Neurodegeneration and Inflammation—An Interesting Interplay in Parkinson’s Disease

 ,
,  ,
, 

Abstract
: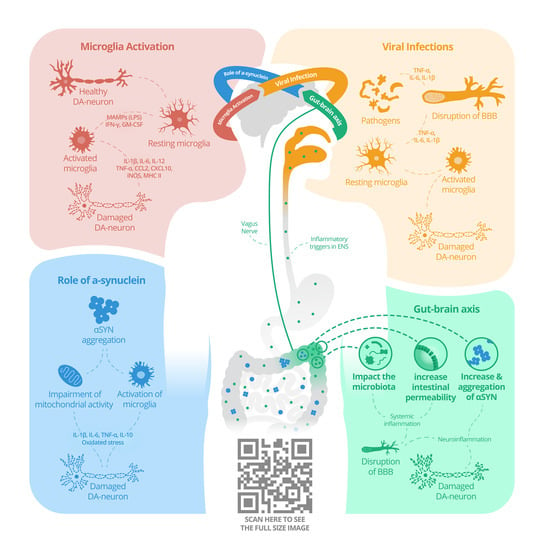
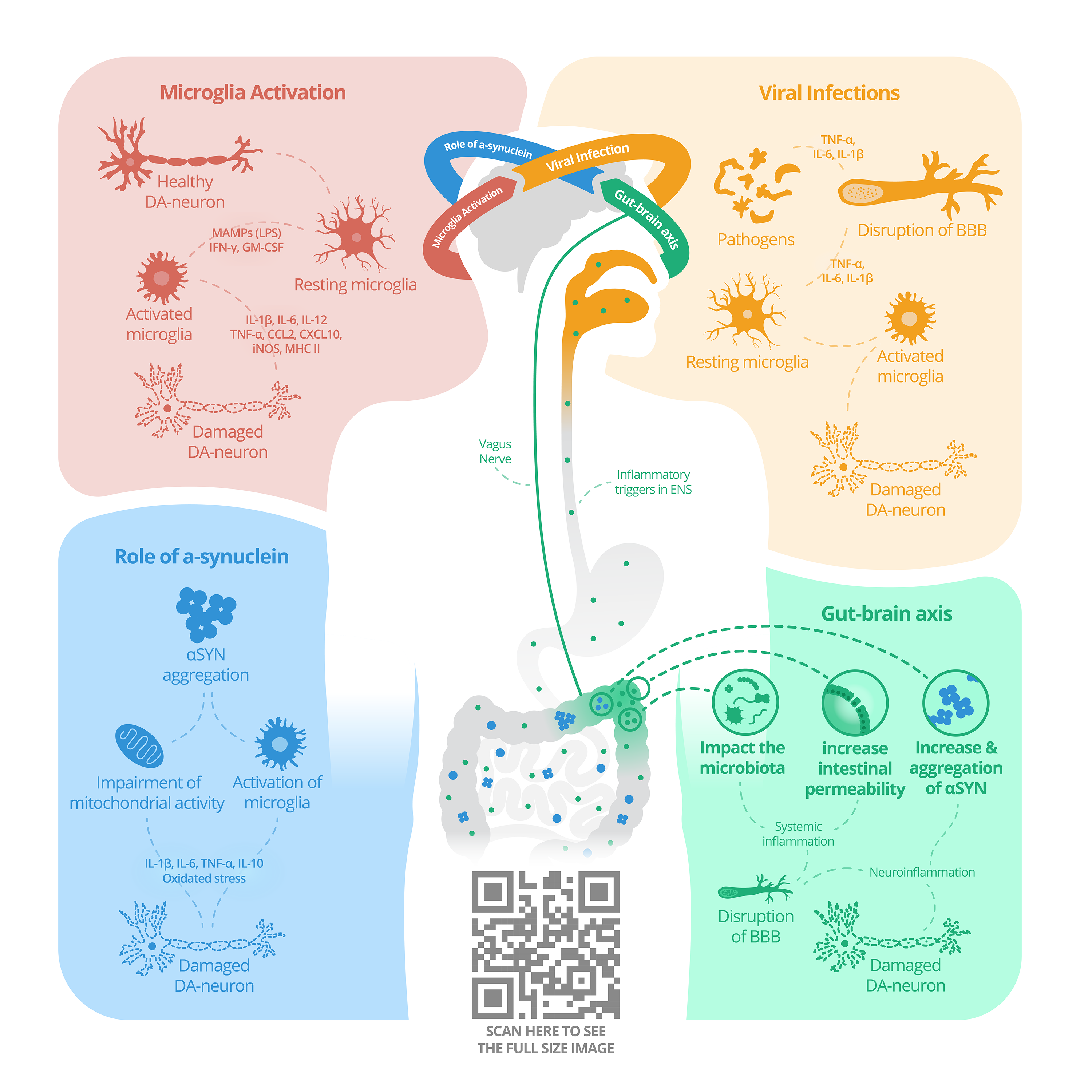{kind=link}
1. Introduction
2. Inflammation in Parkinson’s Disease (PD)
2.1. Microglia Activation
2.2. Specific Cytokine Signaling in Parkinson’s Disease (PD)
2.3. Viral Infections
2.4. Autoimmunity and the Role of A-Synuclein
2.5. Gut–Brain Axis in PD
3. PD-Associated Genes and Neuroinflammation
4. Molecular Biomarkers, Therapeutic Approaches, and Neuroinflammation
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dorsey, E.; Constantinescu, R.; Thompson, J.P.; Biglan, K.M.; Holloway, R.G.; Kieburtz, K.; Marshall, F.J.; Ravina, B.M.; Schifitto, G.; Siderowf, A.; et al. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology 2007, 68, 384–386. [Google Scholar] [CrossRef] [PubMed]
- Tanner, C.M.; Goldman, S.M. Epidemiology of Parkinson’s disease. Neurol. Clin. 1996, 14, 317–335. [Google Scholar] [CrossRef]
- Liu, B.; Gao, H.M.; Hong, J.S. Parkinson’s disease and exposure to infectious agents and pesticides and the occurrence of brain injury: Role of neuroinflammation. Environ. Health Perspect. 2003, 111, 1065–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyko, A.A.; Troyanova, N.I.; Kovalenko, E.I.; Sapozhnikov, A.M. Similarity and Differences in Inflammation-Related Characteristics of the Peripheral Immune System of Patients with Parkinson’s and Alzheimer’s Diseases. Int. J. Mol. Sci. 2017, 18, 2633. [Google Scholar] [CrossRef] [Green Version]
- Nandipati, S.; Litvan, I. Environmental Exposures and Parkinson’s Disease. Int. J. Environ. Res. Public Health 2016, 13, 881. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Hensley, K. Neuroinflammation in Alzheimer’s disease: Mechanisms, pathologic consequences, and potential for therapeutic manipulation. J. Neurol. Dis. 2010, 21, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Phani, S.; Loike, J.D.; Przedborski, S. Neurodegeneration and inflammation in Parkinson’s disease. Parkinsonism Relat. Disord. 2012, 18 (Suppl. 1), S207–S209. [Google Scholar] [CrossRef]
- Moore, D.J.; West, A.B.; Dawson, V.L.; Dawson, T.M. Molecular pathophysiology of Parkinson’s disease. Ann. Rev. Neurosci. 2005, 28, 57–87. [Google Scholar] [CrossRef] [Green Version]
- Schneider, S.A.; Obeso, J.A. Clinical and pathological features of Parkinson’s disease. Curr. Top. Behav. Neurosci. 2005, 22, 205–220. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- More, S.V.; Kumar, H.; Kim, I.S.; Song, S.-Y.; Choi, D.-K. Cellular and molecular mediators of neuroinflammation in the pathogenesis of Parkinson’s disease. Mediat. Inflamm 2013. [Google Scholar] [CrossRef] [PubMed]
- Caggiu, E.; Paulus, K.; Galleri, G.; Arru, G.; Manetti, R.; Sechi, G.P.; Sechi, L.A. Homologous HSV1 and alpha-synuclein peptides stimulate a T cell response in Parkinson’s disease. J. Neuroimmunol. 2017, 310, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Caggiu, E.; Paulus, K.; Arru, G.; Piredda, R.; Sechi, G.P.; Sechi, L.A. Humoral cross reactivity between α-synuclein and herpes simplex-1 epitope in Parkinson’s disease, a triggering role in the disease? J. Neuroimmunol. 2016, 291, 110–114. [Google Scholar] [CrossRef]
- Scheperjans, F.; Aho, V.; Pereira, P.A.; Koskinen, K.; Paulin, L.; Pekkonen, E.; Haapaniemi, E.; Kaakkola, S.; Eerola-Rautio, J.; Pohja, M.; et al. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov. Disord. 2015, 30, 350–358. [Google Scholar] [CrossRef]
- International Parkinson Disease Genomics Consortium. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet 2011, 377, 641–649. [Google Scholar] [CrossRef] [Green Version]
