Interferon-Mediated Long Non-Coding RNA Response in Macrophages in the Context of HIV

 , ,
, ,
Abstract
:1. Introduction
2. Results
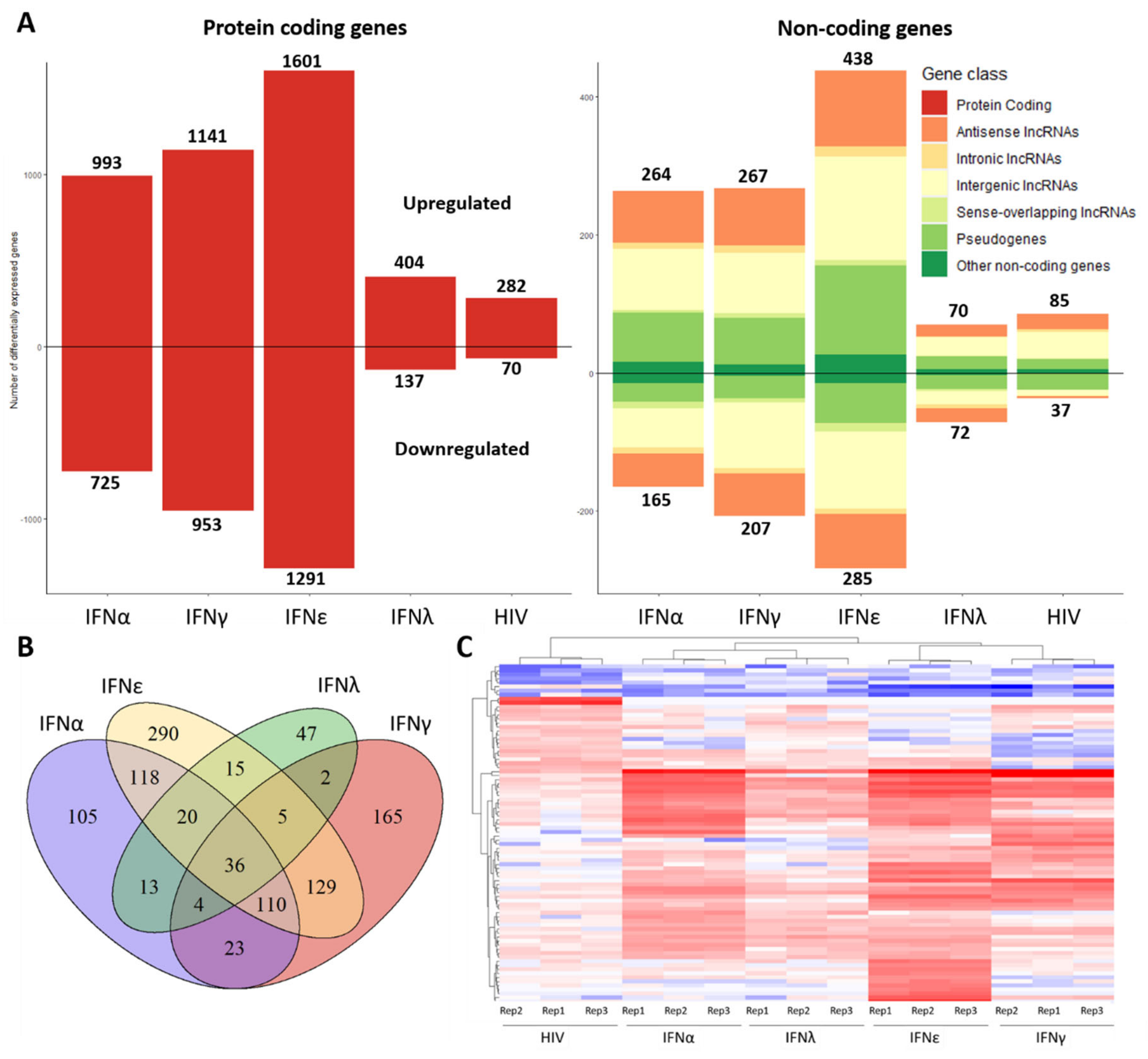
2.1. Transcriptome of Macrophages Stimulated with Interferons or Subjected to HIV-1 Infection
2.2. Gene Enrichment Analysis
2.3. Weighted Gene Co-Expression Network Analysis (WGCNA) to Identify IFN Related lncRNA Hub Genes
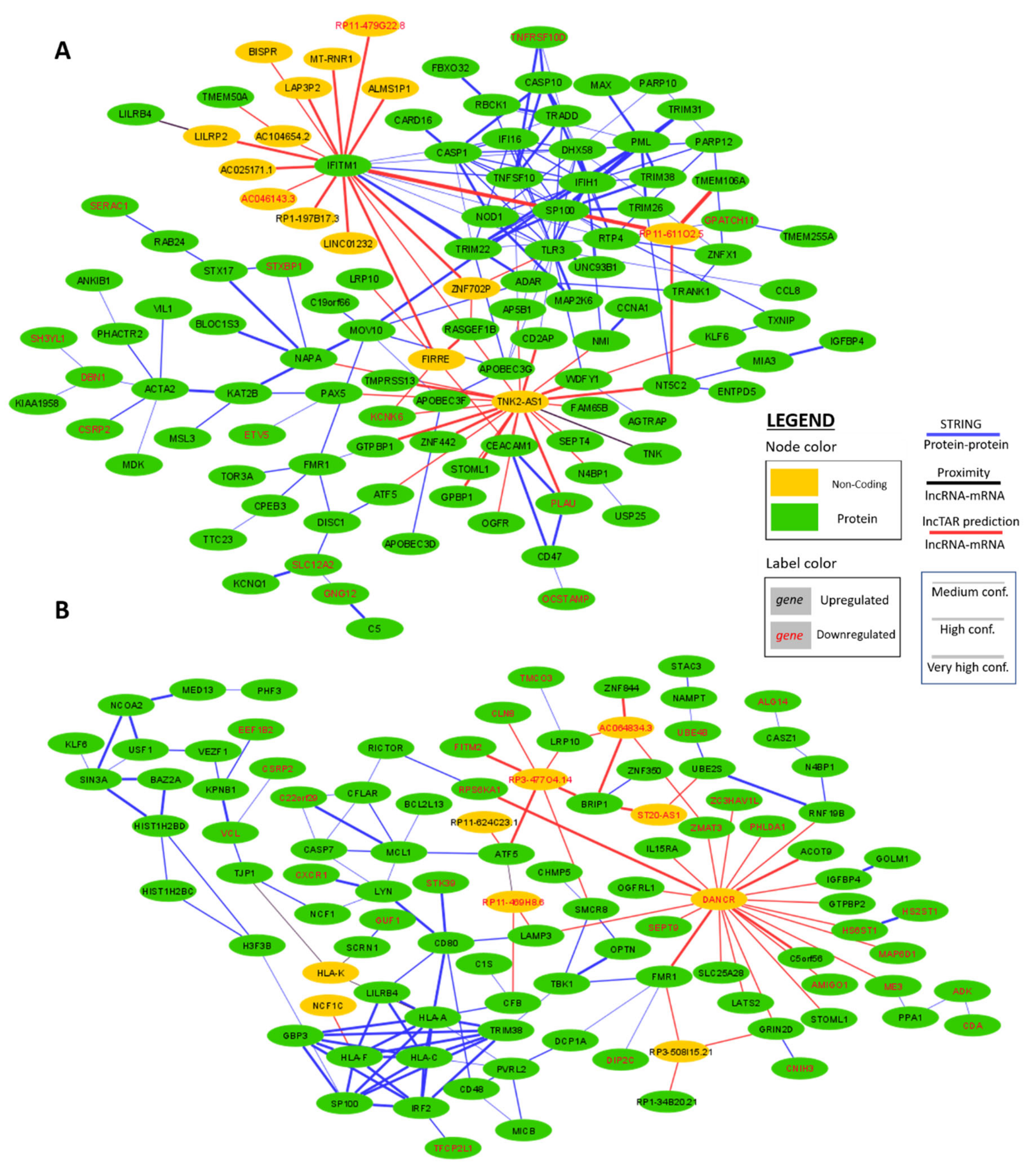
2.4. Interaction Network Construction of WGCNA Hub Genes in IFNα and IFNε Stimulated Conditions
2.5. Interaction Network Construction of Differentially Expressed HIV-1 Correlated Genes
3. Discussion
4. Materials and Methods
4.1. RNA Sequencing Data Generation
4.2. RNA Sequencing Data Processing and Differential Gene Expression Analysis
4.3. Gene Ontology and Pathway Enrichment Analysis
4.4. Weighted Gene Co-Expression Network Analysis (WGCNA)
4.5. Molecular Interaction Analysis (STRING; LncTAR; Proximity)
4.6. Relative Quantification by qPCR for RNAseq Validation
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ART | antiretroviral therapy |
| DANCR | differentiation antagonizing non-protein coding RNA |
| FC | fold change |
| FIRRE | functional intergenic repeating RNA element |
| GBP1P1 | guanylate binding protein 1 pseudogene 1 |
| GO_BP | Gene Ontology Biological Process |
| GS | gene significance |
| HCV | hepatitis C |
| HIV-1 | human immunodeficiency virus type I |
| IFN | interferon |
| ISGs | interferon stimulated genes |
| JAK-STAT | Janus kinase/signal transducer and activator of transcription |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| lncRNAs | long non-coding RNAs |
| LPS | lipopolysaccharides |
| MDA5 | melanoma differentiation-associated protein 5 |
| MDMs | monocyte-derived macrophages |
| MM | module membership |
| ncRNAs | non-coding RNAs |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NRIR | negative regulator of interferon response |
| PAMP | pathogen-associated molecular pattern |
| PBMCs | peripheral blood mononuclear cells |
| qPCR | quantitative reverse transcription polymerase chain reaction |
| RIG-I | retinoic acid-inducible gene I |
| ROS | reactive oxygen species |
| STAT | signal transducer and activator of transcription proteins |
| TNK2-AS1 | TNK2 antisense RNA 1 |
| WGCNA | Weighted gene co-expression network analysis |
References
- Mesev, E.V.; LeDesma, R.A.; Ploss, A. Decoding type I and III interferon signalling during viral infection. Nat. Microbiol. 2019, 4, 914–924. [Google Scholar] [CrossRef]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern recognition receptors and the innate immune response to viral infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef] [PubMed]
