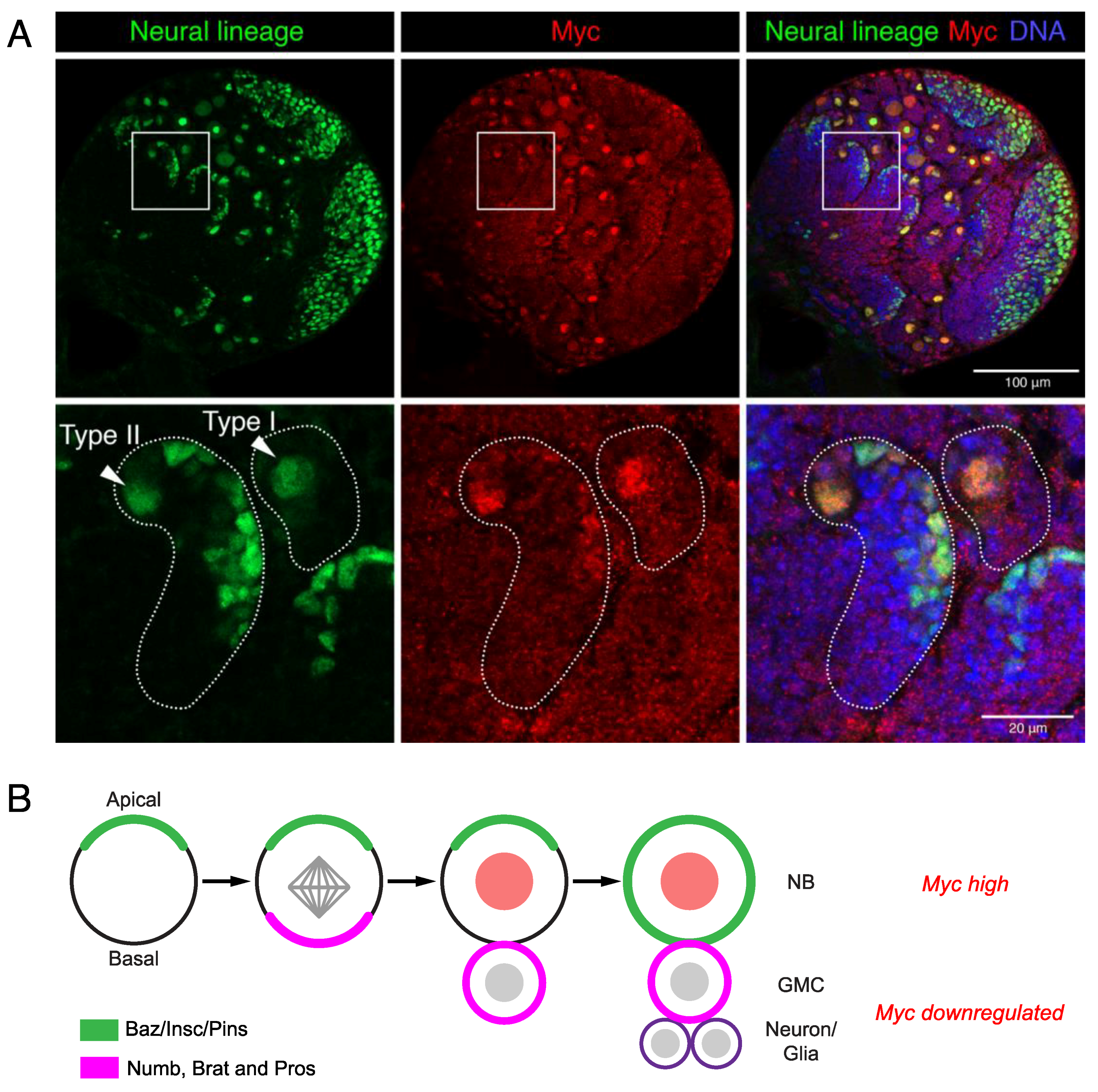
5. Nonvertebrate Models Reveal Myc Roles in Neural Stem Cells
6. Brain Tumours Are Driven by Cancer Stem Cells
7. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| bHLH | basic helix-loop-helix |
| CDKI | Cyclin Dependent Kinase Inhibitor |
| ChIP | Chromatin Immunoprecipitation |
| CNS | central nervous system |
| E | embryonic day |
| GMC | ganglion mother cell |
| NB | neuroblast |
| PNS | peripheral nervous system |
References
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef]
- Zaytseva, O.; Quinn, L. Controlling the Master: Chromatin Dynamics at the MYC Promoter Integrate Developmental Signaling. Genes 2017, 8, 118–124. [Google Scholar] [CrossRef]
- Eilers, M.; Eisenman, R.N. Myc’s broad reach. Genes Dev. 2008, 22, 2755–2766. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Woo, A.J.; Chu, J.; Snow, J.W.; Fujiwara, Y.; Kim, C.G.; Cantor, A.B.; Orkin, S.H. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell 2010, 143, 313–324. [Google Scholar] [CrossRef] [Green Version]
- DePinho, R.A.; Schreiber-Agus, N.; Alt, F.W. MYC Family Oncogenes in the Development of Normal and Neoplastic Cells. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherlands, 1991; Volume 57, pp. 1–46. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Araki, R.; Hoki, Y.; Uda, M.; Nakamura, M.; Jincho, Y.; Tamura, C.; Sunayama, M.; Ando, S.; Sugiura, M.; Yoshida, M.A.; et al. Crucial role of c-Myc in the generation of induced pluripotent stem cells. Stem Cells 2011, 29, 1362–1370. [Google Scholar] [CrossRef]
- Alié, A.; Hayashi, T.; Sugimura, I.; Manuel, M.; Sugano, W.; Mano, A.; Satoh, N.; Agata, K.; Funayama, N. The ancestral gene repertoire of animal stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, E7093–E7100. [Google Scholar] [CrossRef] [Green Version]
- Quinn, L.M.; Secombe, J.; Hime, G.R. Myc in Stem Cell Behaviour: Insights from Drosophila. In Transcriptional and Translational Regulation of Stem Cells; Hime, G., Abud, H., Eds.; Advances in Experimental Medicine and Biology; Springer: Dordrecht, The Netherlands, 2013; Volume 786, pp. 269–285. [Google Scholar] [CrossRef]
- Benassayag, C.; Montero, L.; Colombie, N.; Gallant, P.; Cribbs, D.; Morello, D. Human c-Myc Isoforms Differentially Regulate Cell Growth and Apoptosis in Drosophila melanogaster. Mol. Cell. Biol. 2005, 25, 9897–9909. [Google Scholar] [CrossRef] [Green Version]
- Trumpp, A.; Refaeli, Y.; Oskarsson, T.; Gasser, S.; Murphy, M.; Martin, G.R.; Bishop, J.M. c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 2001, 414, 768–773. [Google Scholar] [CrossRef]