- Witoelar, A.; Jansen, I.; Wang, Y.; Desikan, R.; Gibbs, J.R.; Blauwendraat, C.; Thompson, W.; Hernandez, D.J.; Djurovic, S.; Schork, A.J.; et al. Genome-wide pleiotropy between parkinson disease and autoimmunediseases. JAMA Neurol. 2017, 74, 780–792. [Google Scholar] [CrossRef]
- Blesa, J.; Przedborski, S. Parkinson’s disease: Animal models and dopaminergic cell vulnerability. Front. Neuroanat. 2014. [Google Scholar] [CrossRef] [Green Version]
- Blesa, J.; Phani, S.; Jackson-Lewis, V.; Przedborski, S. Classic and new animal models of Parkinson’s disease. J. Biomed. Biotechnol. 2012, 2012, 845618. [Google Scholar] [CrossRef]
- Varatharaj, A.; Galea, I. The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 2017, 60, 1–12. [Google Scholar] [CrossRef] [Green Version]
- García-Domínguez, I.; Veselá, K.; García-Revilla, J.; Carrillo-Jiménez, A.; Roca-Ceballos, M.A.; Santiago, M.; de Pablos, R.M.; Venero, J.L. Peripheral inflammation enhances microglia response and nigral dopaminergic cell death in an in vivo MPTP model of Parkinson’s disease. Front. Cell. Neurosci. 2018, 12. [Google Scholar] [CrossRef]
- Hanisch, U.-K. Microglia as a source and target of cytokines. Glia 2002, 40, 140–155. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Ann. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef]
- Sawada, M.; Imamura, K.; Nagatsu, T. Role of cytokines in inflammatory process in Parkinson’s disease. J. Neural. Transm. Suppl. 2006, 70, 373–381. [Google Scholar]
- Ezcurra, A.L.D.L.; Chertoff, M.; Ferrari, C.; Graciarena, M.; Pitossi, F. Chronic expression of low levels of tumor necrosis factor-alpha in the substantia nigra elicits progressive neurodegeneration, delayed motor symptoms and microglia/macrophage activation. Neurobiol. Dis. 2010, 37, 630–640. [Google Scholar] [CrossRef]
- Cebrián, C.; Loike, J.D.; Sulzer, D. Neuronal MHC-I expression and its implications in synaptic function, axonal regeneration and Parkinson’s and other brain diseases. Front. Neuroanat. 2014, 8, 114. [Google Scholar] [CrossRef] [Green Version]
- Loane, C.; Politis, M. Positron emission tomography neuroimaging in Parkinson’s disease. Am. J. Transl. Res. 2011, 3, 323–341. [Google Scholar]
- Cerami, C.; Iaccarino, L.; Perani, D. Molecular Imaging of Neuroinflammation in Neurodegenerative Dementias: The Role of in vivo PET Imaging. Int. J. Mol. Sci. 2017, 18, 993. [Google Scholar] [CrossRef] [Green Version]
- Choi, I.; Zhang, Y.; Seegobin, S.P.; Pruvost, M.; Wang, Q.; Purtell, K.; Zhang, B.; Yue, Z. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat. Commun. 2020, 13, 1386. [Google Scholar] [CrossRef] [Green Version]
- Karpenko, M.N.; Vasilishina, A.A.; Gromova, E.A.; Muruzheva, Z.M.; Bernadotte, A. Interleukin-1β, interleukin-1 receptor antagonist, interleukin-6, interleukin-10, and tumor necrosis factor-α levels in CSF and serum in relation to the clinical diversity of Parkinson’s disease. Cell. Immunol. 2018, 327, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Imamura, K.; Hishikawa, N.; Ono, K.; Suzuki, H.; Sawada, M.; Nagatsu, T.; Yoshida, M.; Hashizume, Y. Cytokine production of activated microglia and decrease in neurotrophic factors of neurons in the hippocampus of Lewy body disease brains. Acta Neuropathol. 2005, 109, 141–150. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.K.; Martinez, T.N.; Ruhn, K.A.; Szymkowski, D.E.; Smith, C.G.; Botterman, B.R.; Tansey, K.E.; Tansey, M.G. Blocking Soluble Tumor Necrosis Factor Signaling with Dominant-Negative Tumor Necrosis Factor Inhibitor Attenuates Loss of Dopaminergic Neurons in Models of Parkinson’s Disease. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 9365–9375. [Google Scholar] [CrossRef] [PubMed]