- Crosse, K.M.; Monson, E.A.; Beard, M.R.; Helbig, K.J. Interferon-Stimulated Genes as Enhancers of Antiviral Innate Immune Signaling. J. Innate immun. 2018, 10, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [Green Version]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef]
- Leucci, E.; Vendramin, R.; Spinazzi, M.; Laurette, P.; Fiers, M.; Wouters, J.; Radaelli, E.; Eyckerman, S.; Leonelli, C.; Vanderheyden, K.; et al. Melanoma addiction to the long non-coding RNA SAMMSON. Nature 2016, 531, 518–522. [Google Scholar] [CrossRef]
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, S.; Fitzgerald, K.A. Cytokines and Long Noncoding RNAs. Cold Spring Harb. Perspect. Biol. 2018, 10, a028589. [Google Scholar] [CrossRef]
- Valadkhan, S.; Plasek, L.M. Long Non-Coding RNA-Mediated Regulation of the Interferon Response: A New Perspective on a Familiar Theme. Pathog. Immun. 2018, 3, 126–148. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zheng, Y. lncRNAs Regulate Innate Immune Responses and Their Roles in Macrophage Polarization. Mediat. Inflamm. 2018, 2018, 8050956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, G.; Gong, A.Y.; Wang, Y.; Ma, S.; Chen, X.; Chen, J.; Su, C.J.; Shibata, A.; Strauss-Soukup, J.K.; Drescher, K.M.; et al. LincRNA-Cox2 Promotes Late Inflammatory Gene Transcription in Macrophages through Modulating SWI/SNF-Mediated Chromatin Remodeling. J. Immunol. 2016, 196, 2799–2808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Goswami, S.; Qiu, J.; Chen, Q.; Laverdure, S.; Sherman, B.T.; Imamichi, T. Profiles of Long Non-Coding RNAs and mRNA Expression in Human Macrophages Regulated by Interleukin-27. Int. J. Mol. Sci. 2019, 20, 6207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasker, C.; Subbian, S.; Gao, P.; Couret, J.; Levine, C.; Ghanny, S.; Soteropoulos, P.; Zhao, X.; Landau, N.; Lu, W.; et al. IFN-epsilon protects primary macrophages against HIV infection. JCI Insight 2016, 1, e88255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goujon, C.; Malim, M.H. Characterization of the alpha interferon-induced postentry block to HIV-1 infection in primary human macrophages and T cells. J. Virol. 2010, 84, 9254–9266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganor, Y.; Real, F.; Sennepin, A.; Dutertre, C.A.; Prevedel, L.; Xu, L.; Tudor, D.; Charmeteau, B.; Couedel-Courteille, A.; Marion, S.; et al. HIV-1 reservoirs in urethral macrophages of patients under suppressive antiretroviral therapy. Nat. Microbiol. 2019, 4, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.E.; Jaworowski, A.; Hearps, A.C. The HIV Reservoir in Monocytes and Macrophages. Front. Immunol. 2019, 10, 1435. [Google Scholar] [CrossRef] [Green Version]
