Recycling the Purpose of Old Drugs to Treat Ovarian Cancer


Abstract
: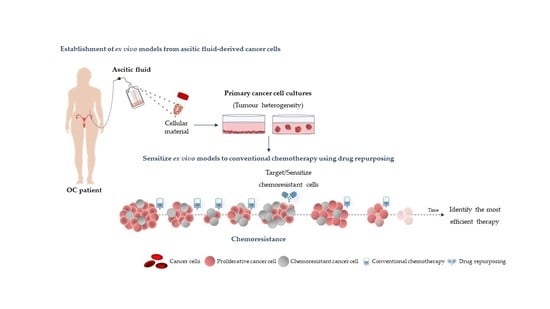
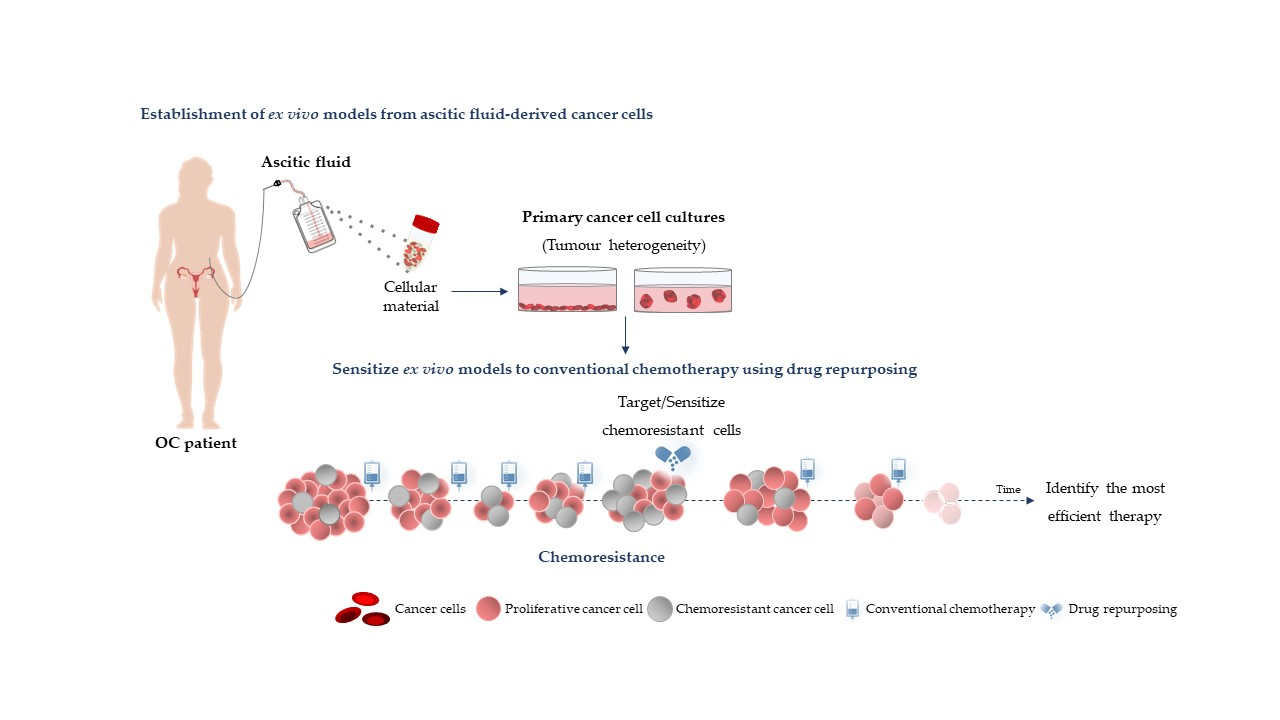{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Drug Repurposing for Ovarian Cancer Therapy
2.1. Statins
2.2. Metformin
2.3. Bisphosphonates
2.4. Ivermectin
2.5. Itraconazole
2.6. Ritonavir
3. Using Ex Vivo Models to Test Individual Drug Repurposing Efficacy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 2D | Two-dimensional |
| 3D | Three-dimensional |
| AIDS | Acquired immunodeficiency syndrome |
| ADP | Adenosine diphosphate |
| AKT | Protein kinase B |
| AMP | Adenosine monophosphate |
| AMPK | Activated protein kinase |
| ATP | Adenosine triphosphate |
| BRCA | Breast cancer |
| CI | Confidential interval |
| CSCs | Cancer stem cells |
| DNA | Deoxyribonucleic acid |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial–mesenchymal transition |
| ERK | Extracellular signal-regulated kinases |
| HAART | Highly active anti-retroviral therapy |
| HGSC | High-grade serous carcinoma |
| HIV | Human immunodeficiency virus |
| HMGCR | 3-Hydroxy-3-methylglutaryl-coenzyme A reductase |
| HR | Hazard ratio |
| iPARPs | PARP inhibitors |
| KPNB1 | Karyopherin subunit beta 1 |
| MDR | Multidrug resistance proteins |
| mTOR | Mammalian target of rapamycin |
| NF-κB | Nuclear factor kappa B |
| OC | Ovarian cancer |
| OD | Odds ratio |
| OS | Overall survival |
| P53/TP53 | Tumour protein |
| PAK-1 | P21–activated kinase |
| PARP | Poly (ADP-ribose) polymerase |
| PDX | Patient derived xenograft |
| PFS | Progression-free survival |
| PI3K | Phosphatidylinositol 3-kinases |
| SMAD2/3 | Mothers against decapentaplegic homolog 2/3 |
| RAS | Rat sarcoma virus |
| ROS | Reactive oxygen species |
| RR | Relative risk |
| TGF-β | Transforming growth factor beta |
| VEGF | Vascular endothelial growth factor |
| VEGFR2 | Vascular endothelial growth factor receptor 2 |
| Wnt/TCF | Wnt/T-cell factor |
| YAP1 | Yes-associated protein 1 |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: Globocan sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferlay, J.C.M.; Soerjomataram, I.; Dyba, T.; Randi, G.; Bettio, M.; Gavin, A.; Visser, O.; Bray, F. Cancer incidence and mortality patterns in europe: Estimates for 40 countries and 25 major cancers in 2018. Eur. J. Cancer Clin. Oncol. 2018, 103, 356–387. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Ueda, Y.; Naka, T.; Enomoto, T. Therapeutic strategies in epithelial ovarian cancer. J. Exp. Clin. Cancer Res. 2012, 31, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, W.F.; Huang, C.Y.; Chang, M.C.; Hu, Y.H.; Chiang, Y.C.; Chen, Y.L.; Hsieh, C.Y.; Chen, C.A. High mesothelin correlates with chemoresistance and poor survival in epithelial ovarian carcinoma. Br. J. Cancer 2009, 100, 1144–1153. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, C.T.; Fuller, A.F. Intensive surgical and chemotherapeutic management of advanced ovarian cancer. Surg. Clin. N. Am. 1978, 58, 131–142. [Google Scholar] [CrossRef]
- Lheureux, S.; Gourley, C.; Vergote, I.; Oza, A.M. Epithelial ovarian cancer. Lancet 2019, 393, 1240–1253. [Google Scholar] [CrossRef] [Green Version]
- Bowtell, D.D.; Bohm, S.; Ahmed, A.A.; Aspuria, P.J.; Bast, R.C., Jr.; Beral, V.; Berek, J.S.; Birrer, M.J.; Blagden, S.; Bookman, M.A.; et al. Rethinking ovarian cancer ii: Reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 2015, 15, 668–679. [Google Scholar] [CrossRef]
- Hille, S.; Rein, D.T.; Riffelmann, M.; Neumann, R.; Sartorius, J.; Pfutzner, A.; Kurbacher, C.M.; Schondorf, T.; Breidenbach, M. Anticancer drugs induce mdr1 gene expression in recurrent ovarian cancer. Anticancer Drugs 2006, 17, 1041–1044. [Google Scholar] [CrossRef]
- Howlader, N.N.A.; Krapcho, M.; Miller, D.; Bishop, K.; Kosary, C.L.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. (Eds.) Seer Cancer Statistics Review, 1975–2014, National Cancer Institute. Bethesda, md. Available online: Https://seer.Cancer.Gov/csr/1975_2014/ (accessed on 1 August 2020).