- Elyaman, W.; Khoury, S.J. Th9 cells in the pathogenesis of EAE and multiple sclerosis. Semin. Immunopathol. 2017, 39, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Elyaman, W.; Bradshaw, E.M.; Uyttenhove, C.; Dardalhon, V.; Awasthi, A.; Imitola, J.; Bettelli, E.; Oukka, M.; van Snick, J.; Renauld, J.-C.; et al. IL-9 induces differentiation of TH17 cells and enhances function of FoxP3+ natural regulatory T cells. Proc. Natl. Acad. Sci. USA 2009, 106, 12885–12890. [Google Scholar] [CrossRef] [Green Version]
- Picca, A.; Guerra, F.; Calvani, R.; Marini, F.; Biancolillo, A.; Landi, G.; Beli, R.; Landi, F.; Bernabei, R.; Bentivoglio, A.R.; et al. Mitochondrial signatures in Circulating Extracellular Vesicles of Older Adults with Parkinson’s Disease: Results from the EXosomes in PArkiNson’s disease (EXPAND) study. J. Clin. Med. 2020, 9, 504. [Google Scholar] [CrossRef] [Green Version]
- Deleidi, M.; Gasser, T. The role of inflammation in sporadic and familial Parkinson’s disease. Cell. Mol. Life Sci. 2013, 70, 4259–4273. [Google Scholar] [CrossRef]
- Collins, L.M.; Toulouse, A.; Connor, T.J.; Nolan, Y.M. Contributions of central and systemic inflammation to the pathophysiology of Parkinson’s disease. Neuropharmacology 2012, 62, 2154–2168. [Google Scholar] [CrossRef] [Green Version]
- Tang, P.; Chong, L.; Li, X.; Liu, Y.; Liu, P.; Hou, C.; Li, R. Correlation between serum RANTES levels and the severity of Parkinson’s disease. Oxid. Med. Cell Longev. 2014, 2014, 208408. [Google Scholar] [CrossRef] [Green Version]
- Von Economo, C. Encephalitis Lethargica: Its Sequelae and Treatment; Oxford University Press: Oxford, UK, 1931. [Google Scholar]
- Vilensky, J.A.; Gilman, S.; McCall, S. A historical analysis of the relationship between encephalitis lethargica and postencephalitic parkinsonism: A complex rather than a direct relationship. Mov. Dis. Off. J. Mov. Dis. Soc. 2010, 25, 1116–1123. [Google Scholar] [CrossRef] [Green Version]
- Olsen, L.K.; Dowd, E.; McKernan, D.P. A role for viral infections in Parkinson’s etiology? Neuronal Signal. 2018, 2, NS20170166. [Google Scholar] [CrossRef] [PubMed]
- Sadasivan, S.; Zanin, M.; O’Brien, K.; Schultz-Cherry, S.; Smeyne, R.J. Induction of microglia activation after infection with the non-neurotropic A/CA/04/2009 H1N1 influenza virus. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadasivan, S.; Sharp, B.; Schultz-Cherry, S.; Smeyne, R.J. Synergistic effects of influenza and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) can be eliminated by the use of influenza therapeutics: Experimental evidence for the multi-hit hypothesis. NPJ Parkinson’s Dis. 2017, 3. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhou, Y.; Yang, Z. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell. Mol. Immunol. 2016, 13, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemling, N.; Röyttä, M.; Rinne, J.; Pöllänen, P.; Broberg, E.; Tapio, V.; Tero, V.; Veijo, H. Herpesviruses in brains in Alzheimer’s and Parkinson’s diseases. Ann. Neurol. 2003, 54, 267–271. [Google Scholar] [CrossRef]
- Bu, X.-L.; Wang, X.; Xiang, Y.; Shen, L.-L.; Wang, Q.-H.; Liu, Y.-H.; Wang, Y.-J. The association between infectious burden and Parkinson’s disease: A case-control study. Parkinsonism Relat. Dis. 2015, 21, 877–881. [Google Scholar] [CrossRef]
- Woulfe, J.M.; Gray, M.T.; Gray, D.A.; Munoz, D.G.; Middeldorp, J.M. Hypothesis: A role for EBV-induced molecular mimicry in Parkinson’s disease. Parkinsonism Relat. Disord. 2014, 20, 685–694. [Google Scholar] [CrossRef]