- Dang, C.V.; O’Donnell, K.A.; Zeller, K.I.; Nguyen, T.; Osthus, R.C.; Li, F. The c-Myc target gene network. Semin. Cancer Biol. 2006, 16, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.H.; Loboda, A.P.; Showe, M.K.; Showe, L.C.; McMahon, S.B. Analysis of genomic targets reveals complex functions of MYC. Nat. Rev. Cancer 2004, 4, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Orian, A.; Grewal, S.S.; Knoepfler, P.S.; Edgar, B.A.; Parkhurst, S.M.; Eisenman, R.N. Genomic binding and transcriptional regulation by the Drosophila Myc and Mnt transcription factors. Cold Spring Harbor Symp. Quant. Biol. 2005, 70, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Orian, A.; van Steensel, B.; Delrow, J.; Bussemaker, H.J.; Li, L.; Sawado, T.; Williams, E.; Loo, L.W.M.; Cowley, S.M.; Yost, C.; et al. Genomic binding by the Drosophila Myc, Max, Mad/Mnt transcription factor network. Genes Dev. 2003, 17, 1101–1114. [Google Scholar] [CrossRef] [Green Version]
- Wolf, E.; Lin, C.Y.; Eilers, M.; Levens, D.L. Taming of the beast: Shaping Myc-dependent amplification. Trends Cell Biol. 2015, 25, 241–248. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.Y.; Lovén, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef] [Green Version]
- Nie, Z.; Hu, G.; Wei, G.; Cui, K.; Yamane, A.; Resch, W.; Wang, R.; Green, D.R.; Tessarollo, L.; Casellas, R.; et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 2012, 151, 68–79. [Google Scholar] [CrossRef] [Green Version]
- Walz, S.; Lorenzin, F.; Morton, J.; Wiese, K.E.; von Eyss, B.; Herold, S.; Rycak, L.; Dumay-Odelot, H.; Karim, S.; Bartkuhn, M.; et al. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 2014, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Sabò, A.; Kress, T.R.; Pelizzola, M.; de Pretis, S.; Gorski, M.M.; Tesi, A.; Morelli, M.J.; Bora, P.; Doni, M.; Verrecchia, A.; et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 2014, 511, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Staller, P.; Peukert, K.; Kiermaier, A.; Seoane, J.; Lukas, J.; Karsunky, H.; Möröy, T.; Bartek, J.; Massagué, J.; Hänel, F.; et al. Repression of p15INK4b expression by Myc through association with Miz-1. Nat. Cell Biol. 2001, 3, 392–399. [Google Scholar] [CrossRef]
- Grandori, C.; Cowley, S.M.; James, L.P.; Eisenman, R.N. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu. Rev. Cell Dev. Biol. 2000, 16, 653–699. [Google Scholar] [CrossRef] [PubMed]
- Lorenzin, F.; Benary, U.; Baluapuri, A.; Walz, S.; Jung, L.A.; von Eyss, B.; Kisker, C.; Wolf, J.; Eilers, M.; Wolf, E. Different promoter affinities account for specificity in MYC-dependent gene regulation. Elife 2016, 5, e15161. [Google Scholar] [CrossRef] [PubMed]
- Tansey, W.P. Mammalian MYC Proteins and Cancer. New J. Sci. 2014, 2014, 1–27. [Google Scholar] [CrossRef] [Green Version]
- Rickman, D.S.; Schulte, J.H.; Eilers, M. The Expanding World of N-MYC–Driven Tumors. Cancer Discov. 2018, 8, 150–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutter, S.; Bolin, S.; Weishaupt, H.; Swartling, F.J. Modeling and Targeting MYC Genes in Childhood Brain Tumors. Genes 2017, 8, 107. [Google Scholar] [CrossRef]
