Methods and Applications of In Silico Aptamer Design and Modeling




Abstract
:1. Introduction
2. Proteins as Targets of Aptamer Design
2.1. Coagulation-Related Proteins
2.2. Infection-Related Proteins
2.3. Cancer-Related Proteins
2.4. Other Proteins
3. Antibiotics as Targets of Aptamer Design
4. Organophosphates as Targets of In Silico Aptamer Modeling
5. Different Low-Molecular-Weight Compounds as Targets of Aptamer Design
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chushak, Y.; Stone, M.O. In silico selection of RNA aptamers. Nucleic Acids Res. 2009, 37, e87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruspe, S.; Mittelberger, F.; Szameit, K.; Hahn, U. Aptamers as drug delivery vehicles. ChemMedChem 2014, 9, 1998–2011. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Yan, J.; Xiong, H.; Liu, Y.; Peng, D.; Liu, Z. Investigations on the interface of nucleic acid aptamers and binding targets. Analyst 2018, 143, 5317–5338. [Google Scholar] [CrossRef] [PubMed]
- Yuce, M.; Kurt, H. How to make nanobiosensors: Surface modification and characterisation of nanomaterials for biosensing applications. RSC Adv. 2017, 7, 49386–49403. [Google Scholar] [CrossRef] [Green Version]
- Ren, Q.; Ga, L.; Lu, Z.; Ai, J.; Wang, T. Aptamer-functionalized nanomaterials for biological applications. Mater. Chem. Front. 2020, 4, 1569–1585. [Google Scholar] [CrossRef]
- Villalonga, A.; Pérez-Calabuig, A.M.; Villalonga, R. Electrochemical biosensors based on nucleic acid aptamers. Anal. Bioanal. Chem. 2020, 412, 55–72. [Google Scholar] [CrossRef]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef]
- Zhuo, Z.; Yu, Y.; Wang, M.; Li, J.; Zhang, Z.; Liu, J.; Wu, X.; Lu, A.; Zhang, G.; Zhang, B. Recent advances in SELEX technology and aptamer applications in biomedicine. Int. J. Mol. Sci. 2017, 18, 2142. [Google Scholar] [CrossRef] [Green Version]
- Komarova, N.; Kuznetsov, A. Inside the black box: What makes SELEX better? Molecules 2019, 24, 3598. [Google Scholar] [CrossRef] [Green Version]
- Bayat, P.; Nosrati, R.; Alibolandi, M.; Rafatpanah, H.; Abnous, K.; Khedri, M.; Ramezani, M. SELEX methods on the road to protein targeting with nucleic acid aptamers. Biochimie 2018, 154, 132–155. [Google Scholar] [CrossRef]
- Antipova, O.M.; Zavyalova, E.G.; Golovin, A.V.; Pavlova, G.V.; Kopylov, A.M.; Reshetnikov, R.V. Advances in the application of modified nucleotides in SELEX technology. Biochemistry 2018, 83, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Hoinka, J.; Przytycka, T. AptaPLEX—A dedicated, multithreaded demultiplexer for HT-SELEX data. Methods 2016, 106, 82–85. [Google Scholar] [CrossRef] [PubMed]
- McKeague, M.; Wong, R.S.; Smolke, C.D. Opportunities in the design and application of RNA for gene expression control. Nucleic Acids Res. 2016, 44, 2987–2999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamada, M. In silico approaches to RNA aptamer design. Biochimie 2018, 145, 8–14. [Google Scholar] [CrossRef]
- Emami, N.; Pakchin, P.S.; Ferdousi, R. Computational predictive approaches for interaction and structure of aptamers. J. Theor. Biol. 2020, 497, 110268. [Google Scholar] [CrossRef]
- Yan, Z.; Wang, J. SPA-LN: A scoring function of ligand-nucleic acid interactions via optimizing both specificity and affinity. Nucleic Acids Res. 2017, 45, e110. [Google Scholar] [CrossRef]
- Li, X.; Chung, L.W.; Li, G. Multiscale simulations on spectral tuning and the photoisomerization mechanism in fluorescent RNA spinach. J. Chem. Theory Comput. 2016, 12, 5453–5464. [Google Scholar] [CrossRef]
- Kumar, P.; Kumar, A. Nucleobase sequence based building up of reliable QSAR models with the index of ideality correlation using Monte Carlo method. J. Biomol. Struct. Dyn. 2020, 38, 3296–3306. [Google Scholar] [CrossRef]
- Boushaba, K.; Levine, H.; Hamilton, M.N. A mathematical feasibility argument for the use of aptamers in chemotherapy and imaging. Math. Biosci. 2009, 220, 131–142. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Ellington, A.D. Design principles for ligand-sensing, conformation-switching ribozymes. PLoS Comput. Biol. 2009, 5, e1000620. [Google Scholar] [CrossRef] [Green Version]
- Avihoo, A.; Gabdank, I.; Shapira, M.; Barash, D. In silico design of small RNA switches. IEEE Trans. Nanobiosci. 2007, 6, 4–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domin, G.; Findeiß, S.; Wachsmuth, M.; Will, S.; Stadler, P.F.; Mörl, M. Applicability of a computational design approach for synthetic riboswitches. Nucleic Acids Res. 2017, 45, 4108–4119. [Google Scholar] [CrossRef] [Green Version]