- Szaniawski, M.A.; Spivak, A.M.; Cox, J.E.; Catrow, J.L.; Hanley, T.; Williams, E.; Tremblay, M.J.; Bosque, A.; Planelles, V. SAMHD1 Phosphorylation Coordinates the Anti-HIV-1 Response by Diverse Interferons and Tyrosine Kinase Inhibition. mBio 2018, 9, e00819-18. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucl. Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef]
- Gene Ontology, C. Gene Ontology Consortium: Going forward. Nucl. Acids Res. 2015, 43, D1049–D1056. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucl. Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucl. Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Ma, W.; Zeng, P.; Wang, J.; Geng, B.; Yang, J.; Cui, Q. LncTar: A tool for predicting the RNA targets of long noncoding RNAs. Brief. Bioinform. 2015, 16, 806–812. [Google Scholar] [CrossRef]
- Mattick, J.S.; Gagen, M.J. The evolution of controlled multitasked gene networks: The role of introns and other noncoding RNAs in the development of complex organisms. Mol. Biol. Evol. 2001, 18, 1611–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scacalossi, K.R.; van Solingen, C.; Moore, K.J. Long non-coding RNAs regulating macrophage functions in homeostasis and disease. Vasc. Pharmacol. 2019, 114, 122–130. [Google Scholar] [CrossRef]
- Xie, Y.; Wang, M.; Tian, J.; Li, X.; Yang, M.; Zhang, K.; Tan, S.; Luo, L.; Luo, C.; Peng, L.; et al. Long non-coding RNA expressed in macrophage co-varies with the inflammatory phenotype during macrophage development and polarization. J. Cell Mol. Med. 2019, 23, 6530–6542. [Google Scholar] [CrossRef]
- Huang, J.K.; Ma, L.; Song, W.H.; Lu, B.Y.; Huang, Y.B.; Dong, H.M.; Ma, X.K.; Zhu, Z.Z.; Zhou, R. LncRNA-MALAT1 Promotes Angiogenesis of Thyroid Cancer by Modulating Tumor-Associated Macrophage FGF2 Protein Secretion. J. Cell Biochem. 2017, 118, 4821–4830. [Google Scholar] [CrossRef]
- Atri, C.; Guerfali, F.Z.; Laouini, D. Role of Human Macrophage Polarization in Inflammation during Infectious Diseases. Int. J. Mol. Sci. 2018, 19, 1801. [Google Scholar] [CrossRef] [Green Version]
- Read, S.A.; Wijaya, R.; Ramezani-Moghadam, M.; Tay, E.; Schibeci, S.; Liddle, C.; Lam, V.W.T.; Yuen, L.; Douglas, M.W.; Booth, D.; et al. Macrophage Coordination of the Interferon Lambda Immune Response. Front. Immunol. 2019, 10, 2674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wentker, P.; Eberhardt, M.; Dreyer, F.S.; Bertrams, W.; Cantone, M.; Griss, K.; Schmeck, B.; Vera, J. An Interactive Macrophage Signal Transduction Map Facilitates Comparative Analyses of High-Throughput Data. J. Immunol. 2017, 198, 2191–2201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.; Nie, F.; Wang, Z.; Sun, M. Pseudogene-expressed RNAs: A new frontier in cancers. Tumour Biol. 2016, 37, 1471–1478. [Google Scholar] [CrossRef]