- Swartling, F.J. Myc proteins in brain tumor development and maintenance. Ups. J. Med. Sci. 2012, 117, 122–131. [Google Scholar] [CrossRef]
- Martínez-Cerdeño, V.; Noctor, S.C. Neural Progenitor Cell Terminology. Front. Neuroanat. 2018, 12, 664–668. [Google Scholar] [CrossRef]
- Malatesta, P.; Hartfuss, E.; Götz, M. Isolation of radial glial cells by fluorescent-activated cell sorting reveals a neuronal lineage. Development 2000, 127, 5253–5263. [Google Scholar]
- Götz, M.; Huttner, W.B. The cell biology of neurogenesis. Nat. Rev. Mol. Cell Biol. 2005, 6, 777–788. [Google Scholar] [CrossRef]
- Downs, K.M.; Martin, G.R.; Bishop, J.M. Contrasting patterns of myc and N-myc expression during gastrulation of the mouse embryo. Genes Dev. 1989, 3, 860–869. [Google Scholar] [CrossRef] [Green Version]
- Knoepfler, P.S.; Cheng, P.F.; Eisenman, R.N. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev. 2002, 16, 2699–2712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatton, K.S.; Mahon, K.; Chin, L.; Chiu, F.C.; Lee, H.W.; Peng, D.; Morgenbesser, S.D.; Horner, J.; DePinho, R.A. Expression and activity of L-Myc in normal mouse development. Mol. Cell. Biol. 1996, 16, 1794–1804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.-L.; Ma, Y.-X.; Xu, R.-J.; Ma, J.-J.; Zhang, H.-C.; Qi, S.-B.; Xu, J.-H.; Qin, X.-Z.; Zhang, H.-N.; Liu, C.-M.; et al. c-Myc controls the fate of neural progenitor cells during cerebral cortex development. J. Cell. Physiol. 2019, 235, 4011–4021. [Google Scholar] [CrossRef] [PubMed]
- Bhaduri, A.; Di Lullo, E.; Jung, D.; Müller, S.; Crouch, E.E.; Espinosa, C.S.; Ozawa, T.; Alvarado, B.; Spatazza, J.; Cadwell, C.R.; et al. Outer Radial Glia-like Cancer Stem Cells Contribute to Heterogeneity of Glioblastoma. Cell Stem Cell 2020, 26, 48–63.e6. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.C.; Wims, M.; Spotts, G.D.; Hann, S.R.; Bradley, A. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 1993, 7, 671–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubois, N.C.; Adolphe, C.; Ehninger, A.; Wang, R.A.; Robertson, E.J.; Trumpp, A. Placental rescue reveals a sole requirement for c-Myc in embryonic erythroblast survival and hematopoietic stem cell function. Development 2008, 135, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Stanton, B.R.; Perkins, A.S.; Tessarollo, L.; Sassoon, D.A.; Parada, L.F. Loss of N-myc function results in embryonic lethality and failure of the epithelial component of the embryo to develop. Genes Dev. 1992, 6, 2235–2247. [Google Scholar] [CrossRef]
- Sawai, S.; Shimono, A.; Wakamatsu, Y.; Palmes, C.; Hanaoka, K.; Kondoh, H. Defects of embryonic organogenesis resulting from targeted disruption of the N-myc gene in the mouse. Development 1993, 117, 1445–1455. [Google Scholar]