- Weidle, U.H.; Birzele, F.; Kollmorgen, G.; Rueger, R. Mechanisms and targets involved in dissemination of ovarian cancer. Cancer Genom. Proteom. 2016, 13, 407–423. [Google Scholar] [CrossRef] [Green Version]
- Aghajanian, C.; Blank, S.V.; Goff, B.A.; Judson, P.L.; Teneriello, M.G.; Husain, A.; Sovak, M.A.; Yi, J.; Nycum, L.R. Oceans: A randomized, double-blind, placebo-controlled phase iii trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J. Clin. Oncol. 2012, 30, 2039–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, R.L.; Brady, M.F.; Herzog, T.J.; Sabbatini, P.; Armstrong, D.K.; Walker, J.L.; Kim, B.G.; Fujiwara, K.; Tewari, K.S.; O’Malley, D.M.; et al. Bevacizumab and paclitaxel-carboplatin chemotherapy and secondary cytoreduction in recurrent, platinum-sensitive ovarian cancer (nrg oncology/gynecologic oncology group study gog-0213): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 779–791. [Google Scholar] [CrossRef] [Green Version]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ariel3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef] [Green Version]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a brca1/2 mutation (solo2/engot-ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef] [Green Version]
- Armando, R.G.; Mengual Gomez, D.L.; Gomez, D.E. New drugs are not enoughdrug repositioning in oncology: An update. Int. J. Oncol. 2020, 56, 651–684. [Google Scholar]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, F.; Sukhatme, V.P.; Bouche, G. Drug repurposing in oncology--patient and health systems opportunities. Nat. Rev. Clin. Oncol. 2015, 12, 732–742. [Google Scholar] [CrossRef]
- Nowak-Sliwinska, P.; Scapozza, L.; Ruiz, I.A.A. Drug repurposing in oncology: Compounds, pathways, phenotypes and computational approaches for colorectal cancer. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 434–454. [Google Scholar] [CrossRef]
- Gunjan, S.; Sharma, T.; Yadav, K.; Chauhan, B.S.; Singh, S.K.; Siddiqi, M.I.; Tripathi, R. Artemisinin derivatives and synthetic trioxane trigger apoptotic cell death in asexual stages of plasmodium. Front. Cell Infect. Microbiol. 2018, 8, 256. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Kaitin, K.I.; DiMasi, J.A. Pharmaceutical innovation in the 21st century: New drug approvals in the first decade, 2000–2009. Clin. Pharmacol. Ther. 2011, 89, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Schachter, M. Chemical, pharmacokinetic and pharmacodynamic properties of statins: An update. Fundam. Clin. Pharmacol. 2005, 19, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Padhy, B.M.; Gupta, Y.K. Drug repositioning: Re-investigating existing drugs for new therapeutic indications. J. Postgrad. Med. 2011, 57, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Akinwunmi, B.; Vitonis, A.F.; Titus, L.; Terry, K.L.; Cramer, D.W. Statin therapy and association with ovarian cancer risk in the new england case control (nec) study. Int. J. Cancer 2019, 144, 991–1000. [Google Scholar] [CrossRef]
- Davies, J.T.; Delfino, S.F.; Feinberg, C.E.; Johnson, M.F.; Nappi, V.L.; Olinger, J.T.; Schwab, A.P.; Swanson, H.I. Current and emerging uses of statins in clinical therapeutics: A review. Lipid Insights 2016, 9, 13–29. [Google Scholar] [CrossRef] [Green Version]
- Knickelbine, T.; Lui, M.; Garberich, R.; Miedema, M.D.; Strauss, C.; VanWormer, J.J. Familial hypercholesterolemia in a large ambulatory population: Statin use, optimal treatment, and identification for advanced medical therapies. J. Clin. Lipidol. 2016, 10, 1182–1187. [Google Scholar] [CrossRef]
- Pletcher, M.J.; Pignone, M.; Jarmul, J.A.; Moran, A.E.; Vittinghoff, E.; Newman, T. Population impact & efficiency of benefit-targeted versus risk-targeted statin prescribing for primary prevention of cardiovascular disease. J. Am. Heart Assoc. 2017, 6, e004316. [Google Scholar]
- Fernandez-Sauze, S.; Grall, D.; Cseh, B.; Van Obberghen-Schilling, E. Regulation of fibronectin matrix assembly and capillary morphogenesis in endothelial cells by rho family gtpases. Exp. Cell Res. 2009, 315, 2092–2104. [Google Scholar] [CrossRef]
- Mizuno, Y.; Jacob, R.F.; Mason, R.P. Inflammation and the development of atherosclerosis. J. Atheroscler. Thromb. 2011, 18, 351–358. [Google Scholar] [CrossRef] [Green Version]
- Spuul, P.; Ciufici, P.; Veillat, V.; Leclercq, A.; Daubon, T.; Kramer, I.J.; Genot, E. Importance of rhogtpases in formation, characteristics, and functions of invadosomes. Small GTPases 2014, 5, e28195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeybek, B.; Costantine, M.; Kilic, G.S.; Borahay, M.A. Therapeutic roles of statins in gynecology and obstetrics: The current evidence. Reprod. Sci. 2018, 25, 802–817. [Google Scholar] [CrossRef] [PubMed]
- Clendening, J.W.; Pandyra, A.; Boutros, P.C.; El Ghamrasni, S.; Khosravi, F.; Trentin, G.A.; Martirosyan, A.; Hakem, A.; Hakem, R.; Jurisica, I.; et al. Dysregulation of the mevalonate pathway promotes transformation. Proc. Natl. Acad. Sci. USA 2010, 107, 15051–15056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martirosyan, A.; Clendening, J.W.; Goard, C.A.; Penn, L.Z. Lovastatin induces apoptosis of ovarian cancer cells and synergizes with doxorubicin: Potential therapeutic relevance. BMC Cancer 2010, 10, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, E.; Nandi, M.; Wilkinson, L.L.; Arrowsmith, D.M.; Curtis, A.D.; Richardson, A. Preclinical evaluation of statins as a treatment for ovarian cancer. Gynecol. Oncol. 2013, 129, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.W.; Dimitroulakos, J.; Minden, M.D.; Penn, L.Z. Hmg-coa reductase inhibitors and the malignant cell: The statin family of drugs as triggers of tumor-specific apoptosis. Leukemia 2002, 16, 508–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, H.M.; Fang, Z.; Sun, W.; Clark, L.H.; Stine, J.E.; Tran, A.Q.; Sullivan, S.A.; Gilliam, T.P.; Zhou, C.; Bae-Jump, V.L. Atorvastatin exhibits anti-tumorigenic and anti-metastatic effects in ovarian cancer in vitro. Am. J. Cancer Res. 2017, 7, 2478–2490. [Google Scholar]
- Pich, C.; Teiti, I.; Rochaix, P.; Mariame, B.; Couderc, B.; Favre, G.; Tilkin-Mariame, A.F. Statins reduce melanoma development and metastasis through mica overexpression. Front. Immunol. 2013, 4, 62. [Google Scholar] [CrossRef] [Green Version]
- Kidera, Y.; Tsubaki, M.; Yamazoe, Y.; Shoji, K.; Nakamura, H.; Ogaki, M.; Satou, T.; Itoh, T.; Isozaki, M.; Kaneko, J.; et al. Reduction of lung metastasis, cell invasion, and adhesion in mouse melanoma by statin-induced blockade of the rho/rho-associated coiled-coil-containing protein kinase pathway. J. Exp. Clin. Cancer Res. 2010, 29, 127. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Wang, Z.; Li, Y.; Li, W.; Chen, Y. Simvastatin prevents proliferation and bone metastases of lung adenocarcinoma in vitro and in vivo. Neoplasma 2013, 60, 240–246. [Google Scholar] [CrossRef]
- Yu, X.; Luo, Y.; Zhou, Y.; Zhang, Q.; Wang, J.; Wei, N.; Mi, M.; Zhu, J.; Wang, B.; Chang, H.; et al. Brca1 overexpression sensitizes cancer cells to lovastatin via regulation of cyclin d1-cdk4-p21waf1/cip1 pathway: Analyses using a breast cancer cell line and tumoral xenograft model. Int. J. Oncol. 2008, 33, 555–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Z.; Fan-Minogue, H.; Bellovin, D.I.; Yevtodiyenko, A.; Arzeno, J.; Yang, Q.; Gambhir, S.S.; Felsher, D.W. Myc phosphorylation, activation, and tumorigenic potential in hepatocellular carcinoma are regulated by hmg-coa reductase. Cancer Res. 2011, 71, 2286–2297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sassano, A.; Platanias, L.C. Statins in tumor suppression. Cancer Lett. 2008, 260, 11–19. [Google Scholar] [CrossRef]
- Greenaway, J.B.; Virtanen, C.; Osz, K.; Revay, T.; Hardy, D.; Shepherd, T.; DiMattia, G.; Petrik, J. Ovarian tumour growth is characterized by mevalonate pathway gene signature in an orthotopic, syngeneic model of epithelial ovarian cancer. Oncotarget 2016, 7, 47343–47365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, Y.; Kashima, H.; Wu, R.C.; Jung, J.G.; Kuan, J.C.; Gu, J.; Xuan, J.; Sokoll, L.; Visvanathan, K.; Shih Ie, M.; et al. Mevalonate pathway antagonist suppresses formation of serous tubal intraepithelial carcinoma and ovarian carcinoma in mouse models. Clin. Cancer Res. 2015, 21, 4652–4662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stine, J.E.; Guo, H.; Sheng, X.; Han, X.; Schointuch, M.N.; Gilliam, T.P.; Gehrig, P.A.; Zhou, C.; Bae-Jump, V.L. The hmg-coa reductase inhibitor, simvastatin, exhibits anti-metastatic and anti-tumorigenic effects in ovarian cancer. Oncotarget 2016, 7, 946–960. [Google Scholar] [CrossRef]