- Groen, J.N.; Capraro, D.; Morris, K.V. The emerging role of pseudogene expressed non-coding RNAs in cellular functions. Int. J. Biochem. Cell Biol. 2014, 54, 350–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barriocanal, M.; Carnero, E.; Segura, V.; Fortes, P. Long Non-Coding RNA BST2/BISPR is Induced by IFN and Regulates the Expression of the Antiviral Factor Tetherin. Front. Immunol. 2014, 5, 655. [Google Scholar] [CrossRef] [PubMed]
- Dallagi, A.; Girouard, J.; Hamelin-Morrissette, J.; Dadzie, R.; Laurent, L.; Vaillancourt, C.; Lafond, J.; Carrier, C.; Reyes-Moreno, C. The activating effect of IFN-gamma on monocytes/macrophages is regulated by the LIF-trophoblast-IL-10 axis via Stat1 inhibition and Stat3 activation. Cell Mol. Immunol. 2015, 12, 326–341. [Google Scholar] [CrossRef] [Green Version]
- Cory, T.J.; Schacker, T.W.; Stevenson, M.; Fletcher, C.V. Overcoming pharmacologic sanctuaries. Curr. Opin. HIV AIDS 2013, 8, 190–195. [Google Scholar] [CrossRef] [Green Version]
- Zalar, A.; Figueroa, M.I.; Ruibal-Ares, B.; Bare, P.; Cahn, P.; de Bracco, M.M.; Belmonte, L. Macrophage HIV-1 infection in duodenal tissue of patients on long term HAART. Antivir. Res. 2010, 87, 269–271. [Google Scholar] [CrossRef]
- Harman, A.N.; Nasr, N.; Feetham, A.; Galoyan, A.; Alshehri, A.A.; Rambukwelle, D.; Botting, R.A.; Hiener, B.M.; Diefenbach, E.; Diefenbach, R.J.; et al. HIV Blocks Interferon Induction in Human Dendritic Cells and Macrophages by Dysregulation of TBK1. J. Virol. 2015, 89, 6575–6584. [Google Scholar] [CrossRef] [Green Version]
- Kambara, H.; Niazi, F.; Kostadinova, L.; Moonka, D.K.; Siegel, C.T.; Post, A.B.; Carnero, E.; Barriocanal, M.; Fortes, P.; Anthony, D.D.; et al. Negative regulation of the interferon response by an interferon-induced long non-coding RNA. Nucl. Acids Res. 2014, 42, 10668–10680. [Google Scholar] [CrossRef] [Green Version]
- Carnero, E.; Barriocanal, M.; Segura, V.; Guruceaga, E.; Prior, C.; Borner, K.; Grimm, D.; Fortes, P. Type I Interferon Regulates the Expression of Long Non-Coding RNAs. Front. Immunol. 2014, 5, 548. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, J.; Wang, J.; Wen, Q.; Wang, H.; He, J.; Hu, S.; He, W.; Du, X.; Liu, S.; et al. Microarray analysis of long noncoding RNA and mRNA expression profiles in human macrophages infected with Mycobacterium tuberculosis. Sci. Rep. 2016, 6, 38963. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Han, D.; Pan, L.; Sun, J. The positive feedback between lncRNA TNK2-AS1 and STAT3 enhances angiogenesis in non-small cell lung cancer. Biochem. Biophys. Res. Commun. 2018, 507, 185–192. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, X.; Xie, M.; Liu, M.; Ye, M.; Li, M.; Chen, X.M.; Li, X.; Zhou, R. The NF-kappaB-Responsive Long Noncoding RNA FIRRE Regulates Posttranscriptional Regulation of Inflammatory Gene Expression through Interacting with hnRNPU. J. Immunol. 2017, 199, 3571–3582. [Google Scholar] [CrossRef] [Green Version]
- Zang, Y.; Zhou, X.; Wang, Q.; Li, X.; Huang, H. LncRNA FIRRE/NF-kB feedback loop contributes to OGD/R injury of cerebral microglial cells. Biochem. Biophys. Res. Commun. 2018, 501, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.J.; Jin, M.Z.; Xia, B.R.; Jin, W.L. Long Non-coding RNA DANCR as an Emerging Therapeutic Target in Human Cancers. Front. Oncol. 2019, 9, 1225. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics (Oxf. Engl.) 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics (Oxf. Engl.) 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Ewels, P.; Magnusson, M.; Lundin, S.; Kaller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics (Oxf. Engl.) 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, research0034. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Module | ID | Biological Process | Number of Genes (%) | Padj |
|---|---|---|---|---|
| Black | GO:0030198 | extracellular matrix organization | 27 (6.8) | 1.32 × 10−4 |
| Black | GO:0006954 | inflammatory response | 39 (4.8) | 1.06 × 10−3 |
| Black | GO:0030335 | positive regulation of cell migration | 32 (5.2) | 3.15 × 10−3 |
| Black | GO:0097529 | myeloid leukocyte migration | 18 (7.7) | 5.02 × 10−3 |
| Black | GO:0043062 | extracellular structure organization | 28 (5.5) | 5.61 × 10−3 |
| Blue | GO:0035966 | response to topologically incorrect protein | 66 (32.2) | 4.91 × 10−7 |
| Blue | GO:0070925 | organelle assembly | 205 (21.9) | 6.09 × 10−7 |
| Blue | GO:0034976 | response to endoplasmic reticulum stress | 82 (27.9) | 7.73 × 10−6 |
| Blue | GO:0007049 | cell cycle | 373 (18.9) | 1.17 × 10−5 |
| Blue | GO:0006974 | cellular response to DNA damage stimulus | 189 (21.4) | 3.19 × 10−5 |
| Brown | GO:0016032 | viral process | 213 (25.0) | 2.61 × 10−15 |
| Brown | GO:0044403 | symbiotic process | 222 (24.4) | 9.79 × 10−15 |
| Brown | GO:0044419 | interspecies interaction between organisms | 229 (23.9) | 4.11 × 10−14 |
| Brown | GO:0034341 | response to interferon-gamma | 69 (33.2) | 6.15 × 10−9 |