- Wey, A.; Knoepfler, P.S. c-myc and N-myc promote active stem cell metabolism and cycling as architects of the developing brain. Oncotarget 2010, 1, 120–130. [Google Scholar] [CrossRef]
- Mainwaring, L.A.; Bhatia, B.; Kenney, A.M. Myc on my mind: A transcription factor family’s essential role in brain development. Oncotarget 2010, 1, 86–88. [Google Scholar] [CrossRef]
- Tronche, F.; Kellendonk, C.; Kretz, O.; Gass, P.; Anlag, K.; Orban, P.C.; Bock, R.; Klein, R.; Schutz, G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet. 1999, 23, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Zindy, F.; Knoepfler, P.S.; Xie, S.; Sherr, C.J.; Eisenman, R.N.; Roussel, M.F. N-Myc and the cyclin-dependent kinase inhibitors p18Ink4c and p27Kip1 coordinately regulate cerebellar development. Proc. Natl. Acad. Sci. USA 2006, 103, 11579–11583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zindy, F.; Soares, H.; Herzog, K.H.; Morgan, J.; Sherr, C.J.; Roussel, M.F. Expression of INK4 inhibitors of cyclin D-dependent kinases during mouse brain development. Cell Growth Differ. 1997, 8, 1139–1150. [Google Scholar] [PubMed]
- Peukert, K.; Staller, P.; Schneider, A.; Carmichael, G.; Hänel, F.; Eilers, M. An alternative pathway for gene regulation by Myc. EMBO J. 1997, 16, 5672–5686. [Google Scholar] [CrossRef] [PubMed]
- Vo, B.T.; Wolf, E.; Kawauchi, D.; Gebhardt, A.; Rehg, J.E.; Finkelstein, D.; Walz, S.; Murphy, B.L.; Youn, Y.H.; Han, Y.-G.; et al. The Interaction of Myc with Miz1 Defines Medulloblastoma Subgroup Identity. Cancer Cell 2016, 29, 5–16. [Google Scholar] [CrossRef] [Green Version]
- Dardenne, E.; Beltran, H.; Benelli, M.; Gayvert, K.; Berger, A.; Puca, L.; Cyrta, J.; Sboner, A.; Noorzad, Z.; MacDonald, T.; et al. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell 2016, 30, 563–577. [Google Scholar] [CrossRef] [Green Version]
- Hatton, B.A.; Knoepfler, P.S.; Kenney, A.M.; Rowitch, D.H.; de Alborán, I.M.; Olson, J.M.; Eisenman, R.N. N-myc is an essential downstream effector of Shh signaling during both normal and neoplastic cerebellar growth. Cancer Res. 2006, 66, 8655–8661. [Google Scholar] [CrossRef] [Green Version]
- Chau, K.F.; Shannon, M.L.; Fame, R.M.; Fonseca, E.; Mullan, H.; Johnson, M.B.; Sendamarai, A.K.; Springel, M.W.; Laurent, B.; Lehtinen, M.K. Downregulation of ribosome biogenesis during early forebrain development. Elife 2018, 7, 6366. [Google Scholar] [CrossRef] [Green Version]
- Fame, R.M.; Shannon, M.L.; Chau, K.F.; Head, J.P.; Lehtinen, M.K. A concerted metabolic shift in early forebrain alters the CSF proteome and depends on MYC downregulation for mitochondrial maturation. Development 2019, 146, dev182857. [Google Scholar] [CrossRef]
- Khudyakov, J.; Bronner-Fraser, M. Comprehensive spatiotemporal analysis of early chick neural crest network genes. Dev. Dyn. 2009, 238, 716–723. [Google Scholar] [CrossRef] [Green Version]