- Cuello, F.M.; Kato, S.C.; Díaz, S.D.; Owen, G. Effects of statins in cancer. Rev. Med. Chil. 2013, 141, 227–236. [Google Scholar]
- Laezza, C.; Malfitano, A.M.; Proto, M.C.; Esposito, I.; Gazzerro, P.; Formisano, P.; Pisanti, S.; Santoro, A.; Caruso, M.G.; Bifulco, M. Inhibition of 3-hydroxy-3-methylglutaryl-coenzyme a reductase activity and of ras farnesylation mediate antitumor effects of anandamide in human breast cancer cells. Endocr. Relat. Cancer 2010, 17, 495–503. [Google Scholar] [CrossRef]
- Horiuchi, A.; Kikuchi, N.; Osada, R.; Wang, C.; Hayashi, A.; Nikaido, T.; Konishi, I. Overexpression of rhoa enhances peritoneal dissemination: Rhoa suppression with lovastatin may be useful for ovarian cancer. Cancer Sci. 2008, 99, 2532–2539. [Google Scholar] [CrossRef]
- Liu, H.; Liang, S.L.; Kumar, S.; Weyman, C.M.; Liu, W.; Zhou, A. Statins induce apoptosis in ovarian cancer cells through activation of jnk and enhancement of bim expression. Cancer Chemother. Pharmacol. 2009, 63, 997–1005. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, M.; Suzuki, T.; Suzuki, M.; Tanaka, R.; Ito, E.; Saito, T. Statin-mediated reduction of osteopontin expression induces apoptosis and cell growth arrest in ovarian clear cell carcinoma. Oncol. Rep. 2011, 25, 41–47. [Google Scholar] [CrossRef] [PubMed]
- de Wolf, E.; Abdullah, M.I.; Jones, S.M.; Menezes, K.; Moss, D.M.; Drijfhout, F.P.; Hart, S.R.; Hoskins, C.; Stronach, E.A.; Richardson, A. Dietary geranylgeraniol can limit the activity of pitavastatin as a potential treatment for drug-resistant ovarian cancer. Sci. Rep. 2017, 7, 5410. [Google Scholar] [CrossRef] [PubMed]
- Bischof, K.; Knappskog, S.; Hjelle, S.M.; Stefansson, I.; Woie, K.; Salvesen, H.B.; Gjertsen, B.T.; Bjorge, L. Influence of p53 isoform expression on survival in high-grade serous ovarian cancers. Sci. Rep. 2019, 9, 5244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef]
- Brennan, D.J.; Brandstedt, J.; Rexhepaj, E.; Foley, M.; Ponten, F.; Uhlen, M.; Gallagher, W.M.; O’Connor, D.P.; O’Herlihy, C.; Jirstrom, K. Tumour-specific hmg-coar is an independent predictor of recurrence free survival in epithelial ovarian cancer. BMC Cancer 2010, 10, 125. [Google Scholar] [CrossRef] [Green Version]
- Xie, W.; Ning, L.; Huang, Y.; Liu, Y.; Zhang, W.; Hu, Y.; Lang, J.; Yang, J. Statin use and survival outcomes in endocrine-related gynecologic cancers: A systematic review and meta-analysis. Oncotarget 2017, 8, 41508–41517. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhou, J. Impact of postdiagnostic statin use on ovarian cancer mortality: A systematic review and meta-analysis of observational studies. Br. J. Clin. Pharmacol. 2018, 84, 1109–1120. [Google Scholar] [CrossRef] [Green Version]
- Urpilainen, E.; Marttila, M.; Hautakoski, A.; Arffman, M.; Sund, R.; Ilanne-Parikka, P.; Arima, R.; Kangaskokko, J.; Puistola, U.; Laara, E.; et al. The role of metformin and statins in the incidence of epithelial ovarian cancer in type 2 diabetes: A cohort and nested case-control study. BJOG 2018, 125, 1001–1008. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, S.F.; Nordestgaard, B.G.; Bojesen, S.E. Statin use and reduced cancer-related mortality. N. Engl. J. Med. 2012, 367, 1792–1802. [Google Scholar] [CrossRef] [Green Version]
- Couttenier, A.; Lacroix, O.; Vaes, E.; Cardwell, C.R.; De Schutter, H.; Robert, A. Statin use is associated with improved survival in ovarian cancer: A retrospective population-based study. PLoS ONE 2017, 12, e0189233. [Google Scholar] [CrossRef] [PubMed]
- Graaf, M.R.; Beiderbeck, A.B.; Egberts, A.C.; Richel, D.J.; Guchelaar, H.J. The risk of cancer in users of statins. J. Clin. Oncol. 2004, 22, 2388–2394. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.; Murphy, L.; Zgaga, L.; Barron, T.I.; Bennett, K. Pre-diagnostic statin use, lymph node status and mortality in women with stages i-iii breast cancer. Br. J. Cancer 2017, 117, 588–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardwell, C.R.; Hicks, B.M.; Hughes, C.; Murray, L.J. Statin use after colorectal cancer diagnosis and survival: A population-based cohort study. J. Clin. Oncol. 2014, 32, 3177–3183. [Google Scholar] [CrossRef]
- Yu, O.; Eberg, M.; Benayoun, S.; Aprikian, A.; Batist, G.; Suissa, S.; Azoulay, L. Use of statins and the risk of death in patients with prostate cancer. J. Clin. Oncol. 2014, 32, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Vogel, T.J.; Goodman, M.T.; Li, A.J.; Jeon, C.Y. Statin treatment is associated with survival in a nationally representative population of elderly women with epithelial ovarian cancer. Gynecol. Oncol. 2017, 146, 340–345. [Google Scholar] [CrossRef]
- Knox, J.J.; Siu, L.L.; Chen, E.; Dimitroulakos, J.; Kamel-Reid, S.; Moore, M.J.; Chin, S.; Irish, J.; LaFramboise, S.; Oza, A.M. A phase i trial of prolonged administration of lovastatin in patients with recurrent or metastatic squamous cell carcinoma of the head and neck or of the cervix. Eur. J. Cancer 2005, 41, 523–530. [Google Scholar] [CrossRef]
- Kornblau, S.M.; Banker, D.E.; Stirewalt, D.; Shen, D.; Lemker, E.; Verstovsek, S.; Estrov, Z.; Faderl, S.; Cortes, J.; Beran, M.; et al. Blockade of adaptive defensive changes in cholesterol uptake and synthesis in aml by the addition of pravastatin to idarubicin + high-dose ara-c: A phase 1 study. Blood 2007, 109, 2999–3006. [Google Scholar] [CrossRef]
- Minden, M.D.; Dimitroulakos, J.; Nohynek, D.; Penn, L.Z. Lovastatin induced control of blast cell growth in an elderly patient with acute myeloblastic leukemia. Leuk. Lymphoma 2001, 40, 659–662. [Google Scholar] [CrossRef]
- Schmidmaier, R.; Baumann, P.; Bumeder, I.; Meinhardt, G.; Straka, C.; Emmerich, B. First clinical experience with simvastatin to overcome drug resistance in refractory multiple myeloma. Eur. J. Haematol. 2007, 79, 240–243. [Google Scholar] [CrossRef]
- Van der Spek, E.; Bloem, A.C.; van de Donk, N.W.; Bogers, L.H.; van der Griend, R.; Kramer, M.H.; de Weerdt, O.; Wittebol, S.; Lokhorst, H.M. Dose-finding study of high-dose simvastatin combined with standard chemotherapy in patients with relapsed or refractory myeloma or lymphoma. Haematologica 2006, 91, 542–545. [Google Scholar] [PubMed]
- Cusi, K.; Consoli, A.; DeFronzo, R.A. Metabolic effects of metformin on glucose and lactate metabolism in noninsulin-dependent diabetes mellitus. J. Clin. Endocrinol. Metab. 1996, 81, 4059–4067. [Google Scholar] [PubMed] [Green Version]
- Mues, C.; Zhou, J.; Manolopoulos, K.N.; Korsten, P.; Schmoll, D.; Klotz, L.O.; Bornstein, S.R.; Klein, H.H.; Barthel, A. Regulation of glucose-6-phosphatase gene expression by insulin and metformin. Horm. Metab. Res. 2009, 41, 730–735. [Google Scholar] [CrossRef]
- Stephenne, X.; Foretz, M.; Taleux, N.; van der Zon, G.C.; Sokal, E.; Hue, L.; Viollet, B.; Guigas, B. Metformin activates amp-activated protein kinase in primary human hepatocytes by decreasing cellular energy status. Diabetologia 2011, 54, 3101–3110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viollet, B.; Guigas, B.; Sanz Garcia, N.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and molecular mechanisms of metformin: An overview. Clin. Sci. 2012, 122, 253–270. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.O.; Lee, S.K.; Kim, J.H.; Kim, N.; You, G.Y.; Moon, J.W.; Kim, S.J.; Park, S.H.; Kim, H.S. Metformin regulates glucose transporter 4 (glut4) translocation through amp-activated protein kinase (ampk)-mediated cbl/cap signaling in 3t3-l1 preadipocyte cells. J. Biol. Chem. 2012, 287, 44121–44129. [Google Scholar] [CrossRef] [Green Version]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex i of cancer cells to reduce tumorigenesis. Elife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Pernicova, I.; Korbonits, M. Metformin--mode of action and clinical implications for diabetes and cancer. Nat. Rev. Endocrinol. 2014, 10, 143–156. [Google Scholar] [CrossRef]
- Pollak, M. The insulin and insulin-like growth factor receptor family in neoplasia: An update. Nat. Rev. Cancer 2012, 12, 159–169. [Google Scholar] [CrossRef]
- Pollak, M.N. Investigating metformin for cancer prevention and treatment: The end of the beginning. Cancer Discov. 2012, 2, 778–790. [Google Scholar] [CrossRef] [Green Version]
