Engineering Next-Generation CAR-T Cells for Better Toxicity Management


Abstract
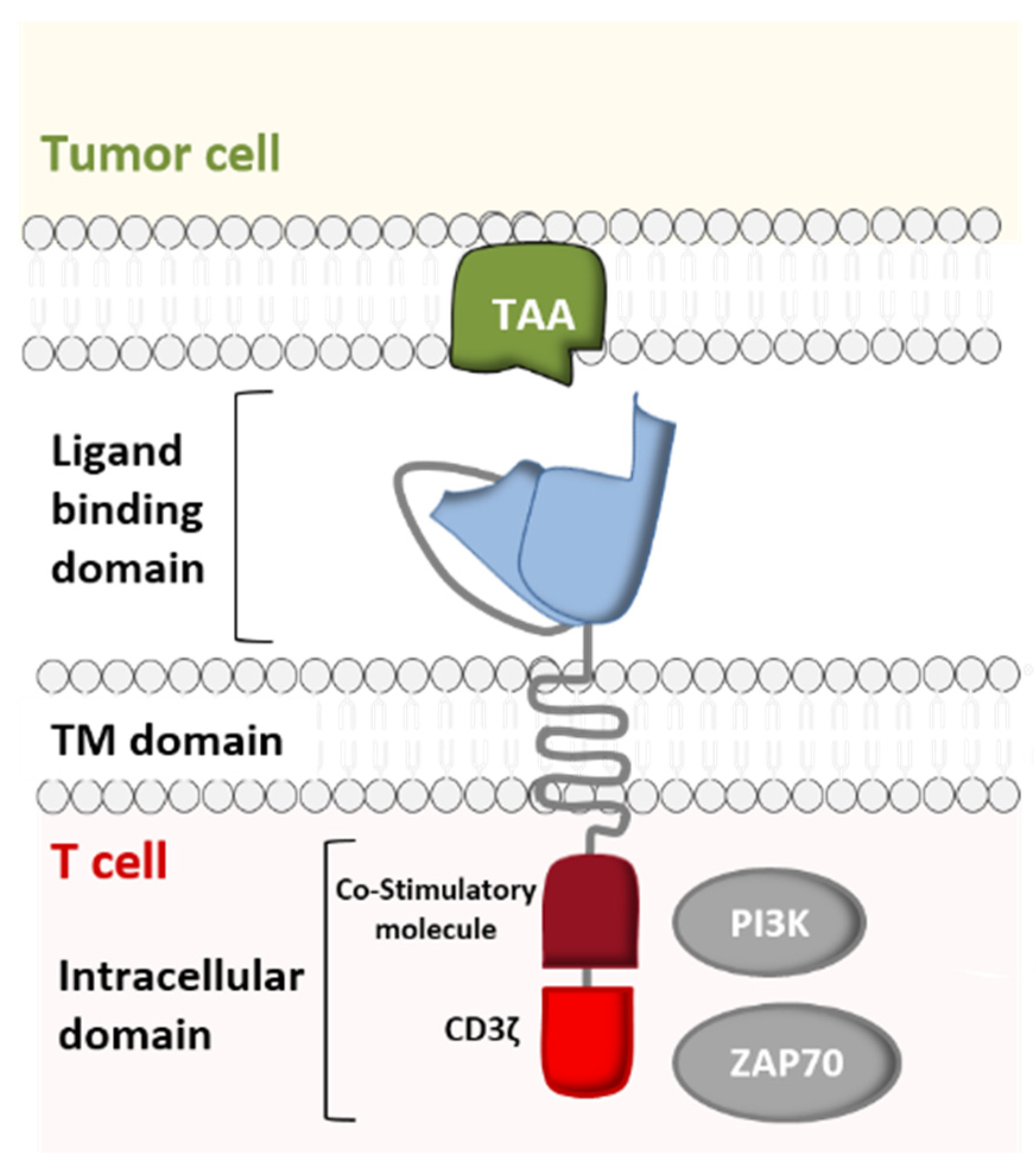