- Kerosuo, L.; Bronner, M.E. cMyc Regulates the Size of the Premigratory Neural Crest Stem Cell Pool. Cell Rep. 2016, 17, 2648–2659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambros, P.F.; Ambros, I.M.; Brodeur, G.M.; Haber, M.; Khan, J.; Nakagawara, A.; Schleiermacher, G.; Speleman, F.; Spitz, R.; London, W.B.; et al. International consensus for neuroblastoma molecular diagnostics: Report from the International Neuroblastoma Risk Group (INRG) Biology Committee. Br. J. Cancer 2009, 100, 1471–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerosuo, L.; Neppala, P.; Hsin, J.; Mohlin, S.; Vieceli, F.M.; Török, Z.; Laine, A.; Westermarck, J.; Bronner, M.E. Enhanced expression of MycN/CIP2A drives neural crest toward a neural stem cell-like fate: Implications for priming of neuroblastoma. Proc. Natl. Acad. Sci. USA 2018, 115, E7351–E7360. [Google Scholar] [CrossRef] [Green Version]
- Kerosuo, L.; Piltti, K.; Fox, H.; Angers-Loustau, A.; Häyry, V.; Eilers, M.; Sariola, H.; Wartiovaara, K. Myc increases self-renewal in neural progenitor cells through Miz-1. J. Cell. Sci. 2008, 121, 3941–3950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinin, N.; Adameyko, I.; Wilhelm, M.; Fritz, N.; Uhlén, P.; Ernfors, P.; Henriksson, M.A. MYC proteins promote neuronal differentiation by controlling the mode of progenitor cell division. EMBO Rep. 2014, 15, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-K.; Tsang, M.; Dawid, I.B. The mych gene is required for neural crest survival during zebrafish development. PLoS ONE 2008, 3, e2029. [Google Scholar] [CrossRef] [Green Version]
- Bellmeyer, A.; Krase, J.; Lindgren, J.; LaBonne, C. The protooncogene c-myc is an essential regulator of neural crest formation in xenopus. Dev. Cell 2003, 4, 827–839. [Google Scholar] [CrossRef] [Green Version]
- Knoblich, J.A. Asymmetric cell division: Recent developments and their implications for tumour biology. Nat. Rev. Mol. Cell Biol. 2010, 11, 849–860. [Google Scholar] [CrossRef] [Green Version]
- Betschinger, J.; Mechtler, K.; Knoblich, J.A. Asymmetric segregation of the tumor suppressor brat regulates self-renewal in Drosophila neural stem cells. Cell 2006, 124, 1241–1253. [Google Scholar] [CrossRef] [Green Version]
- Gómez-López, S.; Lerner, R.G.; Petritsch, C. Asymmetric cell division of stem and progenitor cells during homeostasis and cancer. Cell. Mol. Life Sci. 2014, 71, 575–597. [Google Scholar] [CrossRef] [Green Version]
- Chia, W.; Somers, W.G.; Wang, H. Drosophila neuroblast asymmetric divisions: Cell cycle regulators, asymmetric protein localization, and tumorigenesis. J. Cell Biol. 2008, 180, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Homem, C.C.F.; Knoblich, J.A. Drosophila neuroblasts: A model for stem cell biology. Development 2012, 139, 4297–4310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, S.K.; Rolland, V.; Betschinger, J.; Kinsey, K.A.; Emery, G.; Knoblich, J.A. The