- Moiseeva, O.; Deschenes-Simard, X.; Pollak, M.; Ferbeyre, G. Metformin, aging and cancer. Aging Albany NY 2013, 5, 330–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearce, E.L.; Walsh, M.C.; Cejas, P.J.; Harms, G.M.; Shen, H.; Wang, L.S.; Jones, R.G.; Choi, Y. Enhancing cd8 t-cell memory by modulating fatty acid metabolism. Nature 2009, 460, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Bardeesy, N.; Manning, B.D.; Lopez, L.; Kosmatka, M.; DePinho, R.A.; Cantley, L.C. The lkb1 tumor suppressor negatively regulates mtor signaling. Cancer Cell 2004, 6, 91–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakikhani, M.; Dowling, R.J.; Sonenberg, N.; Pollak, M.N. The effects of adiponectin and metformin on prostate and colon neoplasia involve activation of amp-activated protein kinase. Cancer Prev. Res. 2008, 1, 369–375. [Google Scholar] [CrossRef] [Green Version]
- Lengyel, E.; Litchfield, L.M.; Mitra, A.K.; Nieman, K.M.; Mukherjee, A.; Zhang, Y.; Johnson, A.; Bradaric, M.; Lee, W.; Romero, I.L. Metformin inhibits ovarian cancer growth and increases sensitivity to paclitaxel in mouse models. Am. J. Obstet. Gynecol. 2015, 212, 479.e1–479.e10. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Romero, I.L.; Litchfield, L.M.; Lengyel, E.; Locasale, J.W. Metformin targets central carbon metabolism and reveals mitochondrial requirements in human cancers. Cell Metab. 2016, 24, 728–739. [Google Scholar] [CrossRef]
- Gui, D.Y.; Sullivan, L.B.; Luengo, A.; Hosios, A.M.; Bush, L.N.; Gitego, N.; Davidson, S.M.; Freinkman, E.; Thomas, C.J.; Vander Heiden, M.G. Environment dictates dependence on mitochondrial complex i for nad+ and aspartate production and determines cancer cell sensitivity to metformin. Cell Metab. 2016, 24, 716–727. [Google Scholar] [CrossRef] [Green Version]
- Kurelac, I.; Umesh Ganesh, N.; Iorio, M.; Porcelli, A.M.; Gasparre, G. The multifaceted effects of metformin on tumor microenvironment. Semin. Cell Dev. Biol. 2020, 98, 90–97. [Google Scholar] [CrossRef]
- Li, X.; Li, B.; Ni, Z.; Zhou, P.; Wang, B.; He, J.; Xiong, H.; Yang, F.; Wu, Y.; Lyu, X.; et al. Metformin synergizes with bcl-xl/bcl-2 inhibitor abt-263 to induce apoptosis specifically in p53-defective cancer cells. Mol. Cancer Ther. 2017, 16, 1806–1818. [Google Scholar] [CrossRef] [Green Version]
- Galdieri, L.; Gatla, H.; Vancurova, I.; Vancura, A. Activation of amp-activated protein kinase by metformin induces protein acetylation in prostate and ovarian cancer cells. J. Biol. Chem. 2016, 291, 25154–25166. [Google Scholar] [CrossRef] [Green Version]
- Rattan, R.; Giri, S.; Hartmann, L.C.; Shridhar, V. Metformin attenuates ovarian cancer cell growth in an amp-kinase dispensable manner. J. Cell. Mol. Med. 2011, 15, 166–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Peng, Z.; Shi, M.; Ji, M.; Guo, H.; Shi, H. Metformin combined with p38 mapk inhibitor improves cisplatin sensitivity in cisplatinresistant ovarian cancer. Mol. Med. Rep. 2014, 10, 2346–2350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Zhang, P.; Wang, H.; Hou, D.; Li, W.; Xiao, G.; Li, C. Inhibitory effects of metformin at low concentration on epithelial-mesenchymal transition of cd44(+)cd117(+) ovarian cancer stem cells. Stem Cell. Res. Ther. 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Zhao, N.; Li, D.; Zou, G.; Chen, Y. Metformin improves the sensitivity of ovarian cancer cells to chemotherapeutic agents. Oncol. Lett. 2019, 18, 2404–2411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Shi, H.R.; Ren, F.; Wang, J.L.; Wu, Q.H.; Li, X.; Zhang, R.T. Inhibition of the igf signaling pathway reverses cisplatin resistance in ovarian cancer cells. BMC Cancer 2017, 17, 851. [Google Scholar] [CrossRef]
- Gotlieb, W.H.; Saumet, J.; Beauchamp, M.C.; Gu, J.; Lau, S.; Pollak, M.N.; Bruchim, I. In vitro metformin anti-neoplastic activity in epithelial ovarian cancer. Gynecol. Oncol. 2008, 110, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Feng, Y.; Liu, H.; Wu, J.; Tang, Y.; Wang, Q. Real-time assessment of platinum sensitivity of primary culture from a patient with ovarian cancer with extensive metastasis and the platinum sensitivity enhancing effect by metformin. Oncol. Lett. 2018, 16, 4253–4262. [Google Scholar] [CrossRef] [Green Version]
- Rattan, R.; Graham, R.P.; Maguire, J.L.; Giri, S.; Shridhar, V. Metformin suppresses ovarian cancer growth and metastasis with enhancement of cisplatin cytotoxicity in vivo. Neoplasia 2011, 13, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Li, S.; Sheng, L.; Zhu, J.; Gu, L.; Shen, H.; La, D.; Hambly, B.D.; Bao, S.; Di, W. Metformin inhibits the development and metastasis of ovarian cancer. Oncol. Rep. 2012, 28, 903–908. [Google Scholar] [CrossRef] [Green Version]
- Yasmeen, A.; Beauchamp, M.C.; Piura, E.; Segal, E.; Pollak, M.; Gotlieb, W.H. Induction of apoptosis by metformin in epithelial ovarian cancer: Involvement of the bcl-2 family proteins. Gynecol. Oncol. 2011, 121, 492–498. [Google Scholar] [CrossRef]
- Patel, S.; Kumar, L.; Singh, N. Metformin and epithelial ovarian cancer therapeutics. Cell. Oncol. 2015, 38, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Shank, J.J.; Yang, K.; Ghannam, J.; Cabrera, L.; Johnston, C.J.; Reynolds, R.K.; Buckanovich, R.J. Metformin targets ovarian cancer stem cells in vitro and in vivo. Gynecol. Oncol. 2012, 127, 390–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, J.H.; Jin, Z.J.; Liu, X.J.; Hu, D.; Wang, J.; Luo, Y.; Li, L.L. Metformin in combination with cisplatin inhibits cell viability and induces apoptosis of human ovarian cancer cells by inactivating erk 1/2. Oncol. Lett. 2017, 14, 7557–7564. [Google Scholar] [CrossRef] [Green Version]
- Hijaz, M.; Chhina, J.; Mert, I.; Taylor, M.; Dar, S.; Al-Wahab, Z.; Ali-Fehmi, R.; Buekers, T.; Munkarah, A.R.; Rattan, R. Preclinical evaluation of olaparib and metformin combination in brca1 wildtype ovarian cancer. Gynecol. Oncol. 2016, 142, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Franciosi, M.; Lucisano, G.; Lapice, E.; Strippoli, G.F.; Pellegrini, F.; Nicolucci, A. Metformin therapy and risk of cancer in patients with type 2 diabetes: Systematic review. PLoS ONE 2013, 8, e71583. [Google Scholar] [CrossRef]
- Noto, H.; Goto, A.; Tsujimoto, T.; Noda, M. Cancer risk in diabetic patients treated with metformin: A systematic review and meta-analysis. PLoS ONE 2012, 7, e33411. [Google Scholar] [CrossRef]
- Chu, D.; Wu, J.; Wang, K.; Zhao, M.; Wang, C.; Li, L.; Guo, R. Effect of metformin use on the risk and prognosis of endometrial cancer: A systematic review and meta-analysis. BMC Cancer 2018, 18, 438. [Google Scholar] [CrossRef] [Green Version]
- Hanna, R.K.; Zhou, C.; Malloy, K.M.; Sun, L.; Zhong, Y.; Gehrig, P.A.; Bae-Jump, V.L. Metformin potentiates the effects of paclitaxel in endometrial cancer cells through inhibition of cell proliferation and modulation of the mtor pathway. Gynecol. Oncol. 2012, 125, 458–469. [Google Scholar] [CrossRef] [Green Version]
- Bodmer, M.; Becker, C.; Meier, C.; Jick, S.S.; Meier, C.R. Use of metformin and the risk of ovarian cancer: A case-control analysis. Gynecol. Oncol. 2011, 123, 200–204. [Google Scholar] [CrossRef]
- Dilokthornsakul, P.; Chaiyakunapruk, N.; Termrungruanglert, W.; Pratoomsoot, C.; Saokaew, S.; Sruamsiri, R. The effects of metformin on ovarian cancer: A systematic review. Int. J. Gynecol. Cancer 2013, 23, 1544–1551. [Google Scholar] [CrossRef]
- Romero, I.L.; McCormick, A.; McEwen, K.A.; Park, S.; Karrison, T.; Yamada, S.D.; Pannain, S.; Lengyel, E. Relationship of type ii diabetes and metformin use to ovarian cancer progression, survival, and chemosensitivity. Obstet. Gynecol. 2012, 119, 61–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Liu, B.; Wang, H.; Zhang, T.; Yang, L. Association of metformin use with ovarian cancer incidence and prognosis: A systematic review and meta-analysis. Int. J. Gynecol. Cancer 2019, 29, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.B.; Lei, K.J.; Liu, J.P.; Jia, Y.M. Continuous use of metformin can improve survival in type 2 diabetic patients with ovarian cancer: A retrospective study. Medicine 2017, 96, e7605. [Google Scholar] [CrossRef]
- Kumar, S.; Meuter, A.; Thapa, P.; Langstraat, C.; Giri, S.; Chien, J.; Rattan, R.; Cliby, W.; Shridhar, V. Metformin intake is associated with better survival in ovarian cancer: A case-control study. Cancer 2013, 119, 555–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.J.; Li, S. The prognostic value of metformin for cancer patients with concurrent diabetes: A systematic review and meta-analysis. Diabetes Obes. Metab. 2014, 16, 707–710. [Google Scholar] [CrossRef]