- Findeiß, S.; Etzel, M.; Will, S.; Mörl, M.; Stadler, P.F. Design of artificial riboswitches as biosensors. Sensors 2017, 17, 1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, S.; Wang, Y.; Wang, Z.; Zhang, W. Computational methods for modeling aptamers and designing riboswitches. Int. J. Mol. Sci. 2017, 18, 2442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boussebayle, A.; Torka, D.; Ollivaud, S.; Braun, J.; Bofill-Bosch, C.; Dombrowski, M.; Groher, F.; Hamacher, K.; Suess, B. Next-level riboswitch development-implementation of Capture-SELEX facilitates identification of a new synthetic riboswitch. Nucleic Acids Res. 2019, 47, 4883–4895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, C.Y.; Ashrafuzzaman, M.; Mane, J.Y.; Kapty, J.; Mercer, J.R.; Tuszynski, J.A. Entropic fragment-based approach to aptamer design. Chem. Biol. Drug Des. 2011, 78, 1–13. [Google Scholar] [CrossRef]
- Zavyalova, E.; Golovin, A.; Reshetnikov, R.; Mudrik, N.; Panteleyev, D.; Pavlova, G.; Kopylov, A. Novel modular DNA aptamer for human thrombin with high anticoagulant activity. Curr. Med. Chem. 2011, 18, 3343–3350. [Google Scholar] [CrossRef]
- Varizhuk, A.M.; Tsvetkov, V.B.; Tatarinova, O.N.; Kaluzhny, D.N.; Florentiev, V.L.; Timofeev, E.N.; Shchyolkina, A.K.; Borisova, O.F.; Smirnov, I.P.; Grokhovsky, S.L.; et al. Synthesis, characterization and in vitro activity of thrombin-binding DNA aptamers with triazole internucleotide linkages. Eur. J. Med. Chem. 2013, 67, 90–97. [Google Scholar] [CrossRef]
- Tatarinova, O.; Tsvetkov, V.; Basmanov, D.; Barinov, N.; Smirnov, I.; Timofeev, E.; Kaluzhny, D.; Chuvilin, A.; Klinov, D.; Varizhuk, A.; et al. Comparison of the ‘chemical’ and ‘structural’ approaches to the optimization of the thrombin-binding aptamer. PLoS ONE 2014, 9, e89383. [Google Scholar] [CrossRef] [Green Version]
- Mahmood, M.A.; Ali, W.; Adnan, A.; Iqbal, S.M. 3D structural integrity and interactions of single-stranded protein-binding DNA in a functionalized nanopore. J. Phys. Chem. B 2014, 118, 5799–5806. [Google Scholar] [CrossRef]
- Rangnekar, A.; Nash, J.A.; Goodfred, B.; Yingling, Y.G.; LaBean, T.H. Design of potent and controllable anticoagulants using DNA aptamers and nanostructures. Molecules 2016, 21, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Riesen, A.J.; Fadock, K.L.; Deore, P.S.; Desoky, A.; Manderville, R.A.; Sowlati-Hashjin, S.; Wetmore, S.D. Manipulation of a DNA aptamer-protein binding site through arylation of internal guanine residues. Org. Biomol. Chem. 2018, 16, 3831–3840. [Google Scholar] [CrossRef] [PubMed]
- Sgobba, M.; Olubiyi, O.; Ke, S.; Haider, S. Molecular dynamics of HIV1-integrase in complex with 93del—A structural perspective on the mechanism of inhibition. J. Biomol. Struct. Dyn. 2012, 29, 863–877. [Google Scholar] [CrossRef] [PubMed]
- Do, N.Q.; Lim, K.W.; Teo, M.H.; Heddi, B.; Phan, A.T. Stacking of G-quadruplexes: NMR structure of a G-rich oligonucleotide with potential anti-HIV and anticancer activity. Nucleic Acids Res. 2011, 39, 9448–9457. [Google Scholar] [CrossRef]
- Aeksiri, N.; Songtawee, N.; Gleeson, M.P.; Hannongbua, S.; Choowongkomon, K. Insight into HIV-1 reverse transcriptase-aptamer interaction from molecular dynamics simulations. J. Mol. Model. 2014, 20, 2380. [Google Scholar] [CrossRef]
- Nguyen, P.D.M.; Zheng, J.; Gremminger, T.J.; Qiu, L.; Zhang, D.; Tuske, S.; Lange, M.J.; Griffin, P.R.; Arnold, E.; Chen, S.J.; et al. Binding interface and impact on protease cleavage for an RNA aptamer to HIV-1 reverse transcriptase. Nucleic Acids Res. 2020, 48, 2709–2722. [Google Scholar] [CrossRef]
- Song, Y.; Song, J.; Wei, X.; Huang, M.; Sun, M.; Zhu, L.; Lin, B.; Shen, H.; Zhu, Z.; Yang, C. Discovery of aptamers targeting the receptor-binding domain of the SARS-CoV-2 spike glycoprotein. Anal. Chem. 2020, 92, 9895–9900. [Google Scholar] [CrossRef]
- Sabri, M.Z.; Abdul Hamid, A.A.; Sayed Hitam, S.M.; Abdul Rahim, M.Z. In silico screening of aptamers configuration against hepatitis B surface antigen. Adv. Bioinform. 2019, 2019, 6912914. [Google Scholar] [CrossRef] [Green Version]
- Soon, S.; Nordin, N.A. In silico predictions and optimization of aptamers against Streptococcus agalactiae surface protein using computational docking. Mater. Today Proc. 2019, 16, 2096–2100. [Google Scholar] [CrossRef]
- Rockey, W.M.; Hernandez, F.J.; Huang, S.Y.; Cao, S.; Howell, C.A.; Thomas, G.S.; Liu, X.Y.; Lapteva, N.; Spencer, D.M.; McNamara, J.O.; et al. Rational truncation of an RNA aptamer to prostate-specific membrane antigen using computational structural modeling. Nucleic Acid Ther. 2011, 21, 299–314. [Google Scholar] [CrossRef] [Green Version]
- Bavi, R.; Liu, Z.; Han, Z.; Zhang, H.; Gu, Y. In silico designed RNA aptamer against epithelial cell adhesion molecule for cancer cell imaging. Biochem. Biophys. Res. Commun. 2019, 509, 937–942. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.R.; Weber, J.K.; Yin, W.; Huynh, T.; Duan, W.; Zhou, R. In silico design and validation of high-affinity RNA aptamers targeting epithelial cellular adhesion molecule dimers. Proc. Natl. Acad. Sci. USA 2020, 117, 8486–8493. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.L.; Cui, H.F.; Du, J.F.; Lv, Q.Y.; Songa, X. In silico post-SELEX screening and experimental characterizations for acquisition of high affinity DNA aptamers against carcinoembryonic antigen. RSC Adv. 2019, 9, 6328–6334. [Google Scholar] [CrossRef] [Green Version]