tumor suppressors Brat and Numb regulate transit-amplifying neuroblast lineages in Drosophila. Dev. Cell 2008, 14, 535–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.A.; Lau, K.M.; Rahmani, Z.; Dho, S.E.; Brothers, G.; She, Y.M.; Berry, D.M.; Bonneil, E.; Thibault, P.; Schweisguth, F.; et al. aPKC-mediated phosphorylation regulates asymmetric membrane localization of the cell fate determinant Numb. EMBO J. 2007, 26, 468–480. [Google Scholar] [CrossRef] [Green Version]
- Wirtz-Peitz, F.; Nishimura, T.; Knoblich, J.A. Linking cell cycle to asymmetric division: Aurora-A phosphorylates the Par complex to regulate Numb localization. Cell 2008, 135, 161–173. [Google Scholar] [CrossRef] [Green Version]
- Arama, E.; Dickman, D.; Kimchie, Z.; Shearn, A.; Lev, Z. Mutations in the beta-propeller domain of the Drosophila brain tumor (brat) protein induce neoplasm in the larval brain. Oncogene 2000, 19, 3706–3716. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Kong, J.; Tucker-Burden, C.; Anand, M.; Rong, Y.; Rahman, F.; Moreno, C.S.; Van Meir, E.G.; Hadjipanayis, C.G.; Brat, D.J. Human Brat ortholog TRIM3 is a tumor suppressor that regulates asymmetric cell division in glioblastoma. Cancer Res. 2014, 74, 4536–4548. [Google Scholar] [CrossRef] [Green Version]
- Samuels, T.J.; Järvelin, A.I.; Ish-Horowicz, D.; Davis, I. Imp/IGF2BP levels modulate individual neural stem cell growth and division through myc mRNA stability. Elife 2020, 9, 166. [Google Scholar] [CrossRef]
- Weidensdorfer, D.; Stöhr, N.; Baude, A.; Lederer, M.; Köhn, M.; Schierhorn, A.; Buchmeier, S.; Wahle, E.; Hüttelmaier, S. Control of c-myc mRNA stability by IGF2BP1-associated cytoplasmic RNPs. RNA 2009, 15, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Mu, Q.; Wang, L.; Yu, F.; Gao, H.; Lei, T.; Li, P.; Liu, P.; Zheng, X.; Hu, X.; Chen, Y.; et al. Imp2 regulates GBM progression by activating IGF2/PI3K/Akt pathway. Cancer Biol. Ther. 2015, 16, 623–633. [Google Scholar] [CrossRef] [Green Version]
- Yoon, K.; Gaiano, N. Notch signaling in the mammalian central nervous system: Insights from mouse mutants. Nat. Neurosci. 2005, 8, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Louvi, A.; Artavanis-Tsakonas, S. Notch signalling in vertebrate neural development. Nat. Rev. Neurosci. 2006, 7, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Somers, G.W.; Bashirullah, A.; Heberlein, U.; Yu, F.; Chia, W. Aurora-A acts as a tumor suppressor and regulates self-renewal of Drosophila neuroblasts. Genes Dev. 2006, 20, 3453–3463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Lu, B. Regulation of cell growth by Notch signaling and its differential requirement in normal vs. tumor-forming stem cells in Drosophila. Genes Dev. 2011, 25, 2644–2658. [Google Scholar] [CrossRef] [Green Version]