- Gong, T.T.; Wu, Q.J.; Lin, B.; Ruan, S.K.; Kushima, M.; Takimoto, M. Observational studies on the association between post-diagnostic metformin use and survival in ovarian cancer: A systematic review and meta-analysis. Front. Oncol. 2019, 9, 458. [Google Scholar] [CrossRef]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [Green Version]
- Gandini, S.; Puntoni, M.; Heckman-Stoddard, B.M.; Dunn, B.K.; Ford, L.; DeCensi, A.; Szabo, E. Metformin and cancer risk and mortality: A systematic review and meta-analysis taking into account biases and confounders. Cancer Prev. Res. 2014, 7, 867–885. [Google Scholar] [CrossRef] [Green Version]
- Saraei, P.; Asadi, I.; Kakar, M.A.; Moradi-Kor, N. The beneficial effects of metformin on cancer prevention and therapy: A comprehensive review of recent advances. Cancer Manag. Res. 2019, 11, 3295–3313. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Zhong, X.; Gao, P.; Shi, J.; Wu, Z.; Guo, Z.; Wang, Z.; Song, Y. The potential effect of metformin on cancer: An umbrella review. Front. Endocrinol. 2019, 10, 617. [Google Scholar] [CrossRef]
- Zi, F.; Zi, H.; Li, Y.; He, J.; Shi, Q.; Cai, Z. Metformin and cancer: An existing drug for cancer prevention and therapy. Oncol. Lett. 2018, 15, 683–690. [Google Scholar] [CrossRef] [Green Version]
- Bao, B.; Azmi, A.S.; Ali, S.; Zaiem, F.; Sarkar, F.H. Metformin may function as anti-cancer agent via targeting cancer stem cells: The potential biological significance of tumor-associated mirnas in breast and pancreatic cancers. Ann. Transl. Med. 2014, 2, 59. [Google Scholar] [PubMed]
- Chae, J.W.; Baek, I.H.; Lee, B.Y.; Cho, S.K.; Kwon, K.I. Population pk/pd analysis of metformin using the signal transduction model. Br. J. Clin. Pharmacol. 2012, 74, 815–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shackelford, D.B.; Abt, E.; Gerken, L.; Vasquez, D.S.; Seki, A.; Leblanc, M.; Wei, L.; Fishbein, M.C.; Czernin, J.; Mischel, P.S.; et al. Lkb1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell 2013, 23, 143–158. [Google Scholar] [CrossRef] [Green Version]
- Pierotti, M.A.; Berrino, F.; Gariboldi, M.; Melani, C.; Mogavero, A.; Negri, T.; Pasanisi, P.; Pilotti, S. Targeting metabolism for cancer treatment and prevention: Metformin, an old drug with multi-faceted effects. Oncogene 2013, 32, 1475–1487. [Google Scholar] [CrossRef] [PubMed]
- Eikawa, S.; Nishida, M.; Mizukami, S.; Yamazaki, C.; Nakayama, E.; Udono, H. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc. Natl. Acad. Sci. USA 2015, 112, 1809–1814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Barco, S.; Vazquez-Martin, A.; Cufi, S.; Oliveras-Ferraros, C.; Bosch-Barrera, J.; Joven, J.; Martin-Castillo, B.; Menendez, J.A. Metformin: Multi-faceted protection against cancer. Oncotarget 2011, 2, 896–917. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.R.; Chan, D.K.; Shank, J.J.; Griffith, K.A.; Fan, H.; Szulawski, R.; Yang, K.; Reynolds, R.K.; Johnston, C.; McLean, K.; et al. Phase ii clinical trial of metformin as a cancer stem cell-targeting agent in ovarian cancer. JCI Insight 2020, 5, e133247. [Google Scholar]
- Broekman, K.E.; Hof, M.A.J.; Touw, D.J.; Gietema, J.A.; Nijman, H.W.; Lefrandt, J.D.; Reyners, A.K.L.; Jalving, M. Phase i study of metformin in combination with carboplatin/paclitaxel chemotherapy in patients with advanced epithelial ovarian cancer. Invest. New Drugs 2020, 38, 1454–1462. [Google Scholar] [CrossRef] [Green Version]
- Mystakidou, K.; Katsouda, E.; Stathopoulou, E.; Vlahos, L. Approaches to managing bone metastases from breast cancer: The role of bisphosphonates. Cancer Treat. Rev. 2005, 31, 303–311. [Google Scholar] [CrossRef]
- Russell, R.G.; Rogers, M.J. Bisphosphonates: From the laboratory to the clinic and back again. Bone 1999, 25, 97–106. [Google Scholar] [CrossRef]
- Gronich, N.; Rennert, G. Beyond aspirin-cancer prevention with statins, metformin and bisphosphonates. Nat. Rev. Clin. Oncol. 2013, 10, 625–642. [Google Scholar] [CrossRef] [PubMed]
- Muinelo-Romay, L.; Garcia, D.; Alonso-Alconada, L.; Vieito, M.; Carmona, M.; Martinez, N.; Aguin, S.; Abal, M.; Lopez-Lopez, R. Zoledronic acid as an antimetastatic agent for different human tumor cell lines. Anticancer Res. 2013, 33, 5295–5300. [Google Scholar] [PubMed]
- Yuasa, T.; Kimura, S.; Ashihara, E.; Habuchi, T.; Maekawa, T. Zoledronic acid-a multiplicity of anti-cancer action. Curr. Med. Chem. 2007, 14, 2126–2135. [Google Scholar] [CrossRef] [PubMed]
- Gnant, M.; Mlineritsch, B.; Schippinger, W.; Luschin-Ebengreuth, G.; Postlberger, S.; Menzel, C.; Jakesz, R.; Seifert, M.; Hubalek, M.; Bjelic-Radisic, V.; et al. Endocrine therapy plus zoledronic acid in premenopausal breast cancer. N. Engl. J. Med. 2009, 360, 679–691. [Google Scholar] [CrossRef]
- Coleman, R.E.; Winter, M.C.; Cameron, D.; Bell, R.; Dodwell, D.; Keane, M.M.; Gil, M.; Ritchie, D.; Passos-Coelho, J.L.; Wheatley, D.; et al. The effects of adding zoledronic acid to neoadjuvant chemotherapy on tumour response: Exploratory evidence for direct anti-tumour activity in breast cancer. Br. J. Cancer 2010, 102, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Bosch-Barrera, J.; Merajver, S.D.; Menendez, J.A.; Van Poznak, C. Direct antitumour activity of zoledronic acid: Preclinical and clinical data. Clin. Transl. Oncol. 2011, 13, 148–155. [Google Scholar] [CrossRef]
- Senaratne, S.G.; Colston, K.W. Direct effects of bisphosphonates on breast cancer cells. Breast Cancer Res. 2002, 4, 18–23. [Google Scholar] [CrossRef] [Green Version]
- Dumon, J.C.; Journe, F.; Kheddoumi, N.; Lagneaux, L.; Body, J.J. Cytostatic and apoptotic effects of bisphosphonates on prostate cancer cells. Eur. Urol. 2004, 45, 521–528, discussion 528–529. [Google Scholar] [CrossRef]
- Sawada, K.; Morishige, K.; Tahara, M.; Kawagishi, R.; Ikebuchi, Y.; Tasaka, K.; Murata, Y. Alendronate inhibits lysophosphatidic acid-induced migration of human ovarian cancer cells by attenuating the activation of rho. Cancer Res. 2002, 62, 6015–6020. [Google Scholar]
- Hirata, J.; Kikuchi, Y.; Kudoh, K.; Kita, T.; Seto, H. Inhibitory effects of bisphosphonates on the proliferation of human ovarian cancer cell lines and the mechanism. Med. Chem. 2006, 2, 223–226. [Google Scholar] [PubMed]
- Nagasawa, Y.; Chen, J.; Hashimoto, K. Antiarrhythmic properties of a prior oral loading of amiodarone in in vivo canine coronary ligation/reperfusion-induced arrhythmia model: Comparison with other class iii antiarrhythmic drugs. J. Pharmacol. Sci. 2005, 97, 393–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knight, L.A.; Kurbacher, C.M.; Glaysher, S.; Fernando, A.; Reichelt, R.; Dexel, S.; Reinhold, U.; Cree, I.A. Activity of mevalonate pathway inhibitors against breast and ovarian cancers in the atp-based tumour chemosensitivity assay. BMC Cancer 2009, 9, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karabulut, B.; Karaca, B.; Varol, U.; Muslu, U.; Cakar, B.; Atmaca, H.; Kisim, A.; Uzunoglu, S.; Uslu, R. Enhancing cytotoxic and apoptotic effect in ovcar-3 and mdah-2774 cells with all-trans retinoic acid and zoledronic acid: A paradigm of synergistic molecular targeting treatment for ovarian cancer. J. Exp. Clin. Cancer Res. 2010, 29, 102. [Google Scholar] [CrossRef] [Green Version]
- Atmaca, H.; Gorumlu, G.; Karaca, B.; Degirmenci, M.; Tunali, D.; Cirak, Y.; Purcu, D.U.; Uzunoglu, S.; Karabulut, B.; Sanli, U.A.; et al. Combined gossypol and zoledronic acid treatment results in synergistic induction of cell death and regulates angiogenic molecules in ovarian cancer cells. Eur. Cytokine Netw. 2009, 20, 121–130. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Kashima, H.; Rahmanto, Y.S.; Banno, K.; Yu, Y.; Matoba, Y.; Watanabe, K.; Iijima, M.; Takeda, T.; Kunitomi, H.; et al. Drug repositioning of mevalonate pathway inhibitors as antitumor agents for ovarian cancer. Oncotarget 2017, 8, 72147–72156. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, K.; Morishige, K.; Sawada, K.; Tahara, M.; Kawagishi, R.; Ikebuchi, Y.; Sakata, M.; Tasaka, K.; Murata, Y. Alendronate inhibits intraperitoneal dissemination in in vivo ovarian cancer model. Cancer Res. 2005, 65, 540–545. [Google Scholar]
- Ou, Y.J.; Chiu, H.F.; Wong, Y.H.; Yang, C.C.; Yang, Y.H. Bisphosphonate use and the risk of breast cancer: A meta-analysis of observational studies. Pharmacoepidemiol. Drug Saf. 2017, 26, 1286–1295. [Google Scholar] [CrossRef]
- Ou, Y.J.; Chiu, H.F.; Wong, Y.H.; Yang, Y.H. Bisphosphonate use and the risk of endometrial cancer: A meta-analysis of observational studies. Pharmacoepidemiol. Drug Saf. 2016, 25, 1107–1115. [Google Scholar] [CrossRef]