- Bonini, N.M.; Giasson, B.I. Snaring the function of alpha-synuclein. Cell 2005, 123, 359–361. [Google Scholar] [CrossRef]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [Green Version]
- Orr, C.F.; Rowe, D.B.; Mizuno, Y.; Mori, H.; Halliday, G.M. A possible role for humoral immunity in the pathogenesis of Parkinson’s disease. Brain 2005, 128, 2665–2674. [Google Scholar] [CrossRef] [Green Version]
- Klegeris, A.; Pelech, S.; Giasson, B.I.; Maguire, J.; Zhang, H.; McGeer, E.G.; McGeer, P.L. Alpha-synuclein activates stress signaling protein kinases in THP-1 cells and microglia. Neurobiol. Aging 2008, 29, 739–752. [Google Scholar] [CrossRef]
- Atik, A.; Stewart, T.; Zhang, J. Alpha-synuclein as a biomarker for Parkinson’s disease. Brain Pathol. 2016, 26, 410–418. [Google Scholar] [CrossRef]
- Arlehamn, C.S.L.; Dhanwani, R.; Pham, J.; Kuan, R.; Frazier, A.; Dutra, J.R.; Phillips, E.; Mallal, S.; Roederer, M.; Marder, K.S.; et al. α-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat. Commun. 2020, 11, 1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollenhauer, B.; Locascio, J.J.; Schulz-Schaeffer, W.; Sixel-Döring, F.; Trenkwalder, G.; Schlossmacher, M.G. A-Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: A cohort study. Lancet Neurol. 2011, 10, 230–240. [Google Scholar] [CrossRef]
- Fairfoul, G.; McGuire, L.I.; Pal, S.; Ironside, J.W.; Neumann, J.; Christie, S.; Joachim, C.; Esiri, M.; Evetts, S.G.; Rolinski, M.; et al. Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann. Clin. Transl. Neurol. 2016, 3, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Kustrimovic, N.; Comi, C.; Magistrelli, L.; Rasini, E.; Legnaro, M.; Bombelli, R.; Aleksic, I.; Blandini, F.; Minafra, B.; Riboldazzi, G.; et al. Parkinson’s disease patients have a complex phenotypic and functional Th1 bias: Cross-sectional studies of CD4+ Th1/Th2/T17 and Treg in drug-naïve and drug-treated patients. J. Neuroinflamm. 2018, 15, 205. [Google Scholar] [CrossRef]
- Baba, Y.; Kuroiwa, A.; Uitti, R.J.; Wszolek, Z.K.; Yamada, T. Alterations of T-lymphocyte populations in Parkinson disease. Parkinsonism Relat. Disord. 2005, 11, 493–498. [Google Scholar] [CrossRef]
- Brochard, V.; Combadière, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Hunot, S. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J. Clin. Investig. 2009, 119, 182–192. [Google Scholar] [CrossRef]
- Sulzer, D.; Alcalay, R.N.; Garretti, F.; Cote, L.; Kanter, E.; Agin-Liebes, J.P.; Liong, C.; McMurtrey, C.; Hildebrand, W.H.; Mao, X.; et al. T cells of Parkinson’s disease patients recognize α–synuclein peptides. Nature 2017, 546, 656–661. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Gao, H.; Luo, Q.; Wang, P.; Yang, X. The correlation of lymphocyte subsets, natural killer cell, and Parkinson’s disease: A meta-analysis. Neurol. Sci. 2017, 38, 1373–1380. [Google Scholar] [CrossRef]
- Niwa, F.; Kuriyama, N.; Nakagawa, M.; Imanishi, J. Effects of peripheral lymphocyte subpopulations and the clinical correlation with Parkinson’s disease. Geriatr. Gerontol. Int. 2012, 12, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Koudstaal, W.; Fletcher, L.; Costa, M.; van Winsen, M.; Siregar, B.; Inganäs, H.; Kim, J.; Keogh, E.; Jeremy, M.; et al. Naturally occurring antibodies isolated from PD patients inhibit synuclein seeding in vitro and recognize Lewy pathology. Acta Neuropathol. 2019, 137, 825–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, H.-L.; Lin, C.-H. Altered gut microbiome and Intestinal pathology in Parkinson’s disease. J. Mov. Dis. 2019, 12, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 2016, 167, 1469–1480.