- Santini, B.L.; Zúñiga-Bustos, M.; Vidal-Limon, A.; Alderete, J.B.; Águila, S.A.; Jiménez, V.A. In silico design of novel mutant anti-MUC1 aptamers for targeted cancer therapy. J. Chem. Inf. Model. 2020, 60, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Knight, C.G.; Platt, M.; Rowe, W.; Wedge, D.C.; Khan, F.; Day, P.J.; McShea, A.; Knowles, J.; Kell, D.B. Array-based evolution of DNA aptamers allows modelling of an explicit sequence-fitness landscape. Nucleic Acids Res. 2009, 37, e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.P.; Kumar, J.V.; Huang, C.J.; Chen, W.Y. Computational selection of RNA aptamer against angiopoietin-2 and experimental evaluation. Biomed. Res. Int. 2015, 2015, 658712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cataldo, R.; Ciriaco, F.; Alfinito, E. A validation strategy for in silico generated aptamers. Comput. Biol. Chem. 2018, 77, 123–130. [Google Scholar] [CrossRef] [Green Version]
- Shcherbinin, D.S.; Gnedenko, O.V.; Khmeleva, S.A.; Usanov, S.A.; Gilep, A.A.; Yantsevich, A.V.; Shkel, T.V.; Yushkevich, I.V.; Radko, S.P.; Ivanov, A.S.; et al. Computer-aided design of aptamers for cytochrome p450. J. Struct. Biol. 2015, 191, 112–119. [Google Scholar] [CrossRef]
- Ahirwar, R.; Nahar, S.; Aggarwal, S.; Ramachandran, S.; Maiti, S.; Nahar, P. In silico selection of an aptamer to estrogen receptor alpha using computational docking employing estrogen response elements as aptamer-alike molecules. Sci. Rep. 2016, 6, 21285. [Google Scholar] [CrossRef] [Green Version]
- Heiat, M.; Najafi, A.; Ranjbar, R.; Latifi, A.M.; Rasaee, M.J. Computational approach to analyze isolated ssDNA aptamers against angiotensin II. J. Biotechnol. 2016, 230, 34–39. [Google Scholar] [CrossRef]
- Rabal, O.; Pastor, F.; Villanueva, H.; Soldevilla, M.M.; Hervas-Stubbs, S.; Oyarzabal, J. In silico aptamer docking studies: From a retrospective validation to a prospective case study-TIM3 aptamers binding. Mol. Ther. Nucleic Acids. 2016, 5, e376. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Lietard, J.; Abou Assi, H.; Gómez-Pinto, I.; González, C.; Somoza, M.M.; Damha, M.J. Mapping the affinity landscape of Thrombin-binding aptamers on 2′F-ANA/DNA chimeric G-Quadruplex microarrays. Nucleic Acids Res. 2017, 45, 1619–1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Olson, W.K. 3DNA: A software package for the analysis, rebuilding, and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003, 31, 5108–5121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pronk, S.; Pall, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Sorin, E.J.; Pande, V.S. Exploring the helix-coil transition via all-atom equilibrium ensemble simulations. Biophys. J. 2005, 88, 2472–2493. [Google Scholar] [CrossRef] [Green Version]
- Pérez, A.; Marchán, I.; Svozil, D.; Sponer, J.; Cheatham, T.E.; Laughton, C.A.; Orozco, M. Refinement of the AMBER force field for nucleic acids: Improving the description of alpha/gamma conformers. Biophys. J. 2007, 92, 3817–3829. [Google Scholar] [CrossRef] [Green Version]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef] [Green Version]
- Tsvetkov, V.; Varizhuk, A.; Pozmogova, G.; Smirnov, I.; Kolganova, N.; Timofeev, E. A universal base in a specific role: Tuning up a thrombin aptamer with 5-nitroindole. Sci. Rep. 2015, 5, 16337. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kale, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Brooks, B.R.; Brooks, C.L., 3rd; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, D.W.; Venkatraman, V. Ultra-fast FFT protein docking on graphics processors. Bioinformatics 2010, 26, 2398–2405. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery Consortium; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Xu, X.; Zhao, P.; Chen, S.J. Vfold: A web server for RNA structure and folding thermodynamics prediction. PLoS ONE 2014, 9, e107504. [Google Scholar] [CrossRef]
- Zhang, D.; Chen, S.J. IsRNA: An iterative simulated reference state approach to modeling correlated interactions in RNA folding. J. Chem. Theor. Comput. 2018, 14, 2230–2239. [Google Scholar] [CrossRef]
- Xu, X.; Qiu, L.; Yan, C.; Ma, Z.; Grinter, S.Z.; Zou, X. Performance of MDockPP in CAPRI rounds 28–29 and 31–35 including the prediction of water-mediated interactions. Proteins 2017, 85, 424–434. [Google Scholar] [CrossRef] [Green Version]
- Benfenati, E.; Toropov, A.A.; Toropova, A.P.; Manganaro, A.; Gonella Diaza, R. coral software: QSAR for anticancer agents. Chem. Biol. Drug Des. 2011, 77, 471–476. [Google Scholar] [CrossRef]
- Musafia, B.; Oren-Banaroya, R.; Noiman, S. Designing anti-influenza aptamers: Novel quantitative structure activity relationship approach gives insights into aptamer-virus interaction. PLoS ONE 2014, 9, e97696. [Google Scholar] [CrossRef]