| Brown | GO:0045087 | innate immune response | 224 (21.4) | 1.93 × 10−8 |
| Cyan | GO:0098740 | multi organism cell adhesion | 2 (100) | 2.98 × 10−2 |
| Midnightblue | GO:0003009 | skeletal muscle contraction | 4 (8.3) | 3.46 × 10−2 |
| Pink | GO:0006119 | oxidative phosphorylation | 24 (15.7) | 6.17 × 10−15 |
| Pink | GO:0015985 | energy coupled proton transport, down electrochemical gradient | 24 (15.4) | 9.85 × 10−15 |
| Pink | GO:0015986 | ATP synthesis coupled proton transport | 24 (15.4) | 9.85 × 10−15 |
| Pink | GO:0009206 | purine ribonucleoside triphosphate biosynthetic process | 25 (13.0) | 1.31 × 10−13 |
| Pink | GO:0009145 | purine nucleoside triphosphate biosynthetic process | 25 (12.9) | 1.49 × 10−13 |
| Tan | GO:0050859 | negative regulation of B cell receptor signaling pathway | 3 (37.5) | 2.22 × 10−2 |
| Turquoise | GO:0007049 | cell cycle | 509 (25.8) | 7.43 × 10−23 |
| Turquoise | GO:0022402 | cell cycle process | 438 (26.8) | 1.11 × 10−22 |
| Turquoise | GO:0000278 | mitotic cell cycle | 316 (29.2) | 1.58 × 10−21 |
| Turquoise | GO:1903047 | mitotic cell cycle process | 316 (29.2) | 1.58 × 10−21 |
| Turquoise | GO:0140014 | mitotic nuclear division | 316 (29.2) | 1.58 × 10−21 |
| Yellow | GO:0042775 | mitochondrial ATP synthesis coupled electron transport | 19 (19.0) | 2.99 × 10−3 |
| Yellow | GO:0042773 | ATP synthesis coupled electron transport | 19 (18.8) | 3.51 × 10−3 |
| Yellow | GO:0090407 | organophosphate biosynthetic process | 68 (9.1) | 8.61 × 10−3 |
| Yellow | GO:0022900 | electron transport chain | 27 (13.4) | 2.00 × 10−2 |
| Yellow | GO:0019637 | organophosphate metabolic process | 95 (79.2) | 3.04 × 10−2 |
| Module | ID | Biological Process | Source | Number of Genes (%) | Padj |
|---|---|---|---|---|---|
| Blue | 1269649 | Gene expression | REACTOME | 372 (20.2) | 1.63 × 10−8 |
| Blue | 1270038 | Regulation of cholesterol biosynthesis by SREBP | REACTOME | 25 (45.5) | 1.01 × 10−4 |
| Blue | 1270039 | Activation of gene expression by SREBF | REACTOME | 21 (50.0) | 1.51 × 10−4 |
| Blue | 1270037 | Cholesterol biosynthesis | REACTOME | 15 (62.5) | 2.26 × 10−4 |
| Blue | 1268838 | Organelle biogenesis and maintenance | REACTOME | 86 (25.2) | 4.61 × 10−4 |
| Black | 1270244 | Extracellular matrix organization | REACTOME | 24 (8.1) | 1.85 × 10−5 |
| Black | 1470923 | Interleukin-4 and 13 signaling | REACTOME | 12 (10.2) | 5.05 × 10−3 |
| Black | 1270254 | Non-integrin membrane-ECM interactions | REACTOME | 7 (15.2) | 4.68 × 10−2 |
| Brown | 1269311 | Interferon signaling | REACTOME | 65 (32.2) | 2.24 × 10−7 |
| Brown | 377873 | Herpes simplex infection | KEGG | 57 (30.8) | 1.85 × 10−5 |