- Rennert, G.; Rennert, H.S.; Pinchev, M.; Lavie, O. The effect of bisphosphonates on the risk of endometrial and ovarian malignancies. Gynecol. Oncol. 2014, 133, 309–313. [Google Scholar] [CrossRef]
- Gonzalez Canga, A.; Sahagun Prieto, A.M.; Jose Diez Liebana, M.; Martinez, N.F.; Vega, M.S.; Vieitez, J.J. The pharmacokinetics and metabolism of ivermectin in domestic animal species. Vet. J. 2009, 179, 25–37. [Google Scholar] [CrossRef] [PubMed]
- McCavera, S.; Rogers, A.T.; Yates, D.M.; Woods, D.J.; Wolstenholme, A.J. An ivermectin-sensitive glutamate-gated chloride channel from the parasitic nematode haemonchus contortus. Mol. Pharmacol. 2009, 75, 1347–1355. [Google Scholar] [CrossRef] [Green Version]
- Moreno, Y.; Nabhan, J.F.; Solomon, J.; Mackenzie, C.D.; Geary, T.G. Ivermectin disrupts the function of the excretory-secretory apparatus in microfilariae of brugia malayi. Proc. Natl. Acad. Sci. USA 2010, 107, 20120–20125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, W.C. History of avermectin and ivermectin, with notes on the history of other macrocyclic lactone antiparasitic agents. Curr. Pharm. Biotechnol. 2012, 13, 853–865. [Google Scholar] [CrossRef] [PubMed]
- Markowska, A.; Kaysiewicz, J.; Markowska, J.; Huczynski, A. Doxycycline, salinomycin, monensin and ivermectin repositioned as cancer drugs. Bioorg. Med. Chem. Lett. 2019, 29, 1549–1554. [Google Scholar] [CrossRef] [PubMed]
- Drinyaev, V.A.; Mosin, V.A.; Kruglyak, E.B.; Novik, T.S.; Sterlina, T.S.; Ermakova, N.V.; Kublik, L.N.; Levitman, M.; Shaposhnikova, V.V.; Korystov, Y.N. Antitumor effect of avermectins. Eur. J. Pharmacol. 2004, 501, 19–23. [Google Scholar] [CrossRef]
- Hashimoto, H.; Messerli, S.M.; Sudo, T.; Maruta, H. Ivermectin inactivates the kinase pak1 and blocks the pak1-dependent growth of human ovarian cancer and nf2 tumor cell lines. Drug Discov. Ther. 2009, 3, 243–246. [Google Scholar]
- Melotti, A.; Mas, C.; Kuciak, M.; Lorente-Trigos, A.; Borges, I.; Ruiz i Altaba, A. The river blindness drug ivermectin and related macrocyclic lactones inhibit wnt-tcf pathway responses in human cancer. EMBO Mol. Med. 2014, 6, 1263–1278. [Google Scholar] [CrossRef]
- Sharmeen, S.; Skrtic, M.; Sukhai, M.A.; Hurren, R.; Gronda, M.; Wang, X.; Fonseca, S.B.; Sun, H.; Wood, T.E.; Ward, R.; et al. The antiparasitic agent ivermectin induces chloride-dependent membrane hyperpolarization and cell death in leukemia cells. Blood 2010, 116, 3593–3603. [Google Scholar] [CrossRef] [Green Version]
- Dominguez-Gomez, G.; Chavez-Blanco, A.; Medina-Franco, J.L.; Saldivar-Gonzalez, F.; Flores-Torrontegui, Y.; Juarez, M.; Diaz-Chavez, J.; Gonzalez-Fierro, A.; Duenas-Gonzalez, A. Ivermectin as an inhibitor of cancer stemlike cells. Mol. Med. Rep. 2018, 17, 3397–3403. [Google Scholar]
- Didier, A.; Loor, F. The abamectin derivative ivermectin is a potent p-glycoprotein inhibitor. Anticancer Drugs 1996, 7, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Dou, Q.; Chen, H.N.; Wang, K.; Yuan, K.; Lei, Y.; Li, K.; Lan, J.; Chen, Y.; Huang, Z.; Xie, N.; et al. Ivermectin induces cytostatic autophagy by blocking the pak1/akt axis in breast cancer. Cancer Res. 2016, 76, 4457–4469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juarez, M.; Schcolnik-Cabrera, A.; Duenas-Gonzalez, A. The multitargeted drug ivermectin: From an antiparasitic agent to a repositioned cancer drug. Am. J. Cancer Res. 2018, 8, 317–331. [Google Scholar]
- Liu, J.; Liang, H.; Chen, C.; Wang, X.; Qu, F.; Wang, H.; Yang, K.; Wang, Q.; Zhao, N.; Meng, J.; et al. Ivermectin induces autophagy-mediated cell death through the akt/mtor signaling pathway in glioma cells. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef]
- Liu, Y.; Fang, S.; Sun, Q.; Liu, B. Anthelmintic drug ivermectin inhibits angiogenesis, growth and survival of glioblastoma through inducing mitochondrial dysfunction and oxidative stress. Biochem. Biophys. Res. Commun. 2016, 480, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Seth, C.; Mas, C.; Conod, A.; Mueller, J.; Siems, K.; Kuciak, M.; Borges, I.; Ruiz, I.A.A. Long-lasting wnt-tcf response blocking and epigenetic modifying activities of withanolide f in human cancer cells. PLoS ONE 2016, 11, e0168170. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Gao, W.; Dou, Q.; Chen, H.; Li, Q.; Nice, E.C.; Huang, C. Ivermectin induces pak1-mediated cytostatic autophagy in breast cancer. Autophagy 2016, 12, 2498–2499. [Google Scholar] [CrossRef]
- Kwon, Y.J.; Leibovitch, B.A.; Zeng, L.; Mezei, M.; Christova, R.; Yang, S.; Sharma, R.; Aritzia, E.; Bansal, N.; Zhou, M.M.; et al. Selamectin and ivermectin are small molecule inhibitors that interfere with sin3a-pah2 function and exert anti-tumor activity in triple-negative breast cancer. In Proceedings of the 105th Annual Meeting of the American Association for Cancer Research, Atlanta, GA USA, 5–9 April 2014; Volume 74 (Suppl. S19). [Google Scholar]
- Zhu, M.; Li, Y.; Zhou, Z. Antibiotic ivermectin preferentially targets renal cancer through inducing mitochondrial dysfunction and oxidative damage. Biochem. Biophys. Res. Commun. 2017, 492, 373–378. [Google Scholar] [CrossRef]
- Nishio, M.; Sugimachi, K.; Goto, H.; Wang, J.; Morikawa, T.; Miyachi, Y.; Takano, Y.; Hikasa, H.; Itoh, T.; Suzuki, S.O.; et al. Dysregulated yap1/taz and tgf-beta signaling mediate hepatocarcinogenesis in mob1a/1b-deficient mice. Proc. Natl. Acad. Sci. USA 2016, 113, E71–E80. [Google Scholar] [CrossRef] [Green Version]
- Kang, W.; Tong, J.H.; Chan, A.W.; Lee, T.L.; Lung, R.W.; Leung, P.P.; So, K.K.; Wu, K.; Fan, D.; Yu, J.; et al. Yes-associated protein 1 exhibits oncogenic property in gastric cancer and its nuclear accumulation associates with poor prognosis. Clin. Cancer Res. 2011, 17, 2130–2139. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.H.; Kim, Y.K.; Shin, D.H.; Lee, H.J.; Shin, N.; Kim, A.; Lee, J.H.; Choi, K.U.; Kim, J.Y.; Lee, C.H.; et al. Yes associated protein is a poor prognostic factor in well-differentiated lung adenocarcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 15933–15939. [Google Scholar] [PubMed]
- Lee, K.W.; Lee, S.S.; Kim, S.B.; Sohn, B.H.; Lee, H.S.; Jang, H.J.; Park, Y.Y.; Kopetz, S.; Kim, S.S.; Oh, S.C.; et al. Significant association of oncogene yap1 with poor prognosis and cetuximab resistance in colorectal cancer patients. Clin. Cancer Res. 2015, 21, 357–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Li, X.; He, Y.; Li, W.; Wang, Y.; Wang, H.; Jiang, S.; Xin, Y. Yap1 enhances cell proliferation, migration, and invasion of gastric cancer in vitro and in vivo. Oncotarget 2016, 7, 81062–81076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Y.; Chang, T.; Wang, Y.; Liu, Y.; Li, W.; Li, M.; Fan, H.Y. Yap promotes ovarian cancer cell tumorigenesis and is indicative of a poor prognosis for ovarian cancer patients. PLoS ONE 2014, 9, e91770. [Google Scholar] [CrossRef] [PubMed]
- Nambara, S.; Masuda, T.; Nishio, M.; Kuramitsu, S.; Tobo, T.; Ogawa, Y.; Hu, Q.; Iguchi, T.; Kuroda, Y.; Ito, S.; et al. Antitumor effects of the antiparasitic agent ivermectin via inhibition of yes-associated protein 1 expression in gastric cancer. Oncotarget 2017, 8, 107666–107677. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, Y.; Wan, H.; Hu, J. Antibiotic ivermectin selectively induces apoptosis in chronic myeloid leukemia through inducing mitochondrial dysfunction and oxidative stress. Biochem. Biophys. Res. Commun. 2018, 497, 241–247. [Google Scholar] [CrossRef]
- Kodama, M.; Kodama, T.; Newberg, J.Y.; Katayama, H.; Kobayashi, M.; Hanash, S.M.; Yoshihara, K.; Wei, Z.; Tien, J.C.; Rangel, R.; et al. In vivo loss-of-function screens identify kpnb1 as a new druggable oncogene in epithelial ovarian cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E7301–E7310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Qin, T.; Zhu, Z.; Hong, F.; Xu, Y.; Zhang, X.; Xu, X.; Ma, A. Ivermectin augments the in vitro and in vivo efficacy of cisplatin in epithelial ovarian cancer by suppressing akt/mtor signaling. Am. J. Med. Sci. 2020, 359, 123–129. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, P.; Sun, Y.J.; Wu, Y.J. Ivermectin reverses the drug resistance in cancer cells through egfr/erk/akt/nf-kappab pathway. J. Exp. Clin. Cancer Res. 2019, 38, 265. [Google Scholar] [CrossRef]
- Lestner, J.; Hope, W.W. Itraconazole: An update on pharmacology and clinical use for treatment of invasive and allergic fungal infections. Expert Opin. Drug Metab. Toxicol. 2013, 9, 911–926. [Google Scholar] [CrossRef]