- Gingras, A.C.; Raught, B.; Gygi, S.P.; Niedzwiecka, A.; Miron, M.; Burley, S.K.; Polakiewicz, R.D.; Wyslouch-Cieszynska, A.; Aebersold, R.; Sonenberg, N. Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev. 2001, 15, 2852–2864. [Google Scholar] [CrossRef]
- Miron, M.; Verdu, J.; Lachance, P.E.; Birnbaum, M.J.; Lasko, P.F.; Sonenberg, N. The translational inhibitor 4E-BP is an effector of PI(3)K/Akt signalling and cell growth in Drosophila. Nat. Cell Biol 2001, 3, 596–601. [Google Scholar] [CrossRef]
- Rust, K.; Tiwari, M.D.; Mishra, V.K.; Grawe, F.; Wodarz, A. Myc and the Tip60 chromatin remodeling complex control neuroblast maintenance and polarity in Drosophila. EMBO J. 2018, 37, e98659. [Google Scholar] [CrossRef]
- Ravens, S.; Yu, C.; Ye, T.; Stierle, M.; Tora, L. Tip60 complex binds to active Pol II promoters and a subset of enhancers and co-regulates the c-Myc network in mouse embryonic stem cells. Epigenetics Chromatin 2015, 8, 45. [Google Scholar] [CrossRef] [Green Version]
- Bellosta, P.; Hulf, T.; Balla Diop, S.; Usseglio, F.; Pradel, J.; Aragnol, D.; Gallant, P. Myc interacts genetically with Tip48/Reptin and Tip49/Pontin to control growth and proliferation during Drosophila development. Proc. Natl. Acad. Sci. USA 2005, 102, 11799–11804. [Google Scholar] [CrossRef] [Green Version]
- Ignatova, T.N.; Kukekov, V.G.; Laywell, E.D.; Suslov, O.N.; Vrionis, F.D.; Steindler, D.A. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia 2002, 39, 193–206. [Google Scholar] [CrossRef]
- Sanai, N.; Alvarez-Buylla, A.; Berger, M.S. Neural stem cells and the origin of gliomas. N. Engl. J. Med. 2005, 353, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Weathers, S.-P.S.; Gilbert, M.R. Toward Personalized Targeted Therapeutics: An Overview. Neurotherapeutics 2017, 14, 256–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirosh, I.; Venteicher, A.S.; Hebert, C.; Escalante, L.E.; Patel, A.P.; Yizhak, K.; Fisher, J.M.; Rodman, C.; Mount, C.; Filbin, M.G.; et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 2016, 539, 309–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couturier, C.P.; Ayyadhury, S.; Le, P.U.; Nadaf, J.; Monlong, J.; Riva, G.; Allache, R.; Baig, S.; Yan, X.; Bourgey, M.; et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat. Commun. 2020, 11, 3406–3419. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N. Engl. J. Med. 2015, 372, 2481–2498. [CrossRef] [Green Version]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Cancer Genome Atlas Research Network Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Fu, P.; He, Y.-S.; Huang, Q.; Ding, T.; Cen, Y.-C.; Zhao, H.-Y.; Wei, X. Bevacizumab treatment for newly diagnosed glioblastoma: Systematic review and meta-analysis of clinical trials. Mol. Clin. Oncol. 2016, 4, 833–838. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Aldape, K.; Brindle, K.M.; Chesler, L.; Chopra, R.; Gajjar, A.; Gilbert, M.R.; Gottardo, N.; Gutmann, D.H.; Hargrave, D.; Holland, E.C.; et al. Challenges to curing primary brain tumours. Nat. Rev. Clin. Oncol. 2019, 370, 1. [Google Scholar] [CrossRef] [Green Version]