- Song, J.; Zheng, Y.; Huang, M.; Wu, L.; Wang, W.; Zhu, Z.; Song, Y.; Yang, C. A Sequential Multidimensional Analysis Algorithm for Aptamer Identification based on Structure Analysis and Machine Learning. Anal. Chem. 2020, 92, 3307–3314. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wang, J.; Huang, Y.; Xiao, Y. 3dRNA v2.0: An updated Web server for RNA 3D structure prediction. Int. J. Mol. Sci. 2019, 20, 4116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellaousov, S.; Reuter, J.S.; Seetin, M.G.; Mathews, D.H. RNAstructure: Web servers for RNA secondary structure prediction and analysis. Nucleic Acids Res. 2013, 41, 471–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Case, D.A.; Cheatham, T.E., 3rd; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.Y.; Zou, X. MDockPP: A hierarchical approach for protein-protein docking and its application to CAPRI rounds 15-19. Proteins 2010, 78, 3096–3103. [Google Scholar] [CrossRef] [Green Version]
- Patriarca, C.; Macchi, R.M.; Marschner, A.K.; Mellstedt, H. Epithelial cell adhesion molecule expression (CD326) in cancer: A short review. Cancer Treat. Rev. 2012, 38, 68e75. [Google Scholar] [CrossRef]
- Lorenz, R.; Bernhart, S.H.; Höner Zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef]
- Aliev, A.E.; Kulke, M.; Khaneja, H.S.; Chudasama, V.; Sheppard, T.D.; Lanigan, R.M. Motional timescale predictions by molecular dynamics simulations: Case study using proline and hydroxyproline sidechain dynamics. Proteins 2014, 82, 195e215. [Google Scholar] [CrossRef] [Green Version]
- Bavi, R.; Kumar, R.; Choi, L.; Woo Lee, K. Exploration of novel inhibitors for bruton’s tyrosine kinase by 3D QSAR modeling and molecular dynamics simulation. PLoS ONE 2016, 11, e0147190. [Google Scholar] [CrossRef] [Green Version]
- Cheatham, T.E., 3rd; Case, D.A. Twenty-five years of nucleic acid simulations. Biopolymers 2013, 99, 969–977. [Google Scholar] [CrossRef]
- Roberts, V.A.; Thompson, E.E.; Pique, M.E.; Perez, M.S.; Ten Eyck, L.F. DOT2: Macromolecular docking with improved biophysical models. J. Comput. Chem. 2013, 34, 1743–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef] [PubMed]
- Markham, N.R.; Zuker, M. UNAFold: Software for nucleic acid folding and hybridization. Methods Mol. Biol. 2008, 453, 3–31. [Google Scholar] [PubMed]
- Sato, K.; Hamada, M.; Asai, K.; Mituyama, T. CentroidFold: A web server for RNA secondary structure prediction. Nucleic Acids Res. 2009, 37, 277–280. [Google Scholar] [CrossRef] [Green Version]
- Popenda, M.; Szachniuk, M.; Antczak, M.; Purzycka, K.J.; Lukasiak, P.; Bartol, N.; Blazewicz, J.; Adamiak, R.W. Automated 3D structure composition for large RNAs. Nucleic Acids Res. 2012, 40, e112. [Google Scholar] [CrossRef]
- Boniecki, M.J.; Lach, G.; Dawson, W.K.; Tomala, K.; Lukasz, P.; Soltysinski, T.; Rother, K.M.; Bujnicki, J.M. SimRNA: A coarse-grained method for RNA folding simulations and 3D structure prediction. Nucleic Acids Res. 2016, 44, e63. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function: Efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Allen, W.J.; Balius, T.E.; Mukherjee, S.; Brozell, S.R.; Moustakas, D.T.; Lang, P.T.; Case, D.A.; Kuntz, I.D.; Rizzo, R.C. DOCK 6: Impact of new features and current docking performance. J. Comput. Chem. 2015, 36, 1132–1156. [Google Scholar] [CrossRef] [Green Version]
- Vries, S.J.; de Dijk, M.; van Bonvin, A.M. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 2010, 5, 883–897. [Google Scholar] [CrossRef] [Green Version]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, 363–367. [Google Scholar] [CrossRef] [Green Version]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.Y.; Chou, F.C.; Das, R. Modeling complex RNA tertiary folds with Rosetta. Methods Enzymol. 2015, 553, 35–64. [Google Scholar] [PubMed]
- Huang, Y.; Liu, S.; Guo, D.; Li, L.; Xiao, Y. A novel protocol for three-dimensional structure prediction of RNA-protein complexes. Sci. Rep. 2013, 3, 1887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, M.; Strom, M.; Hammond, D.S.; Shigdar, S. Anything you can do, I can do better: Can aptamers replace antibodies in clinical diagnostic applications? Molecules 2019, 24, 4377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilgu, M.; Yan, S.; Khounlo, R.M.; Lamm, M.H.; Nilsen-Hamilton, M. Common secondary and tertiary structural features of aptamer-ligand interaction shared by RNA aptamers with different primary sequences. Molecules 2019, 24, 4535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khoshbin, Z.; Housaindokht, M.R. Computer-aided aptamer design for sulfadimethoxine antibiotic: Step by step mutation based on MD simulation approach. J. Biomol. Struct. Dyn. 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gruber, A.R.; Lorenz, R.; Bernhart, S.H.; Neubock, R.; Hofacker, I.L. The Vienna RNA Websuite. Nucleic Acids Res. 2008, 36, W70–W74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhagwat, M.; Aravind, L. PSI-BLAST tutorial. Methods Mol. Biol. 2007, 395, 177–186. [Google Scholar]