| Brown | 1269171 | Adaptive immune system | REACTOME | 175 (21.2) | 9.65 × 10−5 |
| Brown | 1269312 | Interferon alpha/beta signaling | REACTOME | 28 (40.6) | 2.02 × 10−4 |
| Brown | 1269314 | Interferon gamma signaling | REACTOME | 34 (36.2) | 2.63 × 10−4 |
| Pink | 82942 | Oxidative phosphorylation | KEGG | 22 (16.5) | 8.76 × 10−14 |
| Pink | 1270121 | TCA cycle and respitory electron transport | REACTOME | 24 (14.0) | 1.64 × 10−13 |
| Pink | 83098 | Parkinson’s disease | KEGG | 21 (14.8) | 4.69 × 10−12 |
| Pink | 1270127 | Respiratory electron transport | REACTOME | 20 (15.9) | 5.28 × 10−12 |
| Pink | 83097 | Alzheimer’s disease | KEGG | 21 (12.3) | 2.06 × 10−10 |
| Tan | 1427857 | Regulation of TLR by endogeneous ligand | REACTOME | 3 (18.3) | 3.21 × 10−2 |
| Turquoise | 1269741 | Cell cycle | REACTOME | 215 (34.5) | 7.99 × 10−21 |
| Turquoise | 1269763 | Cell cycle, mitotic | REACTOME | 183 (35.4) | 8.48 × 10−19 |
| Turquoise | 1427846 | rRNA processing in the nucleus and cytosol | REACTOME | 87 (45.1) | 4.25 × 10−15 |
| Turquoise | 1383086 | Major pathway of rRNA processing | REACTOME | 83 (45.9) | 4.25 × 10−15 |
| Turquoise | 1269649 | Gene expression | REACTOME | 468 (25.4) | 9.78 × 10−15 |
| Yellow | 1270128 | Respiratory electron transport | REACTOME | 18 (17.5) | 1.42 × 10−2 |
| Yellow | 82942 | Oxidative phosphorylation | KEGG | 20 (15.0) | 4.66 × 10−2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schynkel, T.; Szaniawski, M.A.; Spivak, A.M.; Bosque, A.; Planelles, V.; Vandekerckhove, L.; Trypsteen, W. Interferon-Mediated Long Non-Coding RNA Response in Macrophages in the Context of HIV. Int. J. Mol. Sci. 2020, 21, 7741. https://doi.org/10.3390/ijms21207741
Schynkel T, Szaniawski MA, Spivak AM, Bosque A, Planelles V, Vandekerckhove L, Trypsteen W. Interferon-Mediated Long Non-Coding RNA Response in Macrophages in the Context of HIV. International Journal of Molecular Sciences. 2020; 21(20):7741. https://doi.org/10.3390/ijms21207741
Chicago/Turabian StyleSchynkel, Tinus, Matthew A. Szaniawski, Adam M. Spivak, Alberto Bosque, Vicente Planelles, Linos Vandekerckhove, and Wim Trypsteen. 2020. "Interferon-Mediated Long Non-Coding RNA Response in Macrophages in the Context of HIV" International Journal of Molecular Sciences 21, no. 20: 7741. https://doi.org/10.3390/ijms21207741
APA StyleSchynkel, T., Szaniawski, M. A., Spivak, A. M., Bosque, A., Planelles, V., Vandekerckhove, L., & Trypsteen, W. (2020). Interferon-Mediated Long Non-Coding RNA Response in Macrophages in the Context of HIV. International Journal of Molecular Sciences, 21(20), 7741. https://doi.org/10.3390/ijms21207741