- Andreotti, J.P.; Silva, W.N.; Costa, A.C.; Picoli, C.C.; Bitencourt, F.C.O.; Coimbra-Campos, L.M.C.; Resende, R.R.; Magno, L.A.V.; Romano-Silva, M.A.; Mintz, A.; et al. Neural stem cell niche heterogeneity. Semin. Cell Dev. Biol. 2019, 95, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Hallal, S.; Mallawaaratchy, D.M.; Wei, H.; Ebrahimkhani, S.; Stringer, B.W.; Day, B.W.; Boyd, A.W.; Guillemin, G.J.; Buckland, M.E.; Kaufman, K.L. Extracellular Vesicles Released by Glioblastoma Cells Stimulate Normal Astrocytes to Acquire a Tumor-Supportive Phenotype Via p53 and MYC Signaling Pathways. Mol. Neurobiol. 2019, 56, 4566–4581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzoyan, Z.; Sollazzo, M.; Allocca, M.; Valenza, A.M.; Grifoni, D.; Bellosta, P. Drosophila melanogaster: A Model Organism to Study Cancer. Front. Genet. 2019, 10, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, M.R. Drosophila Central Nervous System Glia. Cold Spring Harbor Perspect. Biol. 2015, 7, a020552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, N.R.; Shohayeb, B.; Zaytseva, O.; Mitchell, N.; Millard, S.S.; Ng, D.C.H.; Quinn, L.M. Glial-Specific Functions of Microcephaly Protein WDR62 and Interaction with the Mitotic Kinase AURKA Are Essential for Drosophila Brain Growth. Stem Cell Rep. 2017, 9, 32–41. [Google Scholar] [CrossRef] [Green Version]
- Perrin, S.L.; Samuel, M.S.; Koszyca, B.; Brown, M.P.; Ebert, L.M.; Oksdath, M.; Gomez, G.A. Glioblastoma heterogeneity and the tumour microenvironment: Implications for preclinical research and development of new treatments. Biochem. Soc. Trans. 2019, 47, 625–638. [Google Scholar] [CrossRef] [PubMed]
- Linkous, A.; Balamatsias, D.; Snuderl, M.; Edwards, L.; Miyaguchi, K.; Milner, T.; Reich, B.; Cohen-Gould, L.; Storaska, A.; Nakayama, Y.; et al. Modeling Patient-Derived Glioblastoma with Cerebral Organoids. Cell Rep. 2019, 26, 3203–3211. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaytseva, O.; Kim, N.-h.; Quinn, L.M. MYC in Brain Development and Cancer. Int. J. Mol. Sci. 2020, 21, 7742. https://doi.org/10.3390/ijms21207742
Zaytseva O, Kim N-h, Quinn LM. MYC in Brain Development and Cancer. International Journal of Molecular Sciences. 2020; 21(20):7742. https://doi.org/10.3390/ijms21207742
Chicago/Turabian StyleZaytseva, Olga, Nan-hee Kim, and Leonie M. Quinn. 2020. "MYC in Brain Development and Cancer" International Journal of Molecular Sciences 21, no. 20: 7742. https://doi.org/10.3390/ijms21207742
APA StyleZaytseva, O., Kim, N. -h., & Quinn, L. M. (2020). MYC in Brain Development and Cancer. International Journal of Molecular Sciences, 21(20), 7742. https://doi.org/10.3390/ijms21207742