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keshavarzian, A.; Green, S.J.; Engen, P.A.; Voigt, R.M.; Naqib, A.; Forsyth, C.B.; Mutlu, E.; Shannon, K.M. Colonic bacterial composition in Parkinson’s disease. Mov. Disord. 2015, 30, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Maier, S.F.; Goehler, L.E.; Fleshner, M.; Watkins, L.R. The role of the vagus nerve in cytokine-to-brain communication. Ann. N. Y. Acad. Sci. 1998, 840, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Fang, F.; Pedersen, N.L.; Tillander, A.; Ludvigsson, J.F.; Ekbom, A.; Svenningsson, P.; Chen, H.; Wirdefeldt, K. Vagotomy and Parkinson disease: A Swedish register-based matched-cohort study. Neurology 2017, 88, 1996–2002. [Google Scholar] [CrossRef] [Green Version]
- Svensson, E.; Horváth-Puhó, E.; Thomsen, R.W.; Djurhuus, J.C.; Pedersen, L.; Borghammer, P.; Sørensen, H.T. Vagotomy and subsequent risk of Parkinson’s disease. Ann. Neurol. 2015, 78, 522–529. [Google Scholar] [CrossRef]
- Devos, D.; Lebouvier, T.; Lardeux, B.; Biraud, M.; Rouaud, T.; Pouclet, H.; Coron, E.; des Varannes, S.B.; Naveilhan, P.; Nguyen, J.M.; et al. Colonic inflammation in Parkinson’s disease. Neurobiol. Dis. 2013, 50, 42–48. [Google Scholar] [CrossRef]
- Houser, M.C.; Chang, J.; Factor, S.A.; Molho, E.S.; Zabetian, C.P.; Hill-Burns, E.M.; Payami, H.; Hertzberg, V.; Tansey, M. Stool immune profiles evince gastrointestinal inflammation in Parkinson’s disease. Mov. Disord. 2018, 33, 793–804. [Google Scholar] [CrossRef]
- Challis, C.; Hori, A.; Sampson, T.R.; Yoo, B.B.; Challis, R.C.; Hamilton, A.M.; Mazmanian, S.K.; Volpicelli-Daley, L.; Gradinaru, V. Gut-seeded α-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nat. Neurosci. 2020, 23, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Misra, M.K.; Damotte, V.; Hollenbach, J.A. The immunogenetics of neurological disease. Immunology 2018, 153, 399–414. [Google Scholar] [CrossRef] [PubMed]
- Nuytemans, K.; Theuns, J.; Cruts, M.; van Broeckhoven, C. Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: A mutation update. Hum. Mutat. 2010, 31, 763–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; James, W.S.; Cowley, S.A. LRRK2 in peripheral and central nervous system innate immunity: Its link to Parkinson’s disease. Biochem. Soc. Transl. 2017, 45, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Shutinoski, B.; Hakimi, M.; Harmsen, I.E.; Lunn, M.; Rocha, J.; Lengacher, N.; Zhou, Y.Y.; Khan, J.; Nguyen, A.; Hake-Volling, Q.; et al. Lrrk2 alleles modulate inflammation during microbial infection of mice in a sex-dependent manner. Sci. Transl. Med. 2019, 25, 11. [Google Scholar] [CrossRef]
- Kim, B.; Yang, M.S.; Choi, D.; Kim, J.H.; Kim, H.S.; Seol, W.; Choi, S.; Jou, I.; Kim, E.Y.; Joe, E.H. Impaired Inflammatory Responses in Murine Lrrk2-Knockdown Brain Microglia. PLoS ONE 2017, 7, e34693. [Google Scholar] [CrossRef] [Green Version]
- Gardet, A.; Benita, Y.; Li, C.; Sands, B.E.; Ballester, I.; Stevens, C.; Rioux, J.D.; Daly, M.J.; Xavier, R.J.; Podolsky, K. LRRK2 is involved in the IFN-gamma response and host response to pathogens. J. Immunol. 2010, 185, 5577–5585. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Lenardo, M.J. The role of LRRK2 in inflammatory bowel disease. Cell Res. 2012, 22, 1092–1094. [Google Scholar] [CrossRef] [Green Version]
- Hui, K.Y.; Fernandez-Hernandez, H.; Hu, J.; Schaffner, A.; Pankratz, N.; Hsu, N.Y.; Chuang, L.S.; Carmi, S.; Villaverde, N.; Li, X.; et al. Functional variants in LRRK2 confer pleiotropic effects on risk for Crohn’s disease and Parkinson’s disease. Sci. Transl. Med. 2018, 10. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6028002/ (accessed on 5 November 2020).