- Housaindokht, M.R.; Bozorgmehr, M.R.; Bahrololoom, M. Analysis of ligand binding to proteins using molecular dynamics simulations. J. Theor. Biol. 2008, 254, 294–300. [Google Scholar] [CrossRef]
- Kim, N.; Gan, H.H.; Schlick, T. A computational proposal for designing structured RNA pools for in vitro selection of RNAs. RNA 2007, 13, 478–492. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.; McKeague, M.; Pitre, S.; Dumontier, M.; Green, J.; Golshani, A.; Derosa, M.C.; Dehne, F. Computational approaches toward the design of pools for the in vitro selection of complex aptamers. RNA 2010, 16, 2252–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashrafuzzaman, M.; Tseng, C.Y.; Kapty, J.; Mercer, J.R.; Tuszynski, J.A. A computationally designed DNA aptamer template with specific binding to phosphatidylserine. Nucleic Acid Ther. 2013, 23, 418–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jokar, M.; Safaralizadeh, M.H.; Hadizadeh, F.; Rahmani, F.; Kalani, M.R. Apta-nanosensor preparation and in vitro assay for rapid diazinon detection using a computational molecular approach. J. Biomol. Struct. Dyn. 2017, 35, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Ruan, M.; Seydou, M.; Noel, V.; Piro, B.; Maurel, F.; Barbault, F. Molecular dynamics simulation of a RNA aptasensor. J. Phys. Chem. B 2017, 121, 4071–4080. [Google Scholar] [CrossRef]
- Belinskaia, D.A.; Avdonin, P.V.; Avdonin, P.P.; Jenkins, R.O.; Goncharov, N.V. Rational in silico design of aptamers for organophosphates based on the example of paraoxon. Comput. Biol. Chem. 2019, 80, 452–462. [Google Scholar] [CrossRef]
- Carothers, J.M.; Oestreich, S.C.; Davis, J.H.; Szostak, J.W. Informational complexity and functional activity of RNA structures. J. Am. Chem. Soc. 2004, 126, 5130–5137. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.E.I.; Yang, R.; Cieplak, P.; Luo, R.A.Y.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Kikin, O.; D’Antonio, L.; Bagga, P.S. QGRS Mapper: A web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res. 2006, 34, W676–W682. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.H.; Tsai, C.W.; Wu, J.W.; Ruaan, R.C.; Chen, W.Y. Molecular dynamics simulation of the induced-fit binding process of DNA aptamer and L-argininamide. Biotechnol. J. 2012, 7, 1367–1375. [Google Scholar] [CrossRef]
- Albada, H.B.; Golub, E.; Willner, I. Computational docking simulations of a DNA-aptamer for argininamide and related ligands. J. Comput. Aided Mol. Des. 2015, 29, 643–654. [Google Scholar] [CrossRef]
- Verdonck, L.; Buyst, D.; de Vries, A.M.; Gheerardijn, V.; Madder, A.; Martins, J.C. Tethered imidazole mediated duplex stabilization and its potential for aptamer stabilization. Nucleic Acids Res. 2018, 46, 11671–11686. [Google Scholar] [CrossRef]
- Wachsmuth, M.; Findeiß, S.; Weissheimer, N.; Stadler, P.F.; Mörl, M. De novo design of a synthetic riboswitch that regulates transcription termination. Nucleic Acids Res. 2013, 41, 2541–2551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Xia, X.; Luo, Z.; Liang, H.; Shakhnovich, E. Searching the sequence space for potent aptamers using SELEX in silico. J. Chem. Theory Comput. 2015, 11, 5939–5946. [Google Scholar] [CrossRef] [PubMed]
- Jokar, M.; Safaralizadeh, M.H.; Hadizadeh, F.; Rahmani, F.; Kalani, M.R. Design and evaluation of an apta-nano-sensor to detect acetamiprid in vitro and in silico. J. Biomol. Struct. Dyn. 2016, 34, 2505–2517. [Google Scholar] [CrossRef]
- Tomita, Y.; Morita, Y.; Suga, H.; Fujiwara, D. DNA module platform for developing colorimetric aptamer sensors. Biotechniques 2016, 60, 285–292. [Google Scholar] [CrossRef] [Green Version]
- Hilder, T.A.; Hodgkiss, J.M. The bound structures of 17β-estradiol-binding aptamers. Chemphyschem 2017, 18, 1881–1887. [Google Scholar] [CrossRef]
- Zhao, M.; Li, W.; Liu, K.; Li, H.; Lan, X. C4-HSL aptamers for blocking qurom sensing and inhibiting biofilm formation in Pseudomonas aeruginosa and its structure prediction and analysis. PLoS ONE 2019, 14, e0212041. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Source | Target | Computational Methods | Software | Description |
|---|---|---|---|---|
| [26] | Thrombin | Structure prediction, molecular dynamics (MD) | 3D-DART, Amber 10 | Molecular dynamics along with entropic fragment-based approach (EFBA) allowed designing a DNA aptamer, which was surpassed by an aptamer obtained using SELEX prior to it. |
| [27] | Thrombin | Structure prediction, MD | PyMOL 1.1, 3DNA, GROMACS 4.0 | In silico calculations were accompanied by an in vitro thrombin inhibition assay. Two new thrombin aptamers, a 29-mer and a 31-mer with high inhibitory activity, were obtained. |