:1. Introduction
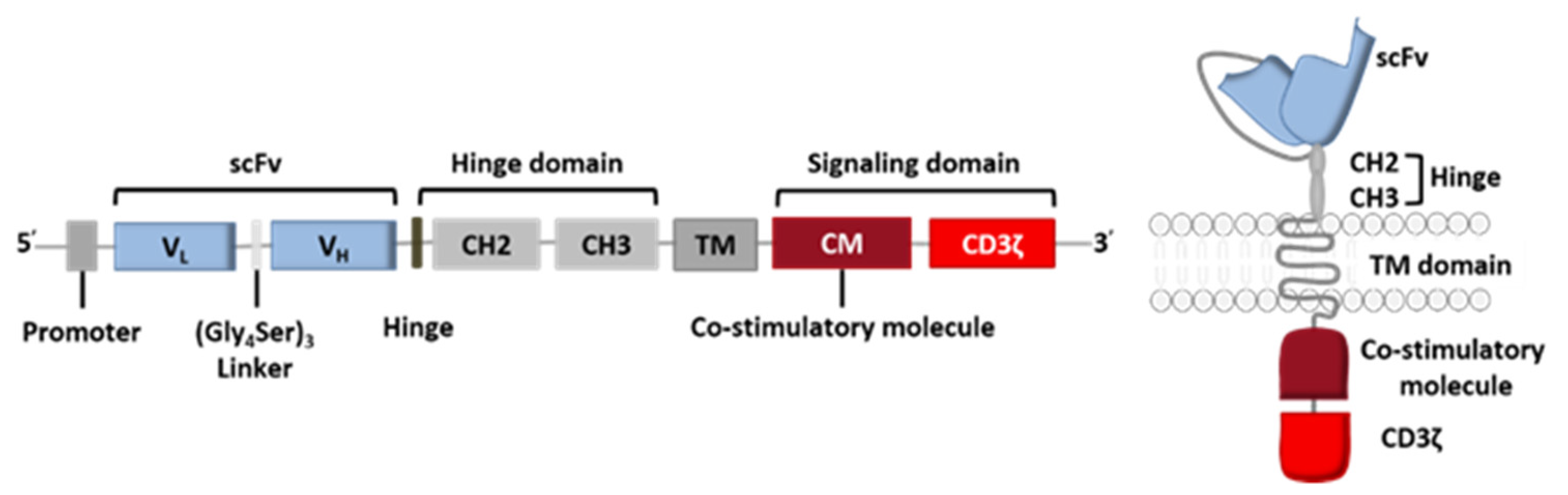
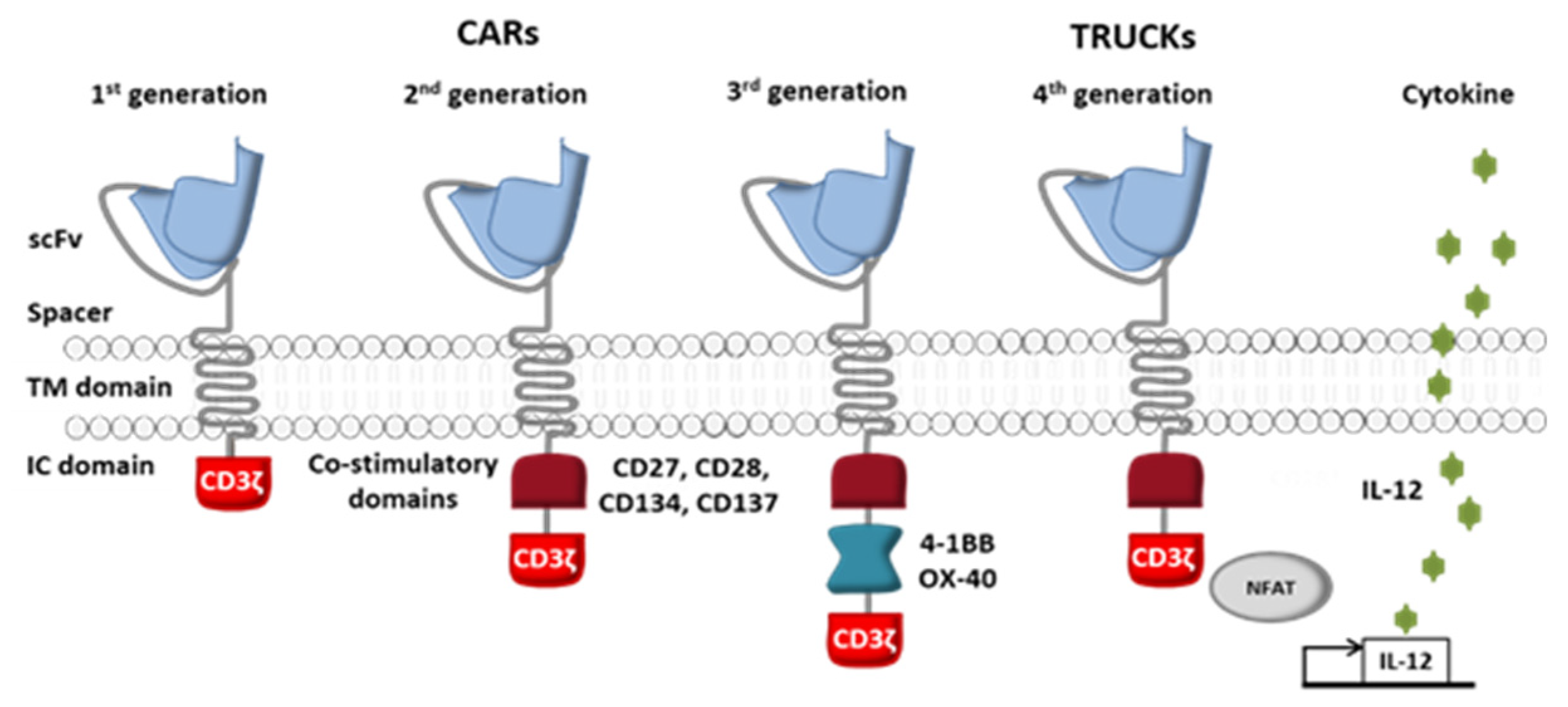