- Pandya, N.A.; Atra, A.A.; Riley, U.; Pinkerton, C.R. Role of itraconazole in haematology/oncology. Arch. Dis. Child. 2003, 88, 258–260. [Google Scholar] [CrossRef] [PubMed]
- Pounds, R.; Leonard, S.; Dawson, C.; Kehoe, S. Repurposing itraconazole for the treatment of cancer. Oncol. Lett. 2017, 14, 2587–2597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shim, J.S.; Liu, J.O. Recent advances in drug repositioning for the discovery of new anticancer drugs. Int. J. Biol. Sci. 2014, 10, 654–663. [Google Scholar] [CrossRef] [Green Version]
- Pantziarka, P.; Sukhatme, V.; Bouche, G.; Meheus, L.; Sukhatme, V.P. Repurposing drugs in oncology (redo)-itraconazole as an anti-cancer agent. Ecancermedicalscience 2015, 9, 521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Aftab, B.T.; Tang, J.Y.; Kim, D.; Lee, A.H.; Rezaee, M.; Kim, J.; Chen, B.; King, E.M.; Borodovsky, A.; et al. Itraconazole and arsenic trioxide inhibit hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell 2013, 23, 23–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Tang, J.Y.; Gong, R.; Kim, J.; Lee, J.J.; Clemons, K.V.; Chong, C.R.; Chang, K.S.; Fereshteh, M.; Gardner, D.; et al. Itraconazole, a commonly used antifungal that inhibits hedgehog pathway activity and cancer growth. Cancer Cell 2010, 17, 388–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Li, J.; Zhang, T.; Zou, L.; Chen, Y.; Wang, K.; Lei, Y.; Yuan, K.; Li, Y.; Lan, J.; et al. Itraconazole suppresses the growth of glioblastoma through induction of autophagy: Involvement of abnormal cholesterol trafficking. Autophagy 2014, 10, 1241–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Head, S.A.; Shi, W.Q.; Yang, E.J.; Nacev, B.A.; Hong, S.Y.; Pasunooti, K.K.; Li, R.J.; Shim, J.S.; Liu, J.O. Simultaneous targeting of npc1 and vdac1 by itraconazole leads to synergistic inhibition of mtor signaling and angiogenesis. ACS Chem. Biol. 2017, 12, 174–182. [Google Scholar] [CrossRef]
- Hu, Q.; Hou, Y.C.; Huang, J.; Fang, J.Y.; Xiong, H. Itraconazole induces apoptosis and cell cycle arrest via inhibiting hedgehog signaling in gastric cancer cells. J. Exp. Clin. Cancer Res. 2017, 36, 50. [Google Scholar] [CrossRef] [Green Version]
- Liang, G.; Liu, M.; Wang, Q.; Shen, Y.; Mei, H.; Li, D.; Liu, W. Itraconazole exerts its anti-melanoma effect by suppressing hedgehog, wnt, and pi3k/mtor signaling pathways. Oncotarget 2017, 8, 28510–28525. [Google Scholar] [CrossRef] [Green Version]
- Tsubamoto, H.; Inoue, K.; Sakata, K.; Ueda, T.; Takeyama, R.; Shibahara, H.; Sonoda, T. Itraconazole inhibits akt/mtor signaling and proliferation in endometrial cancer cells. Anticancer Res. 2017, 37, 515–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, T.; Tsubamoto, H.; Inoue, K.; Sakata, K.; Shibahara, H.; Sonoda, T. Itraconazole modulates hedgehog, wnt/beta-catenin, as well as akt signalling, and inhibits proliferation of cervical cancer cells. Anticancer Res. 2017, 37, 3521–3526. [Google Scholar] [PubMed] [Green Version]
- Chong, C.R.; Xu, J.; Lu, J.; Bhat, S.; Sullivan, D.J., Jr.; Liu, J.O. Inhibition of angiogenesis by the antifungal drug itraconazole. ACS Chem. Biol. 2007, 2, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Ban, L.; Mei, T.; Su, Q.; Li, W.; Huang, Z.; Liu, L.; Wu, Y.; Lv, S.; Wang, A.; Li, S. Anti-fungal drug itraconazole exerts anti-cancer effects in oral squamous cell carcinoma via suppressing hedgehog pathway. Life Sci. 2020, 254, 117695. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Cheng, L.; Qian, W.; Jiang, Z.; Sun, L.; Zhao, Y.; Zhou, Y.; Zhao, L.; Wang, P.; Duan, W.; et al. Itraconazole inhibits invasion and migration of pancreatic cancer cells by suppressing tgf-beta/smad2/3 signaling. Oncol. Rep. 2018, 39, 1573–1582. [Google Scholar]
- Chen, M.B.; Liu, Y.Y.; Xing, Z.Y.; Zhang, Z.Q.; Jiang, Q.; Lu, P.H.; Cao, C. Itraconazole-induced inhibition on human esophageal cancer cell growth requires ampk activation. Mol. Cancer Ther. 2018, 17, 1229–1239. [Google Scholar] [CrossRef] [Green Version]
- Lan, K.; Yan, R.; Zhu, K.; Li, W.; Xu, Z.; Dang, C.; Li, K. Itraconazole inhibits the proliferation of gastric cancer cells in vitro and improves patient survival. Oncol. Lett. 2018, 16, 3651–3657. [Google Scholar] [CrossRef] [Green Version]
- Choi, C.H.; Ryu, J.Y.; Cho, Y.J.; Jeon, H.K.; Choi, J.J.; Ylaya, K.; Lee, Y.Y.; Kim, T.J.; Chung, J.Y.; Hewitt, S.M.; et al. The anti-cancer effects of itraconazole in epithelial ovarian cancer. Sci. Rep. 2017, 7, 6552. [Google Scholar] [CrossRef] [Green Version]
- Ally, M.S.; Ransohoff, K.; Sarin, K.; Atwood, S.X.; Rezaee, M.; Bailey-Healy, I.; Kim, J.; Beachy, P.A.; Chang, A.L.; Oro, A.; et al. Effects of combined treatment with arsenic trioxide and itraconazole in patients with refractory metastatic basal cell carcinoma. JAMA Dermatol. 2016, 152, 452–456. [Google Scholar] [CrossRef]
- Tsubamoto, H.; Sonoda, T.; Ikuta, S.; Tani, S.; Inoue, K.; Yamanaka, N. Combination chemotherapy with itraconazole for treating metastatic pancreatic cancer in the second-line or additional setting. Anticancer Res. 2015, 35, 4191–4196. [Google Scholar]
- Tsubamoto, H.; Sonoda, T.; Yamasaki, M.; Inoue, K. Impact of combination chemotherapy with itraconazole on survival of patients with refractory ovarian cancer. Anticancer Res. 2014, 34, 2481–2487. [Google Scholar]
- Correia, A.; Silva, D.; Correia, A.; Vilanova, M.; Gartner, F.; Vale, N. Study of new therapeutic strategies to combat breast cancer using drug combinations. Biomolecules 2018, 8, 175. [Google Scholar] [CrossRef] [Green Version]
- Hara, M.; Nagasaki, T.; Shiga, K.; Takeyama, H. Suppression of cancer-associated fibroblasts and endothelial cells by itraconazole in bevacizumab-resistant gastrointestinal cancer. Anticancer Res. 2016, 36, 169–177. [Google Scholar]
- Antonarakis, E.S.; Heath, E.I.; Smith, D.C.; Rathkopf, D.; Blackford, A.L.; Danila, D.C.; King, S.; Frost, A.; Ajiboye, A.S.; Zhao, M.; et al. Repurposing itraconazole as a treatment for advanced prostate cancer: A noncomparative randomized phase ii trial in men with metastatic castration-resistant prostate cancer. Oncologist 2013, 18, 163–173. [Google Scholar] [CrossRef]
- Kim, D.J.; Kim, J.; Spaunhurst, K.; Montoya, J.; Khodosh, R.; Chandra, K.; Fu, T.; Gilliam, A.; Molgo, M.; Beachy, P.A.; et al. Open-label, exploratory phase ii trial of oral itraconazole for the treatment of basal cell carcinoma. J. Clin. Oncol. 2014, 32, 745–751. [Google Scholar] [CrossRef]
- Rudin, C.M.; Brahmer, J.R.; Juergens, R.A.; Hann, C.L.; Ettinger, D.S.; Sebree, R.; Smith, R.; Aftab, B.T.; Huang, P.; Liu, J.O. Phase 2 study of pemetrexed and itraconazole as second-line therapy for metastatic nonsquamous non-small-cell lung cancer. J. Thorac. Oncol. 2013, 8, 619–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsubamoto, H.; Sonoda, T.; Inoue, K. Impact of itraconazole on the survival of heavily pre-treated patients with triple-negative breast cancer. Anticancer Res. 2014, 34, 3839–3844. [Google Scholar]
- Mamtani, R.; Yang, Y.X.; Scott, F.I.; Lewis, J.D.; Boursi, B. Association of itraconazole, a hedgehog inhibitor, and bladder cancer. J. Urol. 2016, 196, 343–348. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K.; Tsubamoto, H.; Sakata, K.; Sakane, R.; Hao, H.; Hirota, S.; Sonoda, T.; Shibahara, H. Expression of hedgehog signals and growth inhibition by itraconazole in endometrial cancer. Anticancer Res. 2016, 36, 149–153. [Google Scholar]
- Tsubamoto, H.; Sonoda, T.; Ikuta, S.; Tani, S.; Inoue, K.; Yamanaka, N. Impact of itraconazole after first-line chemotherapy on survival of patients with metastatic biliary tract cancer. Anticancer Res. 2015, 35, 4923–4927. [Google Scholar] [CrossRef] [PubMed]
- Vreugdenhil, G.; Raemaekers, J.M.; van Dijke, B.J.; de Pauw, B.E. Itraconazole and multidrug resistance: Possible effects on remission rate and disease-free survival in acute leukemia. Ann. Hematol. 1993, 67, 107–109. [Google Scholar] [CrossRef]
- Shirakawa, K.; Takara, K.; Tanigawara, Y.; Aoyama, N.; Kasuga, M.; Komada, F.; Sakaeda, T.; Okumura, K. Interaction of docetaxel (“taxotere”) with human p-glycoprotein. Jpn. J. Cancer Res. 1999, 90, 1380–1386. [Google Scholar] [CrossRef]
- Takara, K.; Tanigawara, Y.; Komada, F.; Nishiguchi, K.; Sakaeda, T.; Okumura, K. Cellular pharmacokinetic aspects of reversal effect of itraconazole on p-glycoprotein-mediated resistance of anticancer drugs. Biol. Pharm. Bull. 1999, 22, 1355–1359. [Google Scholar] [CrossRef] [Green Version]