| [28,29] | Thrombin | MD | Amber 8 | Novel triazole-modified and duplex-added aptamers showed potent thrombin-inhibiting activity. |
| [30] | Thrombin | MD | NAMD | DNA-coated nanopore for protein detection was investigated. |
| [31] | Thrombin | MD | Amber | The in silico-designed aptamer demonstrated seven times higher efficiency than previously known anti-thrombin aptamers. |
| [32] | Thrombin | MD | Amber 12 | It was shown that the internal 8-aryl-guanine modification can manipulate the interactions between the DNA bases and the amino acid residues of thrombin. Nevertheless, guanine arylation at the G-tetrad reduced thrombin-binding affinity. |
| [33] | HIV1 integrase | Docking, MD | Hex, GROMACS | MD simulation was performed for the 93del/HIV1 integrase complex. HIV1 integrase interactions with the aptamer inhibited HIV1 integrase interactions with DNA. |
| [34] | HIV1 integrase | MD | Amber | Molecular dynamics were accompanied by nuclear magnetic resonance (NMR) spectroscopy and circular dichroism experiments. T30695 aptamer had a higher interaction energy (−116.4 kcal mol−1) than the previously known 93del aptamer (−103.4 kcal mol−1). |
| [35] | HIV1 reverse transcriptase (HIV1 RT) | MD | GROMACS 4.5 | T1.1 RNA aptamer complex with HIV1 RT was more stable than that with a DNA substrate. |
| [36] | HIV1 RT | Structure prediction, docking, MD | Vfold2D, IsRNA, MDockPR, NAMD | The combination of in silico modeling and NMR allowed the identification of structural RNA elements critical for HIV1 RT inhibition and the determination of the role of UCAA motif in RT–aptamer interaction. |
| [18] | Influenza hemagglutinin | QSAR | CORAL | Experimental pIC50 values were used as a target parameter during QSAR modeling. The study resulted in the design of nine new aptamers with high inhibitory activity. |
| [37] | SARS-CoV-2 spike glycoprotein | Structure prediction, docking, MD | SMART-Aptamer 2, MFold, RNAComposer | Two potent and selective DNA aptamers were designed with equilibrium dissociation constant (Kd) values of 5.8 and 19.9 nM. |
| [38] | Hepatitis B surface antigen (HBsAg) | Structure prediction, docking, MD | Mfold, RNAComposer, AutoDock Vina, GROMACS 5.1 | It was determined that HBsAg/aptamer interactions were stabilized by the dynamic hydrogen bond formation between the active amino acid residues (“a” determinant region) and nucleotides. |
| [39] | Streptococcus agalactiae surface protein | Structure prediction, docking | Mfold, 3dRNA 2.0, AutoDock Vina | All seven RNA aptamers designed carried a hairpin. The best aptamer was a 40-mer with predicted ΔG equal to −14.7 kcal mol−1 and predicted affinity equal to −16.3 kcal mol−1. |
| [40] | Prostate-specific membrane antigen (PSMA) | Structural prediction, docking | RNAstructure 4.6, Amber, MDockPP | Using the “rational truncation” technique, bases were removed from the aptamer to predict the secondary structure of the remaining oligonucleotide. Molecular docking allowed the identification of binding sites of the aptamers on PSMA. |
| [41] | Epithelial adhesion molecule (EpCAM) | Structure prediction, docking, MD | Vienna RNA, Rosetta, AutoDock Vina, GROMACS 5.0 | Flow cytometry and fluorescence microscopy showed that in silico-designed RNA aptamer interacts specifically with the cells that express EpCAM but not with the EpCAM-negative cells. |
| [42] | EpCAM | Structure prediction, docking, MD | Mfold, Dot 2.0, NAMD 2 | The binding modes of aptamers were first predicted and then optimized with MD and docking. Titration calorimetry experiments confirmed that the designed aptamers possessed high affinity to EpCAM. |
| [43] | Carcinoembryonic antigen (CEA) | Structure prediction, docking | Mfold, RNAComposer, ZDOCK | According to ZDOCK, parent sequence with ATG attached to the 3′-end and GAC sequence attached to the 5′-end had the highest score among the designed aptamers. The high affinity of the developed aptamers was confirmed experimentally by bilayer interferometry. |
| [44] | Transmembrane glycoprotein mucin 1 (MUC1) | Docking, MD | AutoDock Vina, Amber 16 | MD, molecular mechanics Generalized Born surface area (MM-GBSA), and conformational analysis revealed novel anti-MUC1 aptamer that might be used in anti-cancer therapy. |
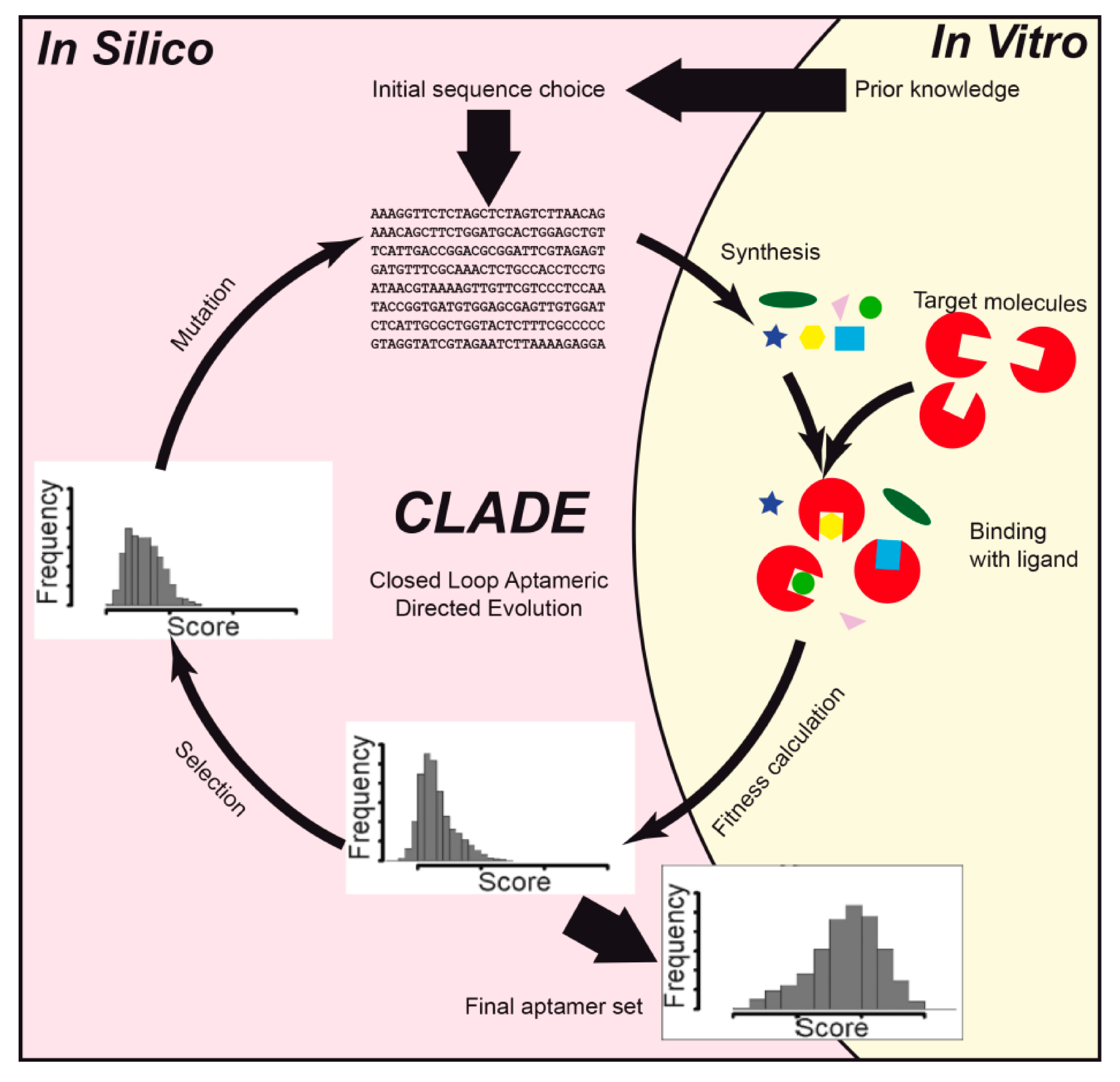
| [45] | Allophycocyanin | Structure prediction, statistical analysis | UNAFold 3.4, R | A joint theoretical-experimental approach, called closed loop aptameric directed evolution (CLADE), was used when 44,131 aptamers were analyzed using the DNA microarray technique. Statistical analysis was done using random forest, regression tree, and genetic programming. |
| [46] | Angiopoietin-2 (Ang2) | Structure prediction, docking | Centroid-Fold, RNAComposer, Discovery Studio 3.5 | Surface plasmon resonance along with Zrank algorithm realized in DS 3.5 allowed finding an RNA aptamer with high target-binding affinity. |