2. From Conventional CAR-T Cells to Next-Generation CAR-T Cells
2.1. CAR-T Cells with an OFF-Switch
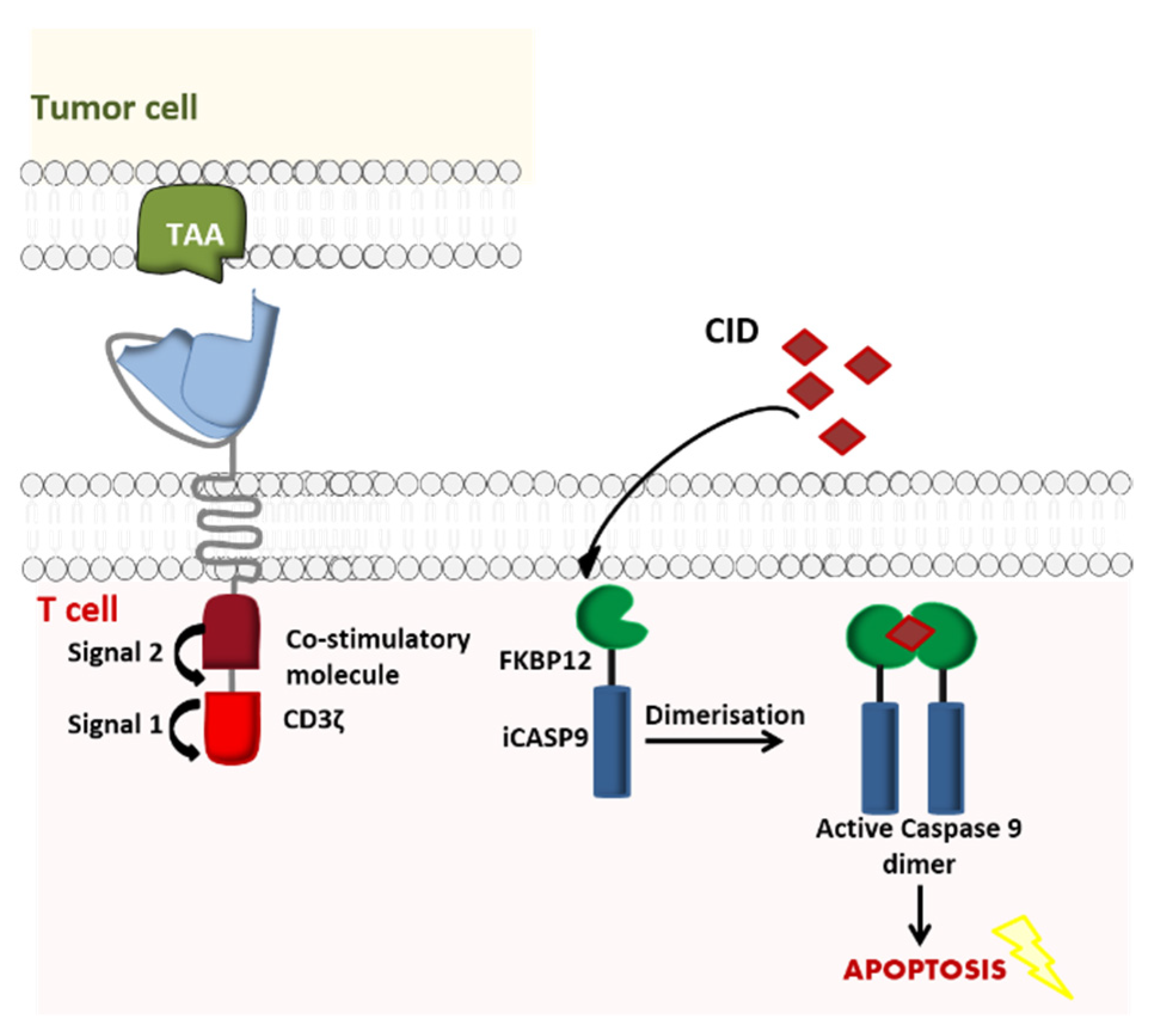
2.1.1. Suicide Gene Switch
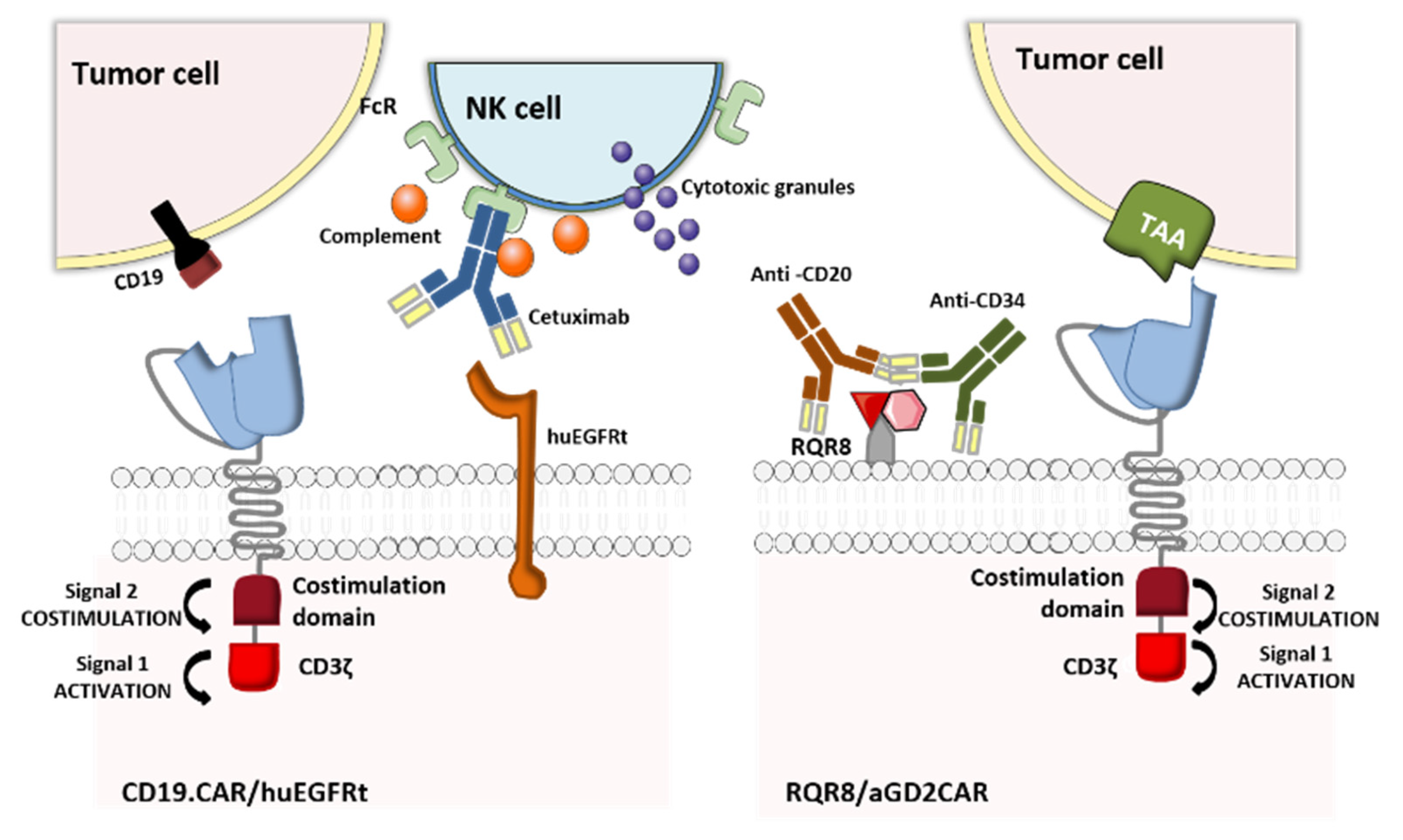