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Zeeshan, A.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [Green Version]
- Pridgeon, J.W.; Olzmann, J.A.; Chin, L.-S.; Li, L. PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol. 2007, 5, e172. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Byun, J.-W.; Choi, I.; Kim, B.; Jeong, H.-K.; Jou, I.; Joe, E. PINK1 deficiency enhances inflammatory cytokine release from acutely prepared brain slices. Exp. Neurobiol. 2013, 22, 38–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lücking, C.B.; Dürr, A.; Bonifati, V.; Vaughan, J.; de Michele, G.; Gasser, T.; Harhangi, B.S.; Meco, G.; Denèfle, P.; Wood, N.W.; et al. Association between Early-Onset Parkinson’s Disease and Mutations in the Parkin Gene. N. Engl. J. Med. 2000, 342, 1560–1567. [Google Scholar] [CrossRef] [PubMed]
- Frank-Cannon, T.C.; Tran, T.; Ruhn, K.A.; Martinez, T.N.; Hong, J.; Marvin, M.; Hartley, M.; Treviño, I.; O’Brien, D.; Casey, B.; et al. Parkin deficiency increases vulnerability to inflammation-related nigral degeneration. J. Neurosci. 2008, 28, 10825–10834. [Google Scholar] [CrossRef] [PubMed]
- Waak, J.; Weber, S.S.; Waldenmaier, A.; Görner, K.; Alunni-Fabbroni, M.; Schell, H.; Vogt-Weisenhorn, D.; Pham, T.-T.; Reumers, V.; Baekelandt, V.; et al. Regulation of astrocyte inflammatory responses by the Parkinson’s disease-associated gene DJ-1. FASEB J. 2009, 23, 2478–2489. [Google Scholar] [CrossRef] [Green Version]
- He, R.; Yan, X.; Guo, J.; Xu, Q.; Tang, B.; Sun, Q. Recent advances in biomarkers for Parkinson’s disease. Front. Aging Neurosci. 2018, 10, 305. [Google Scholar] [CrossRef]
- Fardell, C.; Zettergren, A.; Ran, C.; Belin, A.C.; Ekman, A.; Sydow, O.; Bäckman, L.; Holmberg, B.; Dizdar, N.; Söderkvist, P.; et al. S100B polymorphisms are associated with age of onset of Parkinson’s disease. BMC Med. Genet. 2018. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5848451/ (accessed on 5 November 2020). [CrossRef] [Green Version]
- Grimes, D.A.; Han, F.; Panisset, M.; Racacho, L.; Xiao, F.; Zou, R.; Westaff, K.; Bulman, D.E. Translated mutation in the Nurr1 gene as a cause for Parkinson’s disease. Mov. Disord. 2006, 21, 906–909. [Google Scholar] [CrossRef]
- Betarbet, R.; Sherer, T.B.; Greenamyre, J.T. Ubiquitin-proteasome system and Parkinson’s diseases. Exp. Neurol. 2005, 191 (Suppl. 1), S17–S27. [Google Scholar] [CrossRef]
- Kyratzi, E.; Pavlaki, M.; Stefanis, L. The S18Y polymorphic variant of UCH-L1 confers an antioxidant function to neuronal cells. Hum. Mol. Genet. 2008, 17, 2160–2171. [Google Scholar] [CrossRef]
- Mondello, S.; Constantinescu, R.; Zetterberg, H.; Andreasson, U.; Holmberg, B.; Jeromin, A. CSF α-synuclein and UCH-L1 levels in Parkinson’s disease and atypical parkinsonian disorders. Parkinsonism Relat. Disord. 2014, 20, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Brady, K.R.O.; Kanfer, J.; Shapiro, D. The Metabolism of Glucocerebrosides. I. Purification and properties of a glucocerebroside-cleaving enzyme from spleen tissue. J. Biol. Chem. 1965, 240, 39–43. [Google Scholar] [PubMed]
- Hruska, K.K.S.; Marca, K.M.E.L.; Scott, C.R.; Sidransky, E. Gaucher disease: Mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum. Mutat. 2008, 29, 567–583. [Google Scholar] [CrossRef] [PubMed]
- Tayebi, N.; Walker, J.; Stubblefield, B.; Orvisky, E.; Marca, M.E.L.; Wong, K.; Rosenbaum, H.; Schiffmann, R.; Bembi, B.; Sidransky, E. Gaucher disease with parkinsonian manifestations: Does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol. Genet. Metab. 2003, 79, 104–109. [Google Scholar] [CrossRef]
- Sidransky, E. Gaucher disease and parkinsonism. Mol. Genet. Metab. 2005, 84, 302–304. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Xu, Y.-H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [Green Version]