| [47] | Ang2 | Structure prediction, docking | SimRNA, AutoDock Vina | The calculated effective affinities of the Ang2/aptamer complexes were in agreement with the experiment. |
| [48] | Cytochrome p450 | Docking, molecular dynamics | DOCK 6.5, SYBYL 8.1, Amber 9 | A series of aptamers was designed and showed selective affinity toward cytochrome p450. |
| [49] | Estrogen receptor alpha (ERα) | Docking | AutoDock Vina, Haddock, PatchDock | The aptamer was designed based on independent docking analysis in three different programs and was validated by measuring the thermodynamic parameters of ERα/aptamer interactions using isothermal titration calorimetry. |
| [50] | Angiotensin II | Structure prediction, docking | Mfold 3.1, RNAComposer, ZDOCK 3.0 | The interactions of the aptamers with the protein were analyzed by means of surface plasmon resonance spectroscopy and were consistent with in silico data. |
| [51] | T-cell immunoglobulin mucin-3 (TIM-3) | Structure prediction, docking | RNAstructure 5.3, Rosetta, 3dRPC | Docking scoring parameters were analyzed along with experimental data. Binding sites and binding modes in protein/aptamer complexes were identified. |
| Source | Target | Computational Methods | Software | Description |
|---|---|---|---|---|
| [1] | Gentamicin, neomycin, tobramycin | Structure prediction, docking | Vienna RNA, Rosetta, Amber 10, AutoDock 4.0 | The procedure for the selection of aptamers was rather complicated and included the free energy of secondary structure formation calculation, RNA geometry optimization, and rigid docking. The predicted binding energies were in good agreement with experimental values. |
| [22] | Tetracycline, streptomycin | Structure prediction | RNAFold | Riboswitches were designed using randomly generated spacers with a length from 6 to 20 bases. The in silico design was based on a minimal free energy calculation, which consisted of an antibiotic aptamer, a spacer, a complementary part for the aptamer, and a poly-U sequence at the 3′-end. In the presence of tetracycline, the expression of β-galactosidase was induced in E. coli, resulting in the increase of the enzyme’s activity. |
| [95] | Neomycin-B | Structure prediction, MD | Mfold, GROMACS | Experimental NMR and titration colorimetry studies combined with MD simulations revealed that, despite the difference in nucleotide sequence, the structural and dynamical features of the studied aptamers were similar. The affinity of the aptamers toward other aminoglycosides was shown to be lower compared to the target. |
| [96] | Sulfadimethoxine | Structure prediction, MD | PSI-Blast, GROMACS 5.1 | The aptamer’s affinity to the target was determined through the calculation of binding Gibbs free energy using the MM-PBSA method. The designing procedure was done repeatedly and resulted in a creation of mutant aptamers with the improved affinity to sulfadimethoxine. |
| Source | Target | Computational Methods | Software | Description |
|---|---|---|---|---|
| [100] | Guanosine triphosphate (GTP) | Graph theory, matrix analysis | RAGPOOLS | An approach for engineering RNA pools used an exact set of starting sequences and certain mutation ratios in specific locations within a random region. To produce these key parameters, graph theory and matrix analysis were used. The initial aptamer pools acquired by the described methodology provided the selection of RNAs with higher affinity when compared to the in vitro pools. |
| [1] | Adenosine triphosphate (ATP), flavin mononucleotide (FMN) | Structure prediction, docking | Vienna RNA, Rosetta, Amber 10, AutoDock 4.0 | Both 35- and 40-base RNA aptamers were designed toward FMN and ATP, respectively. The in silico-predicted binding energy of, for example, FMN was in agreement with the experimental binding energy, −7.7 kcal mol−1 and −8.6 kcal mol−1, respectively. |
| [101] | ATP | Structure prediction | Vienna RNA, Mfold | Two methods of improvement of RNA/DNA aptamer complexity were created: random filtering and genetic filtering. One of the obtained 5-way junction aptamers demonstrated improved Kd values compared to those of native ATP aptamers. |
| [28] | Phosphatidylserine (PS) | Molecular dynamics (MD) | Amber 10 | Molecular dynamics along with entropic fragment–based approach (EFBA) allowed designing a DNA 6-mer, which, however, possessed low binding energy. |
| [102] | PS | MD | Amber 11 | The EFBA algorithm was applied to design a DNA aptamer that binds specifically to PS. This study identified the 5′-AAAGAC-3′ sequence as a prospective ssDNA aptamer for PS detection. |
| [103] | Diazinon | Structure prediction, docking, MD | Mfold, RNAComposer, AutoDock 4.2, GROMACS 4.5 | Flexible ligand/receptor docking along with MD calculations under the NVT ensemble (10-ns trajectory) showed that G-quadruplex–forming aptamer is reliable for diazinon sensing. |