- Oldfield, V.; Plosker, G.L. Lopinavir/ritonavir: A review of its use in the management of hiv infection. Drugs 2006, 66, 1275–1299. [Google Scholar] [CrossRef]
- Kumar, S.; Bryant, C.S.; Chamala, S.; Qazi, A.; Seward, S.; Pal, J.; Steffes, C.P.; Weaver, D.W.; Morris, R.; Malone, J.M.; et al. Ritonavir blocks akt signaling, activates apoptosis and inhibits migration and invasion in ovarian cancer cells. Mol. Cancer 2009, 8, 26. [Google Scholar] [CrossRef] [Green Version]
- Carroll, V.; Garzino-Demo, A. Hiv-associated lymphoma in the era of combination antiretroviral therapy: Shifting the immunological landscape. Pathog. Dis. 2015, 73, ftv044. [Google Scholar] [CrossRef]
- Shmakova, A.; Germini, D.; Vassetzky, Y. Hiv-1, haart and cancer: A complex relationship. Int. J. Cancer 2020, 146, 2666–2679. [Google Scholar] [CrossRef]
- Cheung, T.W. Aids-related cancer in the era of highly active antiretroviral therapy (haart): A model of the interplay of the immune system, virus, and cancer. “On the offensive--the trojan horse is being destroyed”--part b: Malignant lymphoma. Cancer Investig. 2004, 22, 787–798. [Google Scholar] [CrossRef]
- Laurence, J. Impact of haart on hiv-linked malignancies. AIDS Read. 2003, 13, 202–205. [Google Scholar]
- Monini, P.; Toschi, E.; Sgadari, C.; Bacigalupo, I.; Palladino, C.; Carlei, D.; Barillari, G.; Ensoli, B. The use of haart for biological tumour therapy. J. HIV Ther. 2006, 11, 53–56. [Google Scholar]
- Ntekim, A.; Campbell, O.; Rothenbacher, D. Optimal management of cervical cancer in hiv-positive patients: A systematic review. Cancer Med. 2015, 4, 1381–1393. [Google Scholar] [CrossRef] [PubMed]
- Clifford, G.M.; Polesel, J.; Rickenbach, M.; Dal Maso, L.; Keiser, O.; Kofler, A.; Rapiti, E.; Levi, F.; Jundt, G.; Fisch, T.; et al. Cancer risk in the swiss hiv cohort study: Associations with immunodeficiency, smoking, and highly active antiretroviral therapy. J. Natl. Cancer Inst. 2005, 97, 425–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kincaid, L. Modern haart decreases cancers in children with hiv. Lancet Oncol. 2007, 8, 103. [Google Scholar] [CrossRef]
- Long, J.L.; Engels, E.A.; Moore, R.D.; Gebo, K.A. Incidence and outcomes of malignancy in the haart era in an urban cohort of hiv-infected individuals. AIDS 2008, 22, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Franzetti, M.; Ricci, E.; Bonfanti, P. The pattern of non-aids-defining cancers in the hiv population: Epidemiology, risk factors and prognosis. A review. Curr. HIV Res. 2019, 17, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Dewan, M.Z.; Uchihara, J.N.; Terashima, K.; Honda, M.; Sata, T.; Ito, M.; Fujii, N.; Uozumi, K.; Tsukasaki, K.; Tomonaga, M.; et al. Efficient intervention of growth and infiltration of primary adult t-cell leukemia cells by an hiv protease inhibitor, ritonavir. Blood 2006, 107, 716–724. [Google Scholar] [CrossRef] [Green Version]
- Ikezoe, T.; Daar, E.S.; Hisatake, J.; Taguchi, H.; Koeffler, H.P. Hiv-1 protease inhibitors decrease proliferation and induce differentiation of human myelocytic leukemia cells. Blood 2000, 96, 3553–3559. [Google Scholar] [CrossRef]
- Labo, N.; Miley, W.; Benson, C.A.; Campbell, T.B.; Whitby, D. Epidemiology of kaposi’s sarcoma-associated herpesvirus in hiv-1-infected us persons in the era of combination antiretroviral therapy. AIDS 2015, 29, 1217–1225. [Google Scholar] [CrossRef]
- Noy, A. Optimizing treatment of hiv-associated lymphoma. Blood 2019, 134, 1385–1394. [Google Scholar] [CrossRef]
- Mazzocchi, A.R.; Rajan, S.A.P.; Votanopoulos, K.I.; Hall, A.R.; Skardal, A. In vitro patient-derived 3d mesothelioma tumor organoids facilitate patient-centric therapeutic screening. Sci. Rep. 2018, 8, 2886. [Google Scholar] [CrossRef] [Green Version]
- Meijer, T.G.; Naipal, K.A.; Jager, A.; van Gent, D.C. Ex vivo tumor culture systems for functional drug testing and therapy response prediction. Future Sci. OA 2017, 3, FSO190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ince, T.A.; Sousa, A.D.; Jones, M.A.; Harrell, J.C.; Agoston, E.S.; Krohn, M.; Selfors, L.M.; Liu, W.; Chen, K.; Yong, M.; et al. Characterization of twenty-five ovarian tumour cell lines that phenocopy primary tumours. Nat. Commun. 2015, 6, 7419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swords, R.T.; Azzam, D.; Al-Ali, H.; Lohse, I.; Volmar, C.H.; Watts, J.M.; Perez, A.; Rodriguez, A.; Vargas, F.; Elias, R.; et al. Ex-vivo sensitivity profiling to guide clinical decision making in acute myeloid leukemia: A pilot study. Leuk. Res. 2018, 64, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Ghani, F.I.; Dendo, K.; Watanabe, R.; Yamada, K.; Yoshimatsu, Y.; Yugawa, T.; Nakahara, T.; Tanaka, K.; Yoshida, H.; Yoshida, M.; et al. An ex-vivo culture system of ovarian cancer faithfully recapitulating the pathological features of primary tumors. Cells 2019, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, L.; Tighe, A.; Golder, A.; Littler, S.; Bakker, B.; Moralli, D.; Murtuza Baker, S.; Donaldson, I.J.; Spierings, D.C.J.; Wardenaar, R.; et al. A living biobank of ovarian cancer ex vivo models reveals profound mitotic heterogeneity. Nat. Commun. 2020, 11, 822. [Google Scholar] [CrossRef] [PubMed]
- Lohse, I.; Al-Ali, H.; Volmar, C.H.; A, D.A.T.; Brothers, S.P.; Capobianco, A.J.; Wahlestedt, C. Ex vivo drug sensitivity testing as a means for drug repurposing in esophageal adenocarcinoma. PLoS ONE 2018, 13, e0203173. [Google Scholar] [CrossRef]
- Murumägi, A.; Ungureanu, D.; Khan, S.; Hirasawa, A.; Arjama, M.; Välimäki, K.; Mikkonen, P.; Niininen, W.; Kumar, A.; Eldfors, S.; et al. Clinical implementation of precision systems oncology in the treatment of ovarian cancer based on ex-vivo drug testing and molecular profiling. In Proceedings of the Annual Meeting of the American Association for Cancer Research: Experimental and Molecular Therapeutics, Atlanta, GA, USA, 29 March–3 April 2019; Volume 79, p. 2945. [Google Scholar]
- Lohmussaar, K.; Boretto, M.; Clevers, H. Human-derived model systems in gynecological cancer research. Trends Cancer 2020. [Google Scholar] [CrossRef]
- Lengyel, E. Ovarian cancer development and metastasis. Am. J. Pathol. 2010, 177, 1053–1064. [Google Scholar] [CrossRef]
- Ahmed, N.; Stenvers, K.L. Getting to know ovarian cancer ascites: Opportunities for targeted therapy-based translational research. Front. Oncol. 2013, 3, 256. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, N.; Riley, C.; Oliva, K.; Rice, G.; Quinn, M. Ascites induces modulation of alpha6beta1 integrin and urokinase plasminogen activator receptor expression and associated functions in ovarian carcinoma. Br. J. Cancer 2005, 92, 1475–1485. [Google Scholar] [CrossRef] [Green Version]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernandez-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [Green Version]
- Weeber, F.; Ooft, S.N.; Dijkstra, K.K.; Voest, E.E. Tumor organoids as a pre-clinical cancer model for drug discovery. Cell Chem. Biol. 2017, 24, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, A.; Plummer, E.R.; Elattar, A.; Soohoo, S.; Uzir, B.; Quinn, J.E.; McCluggage, W.G.; Maxwell, P.; Aneke, H.; Curtin, N.J.; et al. Clinicopathological features of homologous recombination-deficient epithelial ovarian cancers: Sensitivity to parp inhibitors, platinum, and survival. Cancer Res. 2012, 72, 5675–5682. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Sanderson, P.E.; Zheng, W. Drug combination therapy increases successful drug repositioning. Drug Discov. Today 2016, 21, 1189–1195. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nunes, M.; Henriques Abreu, M.; Bartosch, C.; Ricardo, S. Recycling the Purpose of Old Drugs to Treat Ovarian Cancer. Int. J. Mol. Sci. 2020, 21, 7768. https://doi.org/10.3390/ijms21207768
Nunes M, Henriques Abreu M, Bartosch C, Ricardo S. Recycling the Purpose of Old Drugs to Treat Ovarian Cancer. International Journal of Molecular Sciences. 2020; 21(20):7768. https://doi.org/10.3390/ijms21207768
Chicago/Turabian StyleNunes, Mariana, Miguel Henriques Abreu, Carla Bartosch, and Sara Ricardo. 2020. "Recycling the Purpose of Old Drugs to Treat Ovarian Cancer" International Journal of Molecular Sciences 21, no. 20: 7768. https://doi.org/10.3390/ijms21207768
APA StyleNunes, M., Henriques Abreu, M., Bartosch, C., & Ricardo, S. (2020). Recycling the Purpose of Old Drugs to Treat Ovarian Cancer. International Journal of Molecular Sciences, 21(20), 7768. https://doi.org/10.3390/ijms21207768