- Parnetti, L.; Chiasserini, D.; Persichetti, E.; Eusebi, P.; Varghese, S.; Qureshi, M.M.; Dardis, A.; Deganuto, M.; De Carlo, C.; Castrioto, A.; et al. Cerebrospinal fluid lysosomal enzymes and alpha-synuclein in Parkinson’s disease. Mov. Disord. 2014, 29, 1019–1027. [Google Scholar] [CrossRef] [Green Version]
- Mohan, T.; Deng, L.; Wang, B.Z. CCL28 chemokine: An anchoring point bridging innate and adaptive immunity. Int. Immunopharmacol. 2017, 51, 165–170. [Google Scholar] [CrossRef]
- Liu, J.X.; Cao, X.; Liu, Y.; Tang, F.R. CCL28 in the mouse hippocampal CA1 area and the dentate gyrus during and after pilocarpine-induced status epilepticus. Neurochem. Int. 2012, 61, 1094–1101. [Google Scholar] [CrossRef]
- Santaella, A.; Kuiperij, H.B.; van Rumund, A.; Esselink, R.A.; van Gool, A.J.; Bloem, B.R.; Verbeek, M.M. Inflammation biomarker discovery in Parkinson’s disease and atypical parkinsonisms. BMC Neurol. 2020, 26. [Google Scholar] [CrossRef] [Green Version]
- Gur-Wahnon, D.; Mizrachi, T.; Maaravi-Pinto, F.-Y.; Lourbopoulos, A.; Grigoriadis, N.; Higazi, A.-A.R. Talma Brenner The plasminogen activator system: Involvement in central nervous system inflammation and a potential site for therapeutic intervention. J. Neuroinflamm. 2013, 10, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pineda, D.; Ampurdanés, C.; Medina, M.G.; Serratosa, J.; Tusell, J.M.; Saura, J.; Planas, A.; Navarro, P. Tissue plasminogen activator induces microglial inflammation via a noncatalytic molecular mechanism involving activation of mitogen-activated protein kinases and Akt signaling pathways and AnnexinA2 and Galectin-1 receptors. Glia 2012, 60, 526–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, O.; Campion, S.; Perry, V.H.; Murray, C.; Sidenius, N.; Docagne, F.; Cunningham, C. Microglia and the urokinase plasminogen activator receptor/uPA system in innate brain inflammation. Glia 2009, 57, 1802–1814. [Google Scholar] [CrossRef] [Green Version]
- Niaz, A.; Karunia, J.; Mandwie, M.; Keay, K.A.; Musumeci, G.; Al-Badri, G.; Castorina, A. Robust Dopaminergic Differentiation and Enhanced LPS-Induced Neuroinflammatory Response in Serum-Deprived Human SH-SY5Y Cells: Implication for Parkinson’s Disease. J. Mol. Neurosci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kharitonenkov, A.; DiMarchi, R. Fibroblast growth factor 21 night watch: Advances and uncertainties in the field. J. Int. Med. 2017, 281, 233–246. [Google Scholar] [CrossRef]
- Restelli, L.M.; Oettinghaus, B.; Halliday, M.; Agca, C.; Licci, M.; Sironi, L.; Savoia, C.; Hench, J.; Tolnay, M.; Neutzner, A.; et al. Neuronal mitochondrial dysfunction activates the integrated stress response to induce fibroblast growth factor 21. Cell Rep. 2018, 24, 1407–1414. [Google Scholar] [CrossRef] [Green Version]
- Manthripragada, A.D.; Schernhammer, E.S.; Qiu, J.; Friis, S.; Wermuth, L.; Olsen, J.H.; Ritz, B. Non-steroidal anti-inflammatory drug use and the risk of Parkinson’s disease. Neuroepidemiology 2011, 36, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Teema, A.M.; Zaitone, S.A.; Moustafa, Y.M. Ibuprofen or piroxicam protects nigral neurons and delays the development of l-dopa induced dyskinesia in rats with experimental Parkinsonism: Influence on angiogenesis. Neuropharmacology 2016, 107, 432–450. [Google Scholar] [CrossRef]
- Casper, D.; Yaparpalvi, U.; Rempel, N.; Werner, P. Ibuprofen protects dopaminergic neurons against glutamate toxicity in vitro. Neurosci. Lett. 2000, 289, 201–204. [Google Scholar] [CrossRef]
- Poly, T.N.; Islam, M.M.R.; Yang, H.-C.; Li, Y.-C.J. Non-steroidal anti-inflammatory drugs and risk of Parkinson’s disease in the elderly population: A meta-analysis. Eur. J. Clin. Pharmacol. 2019, 75, 99–108. [Google Scholar] [CrossRef]
- Rees, K.; Stowe, R.; Patel, S.; Ives, N.; Breen, K.; Clarke, C.E.; Ben-Shlomo, Y. Non-steroidal anti-inflammatory drugs as disease-modifying agents for Parkinson’s disease: Evidence from observational studies. Cochrane Database Syst. Rev. 2011, 11, CD008454. [Google Scholar] [CrossRef] [PubMed]