| [104] | FMN | MD | Amber 12 | The binding energy of FMN/RNA complex was evaluated using MM-GBSA. FMN/aptamer binding increased significantly when the system was immobilized on the surface of gold, which is in accordance with the experimental data. |
| [105] | Paraoxon | Structure prediction, docking, MD | HyperChem, Discovery Studio, AutoDock 4.2, GROMACS 5.0 | The T17C mutation allowed the improvement of the affinity between aptamer and ligand (from −31.0 kcal mol−1 to −32.3 kcal mol−1), and the T17C-C18T double mutation increased the effectiveness of ligand binding (−32.8 kcal mol−1). |
| Source | Target | Computational Methods | Software | Description |
|---|---|---|---|---|
| [1] | Arginine, codeine, guanine, isoleucine, theophylline | Structure prediction, docking | Vienna RNA, Rosetta, Amber 10, AutoDock 4.0 | Rigid docking binding energy predictions were in good agreement with experimental values, which confirms good performance of the applied aptamer design methodology. |
| [112] | L-Argininamide (L-Arm) | MD | NAMD 2.6 | G10, C16, C9, A12, and C17 bases were significant for aptamer/L-Arm binding, which is important for further aptamer design. |
| [113] | L-Arm, D-Arm, L-Arg, D-Arg, agmatine, ethyl-guanidine, L-Lys, N-methyl L-Arg | Docking | AutoDock 4.0 | The interaction of eight arginine (Arg) like ligands with a DNA aptamer was analyzed. D-Arm possessed the highest affinity toward the aptamer. Theoretically defined binding energies and the Kd of ligands were in good agreement with experimentally determined values. |
| [114] | L-Arm | Structure prediction, MD | Discovery Studio 4.0, Amber 12 | MD simulations of 50 ns were accompanied with UV spectroscopy and NMR. Thermal stabilizing effects occurred upon addition of the imidazole-tethered thymidines. Multiple imidazole moieties also maintained L-Arm binding capacity, which enhanced aptamer efficacy. |
| [115] | Theophylline | Structure prediction | RNAFold 2.0 | The energy difference between the free energy of a riboswitch and a ligand-free aptamer was calculated. Several riboswitches were experimentally tested, and some of them showed ligand-dependent control of gene expression in E. coli, demonstrating that it is possible to design riboswitches for transcription regulation. |
| [116] | Theophylline | Structure prediction, MD | X3DNA, GROMACS 4.5 | Six potent aptamers designed in silico were experimentally determined to bind theophylline with high affinity: Kd was equal to 0.16–0.52 μM, whereas Kd of the original theophylline/RNA complex was equal to 0.32 μM. |
| [117] | Acetamiprid | Structure prediction, docking, MD | Mfold, RNA Composer, AutoDock, NAMD 2.9 | A DNA-based aptasensor was designed for the detection of acetamiprid. Docking revealed two loops as active sites in the aptamer. Circular dichroism spectroscopy and colorimetry confirmed aptamer folding due to stem-loop formation upon acetamiprid binding. |
| [118] | Patulin | Structure prediction | UNAFold 3.8 | Microarray aptamer analysis was combined with in silico secondary structure prediction. In silico studies applied three conditions to the aptamers: (1) presence of a predicted secondary DNA structure producing one hairpin loop without a ligand, (2) hairpin loop with a length from 3 to 7 bases, and (3) stem length from 6 to 9 bases. As a result, a novel patulin aptamer was optimized. |
| [119] | 17β-estradiol (E2) | Structure prediction, docking, MD | Mfold, RNAstructure, ZDOCK, RNAComposer, NAMD 2.10 | Rigid docking of aptamers to E2 was used along with a 30-ns MD. It was demonstrated that E2 binds to a thymine loop region common to all E2-specific aptamers. |
| [120] | N-butanoyl-L-homoserine lactone (C4-HSL) | Structure prediction | RNAstructure 5.6, 3dRNA | The 2-D and 3-D RNA structure predictions showed that SELEX-designed aptamers possessed a conservative Y-shaped structural unit, which is probably responsible for C4-HSL binding. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buglak, A.A.; Samokhvalov, A.V.; Zherdev, A.V.; Dzantiev, B.B. Methods and Applications of In Silico Aptamer Design and Modeling. Int. J. Mol. Sci. 2020, 21, 8420. https://doi.org/10.3390/ijms21228420
Buglak AA, Samokhvalov AV, Zherdev AV, Dzantiev BB. Methods and Applications of In Silico Aptamer Design and Modeling. International Journal of Molecular Sciences. 2020; 21(22):8420. https://doi.org/10.3390/ijms21228420
Chicago/Turabian StyleBuglak, Andrey A., Alexey V. Samokhvalov, Anatoly V. Zherdev, and Boris B. Dzantiev. 2020. "Methods and Applications of In Silico Aptamer Design and Modeling" International Journal of Molecular Sciences 21, no. 22: 8420. https://doi.org/10.3390/ijms21228420
APA StyleBuglak, A. A., Samokhvalov, A. V., Zherdev, A. V., & Dzantiev, B. B. (2020). Methods and Applications of In Silico Aptamer Design and Modeling. International Journal of Molecular Sciences, 21(22), 8420. https://doi.org/10.3390/ijms21228420