- Ferger, B.; Leng, A.; Mura, A.; Hengerer, B.; Feldon, J. Genetic ablation of tumor necrosis factor-alpha (TNF-alpha) and pharmacological inhibition of TNF-synthesis attenuates MPTP toxicity in mouse striatum. J. Neurochem. 2004, 89, 822–833. [Google Scholar] [CrossRef] [PubMed]
- Tomás-Camardiel, M.; Rite, I.; Herrera, A.J.; de Pablos, R.M.; Cano, J.; Machado, A.; Venero, J.L. Minocycline reduces the lipopolysaccharide-induced inflammatory reaction, peroxynitrite-mediated nitration of proteins, disruption of the blood-brain barrier, and damage in the nigral dopaminergic system. Neurobiol. Disease 2004, 16, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Peter, I.; Dubinsky, M.; Bressman, S.; Park, A.; Lu, C.; Chen, N.; Wang, A. Anti–tumor necrosis factor therapy and incidence of Parkinson disease among patients with inflammatory Bowel disease. JAMA Neurol. 2018, 75, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Jing, H.; Wang, S.; Wang, M.; Fu, W.; Zhang, C.; Xu, D. Isobavachalcone attenuates MPTP-induced Parkinson’s disease in mice by inhibition of microglial activation through NF-κB pathway. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Braczynski, A.K.; Schulz, J.B.; Bach, J.-P. Vaccination strategies in tauopathies and synucleinopathies. J. Neurochem. 2017, 143, 467–488. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, D.; Kordower, J.H. Immunotherapy in Parkinson’s disease: Current status and future directions. Neurobiol. Dis. 2019, 132, 104587. [Google Scholar] [CrossRef]
- Mandler, M.; Valera, E.; Rockenstein, E.; Weninger, H.; Patrick, C.; Adame, A.; Santic, R.; Meindl, S.; Vigl, B.; Smrzka, O.; et al. Next-generation active immunization approach for synucleinopathies: Implications for Parkinson’s disease clinical trials. Acta Neuropathol. 2014, 127, 861–879. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Guajardo, V.; Annibali, A.; Jensen, P.H.; Romero-Ramos, M. α-Synuclein vaccination prevents the accumulation of Parkinson Disease like pathologic inclusions in striatum in association with regulatory T Cell recruitment in a rat model. J. Neuropathol. Exp. Neurol. 2013, 72, 624–645. [Google Scholar] [CrossRef] [Green Version]
- Benner, E.J.; Mosley, R.L.; Destache, C.J.; Jackson-Lewis, V.; Nemachek, C.; Green, S.; Przedborski, S.; Gendelman, H. Therapeutic immunization protects dopaminergic neurons in a mouse model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 9435Y40. [Google Scholar] [CrossRef] [Green Version]
- Kirik, D.; Annett, L.E.; Burger, C.; Muzyczka, N.; Mandel, R.; Björklund, A. Nigrostriatal alpha-synucleinopathy induced by viral vector-mediated overexpression of human alpha-synuclein: A new primate model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 2884Y89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marogianni, C.; Sokratous, M.; Dardiotis, E.; Hadjigeorgiou, G.M.; Bogdanos, D.; Xiromerisiou, G. Neurodegeneration and Inflammation—An Interesting Interplay in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 8421. https://doi.org/10.3390/ijms21228421
Marogianni C, Sokratous M, Dardiotis E, Hadjigeorgiou GM, Bogdanos D, Xiromerisiou G. Neurodegeneration and Inflammation—An Interesting Interplay in Parkinson’s Disease. International Journal of Molecular Sciences. 2020; 21(22):8421. https://doi.org/10.3390/ijms21228421
Chicago/Turabian StyleMarogianni, Chrysoula, Maria Sokratous, Efthimios Dardiotis, Georgios M. Hadjigeorgiou, Dimitrios Bogdanos, and Georgia Xiromerisiou. 2020. "Neurodegeneration and Inflammation—An Interesting Interplay in Parkinson’s Disease" International Journal of Molecular Sciences 21, no. 22: 8421. https://doi.org/10.3390/ijms21228421
APA StyleMarogianni, C., Sokratous, M., Dardiotis, E., Hadjigeorgiou, G. M., Bogdanos, D., & Xiromerisiou, G. (2020). Neurodegeneration and Inflammation—An Interesting Interplay in Parkinson’s Disease. International Journal of Molecular Sciences, 21(22), 8421. https://doi.org/10.3390/ijms21228421