2.1.2. Elimination Markers
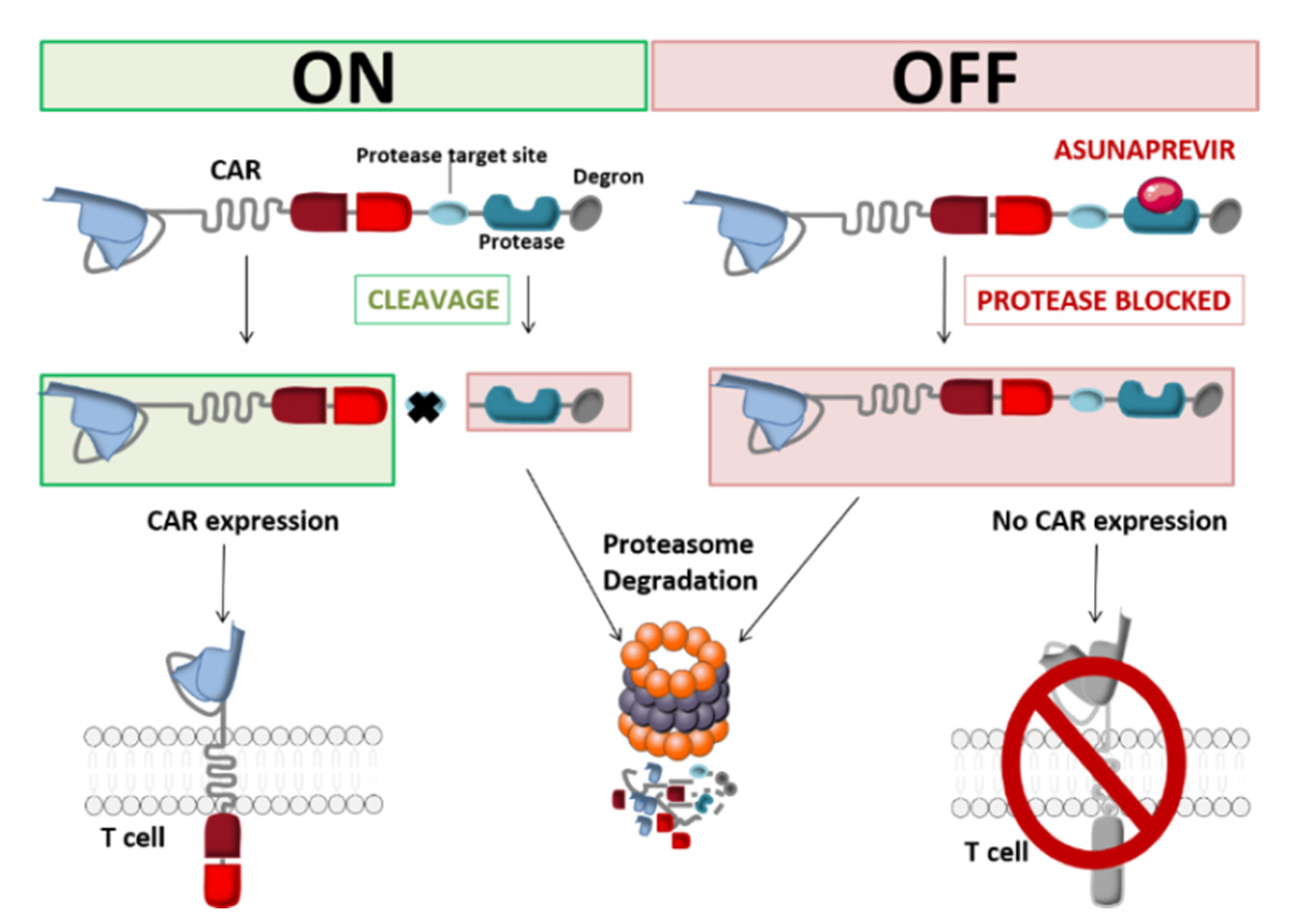
2.1.3. SMASh CARs
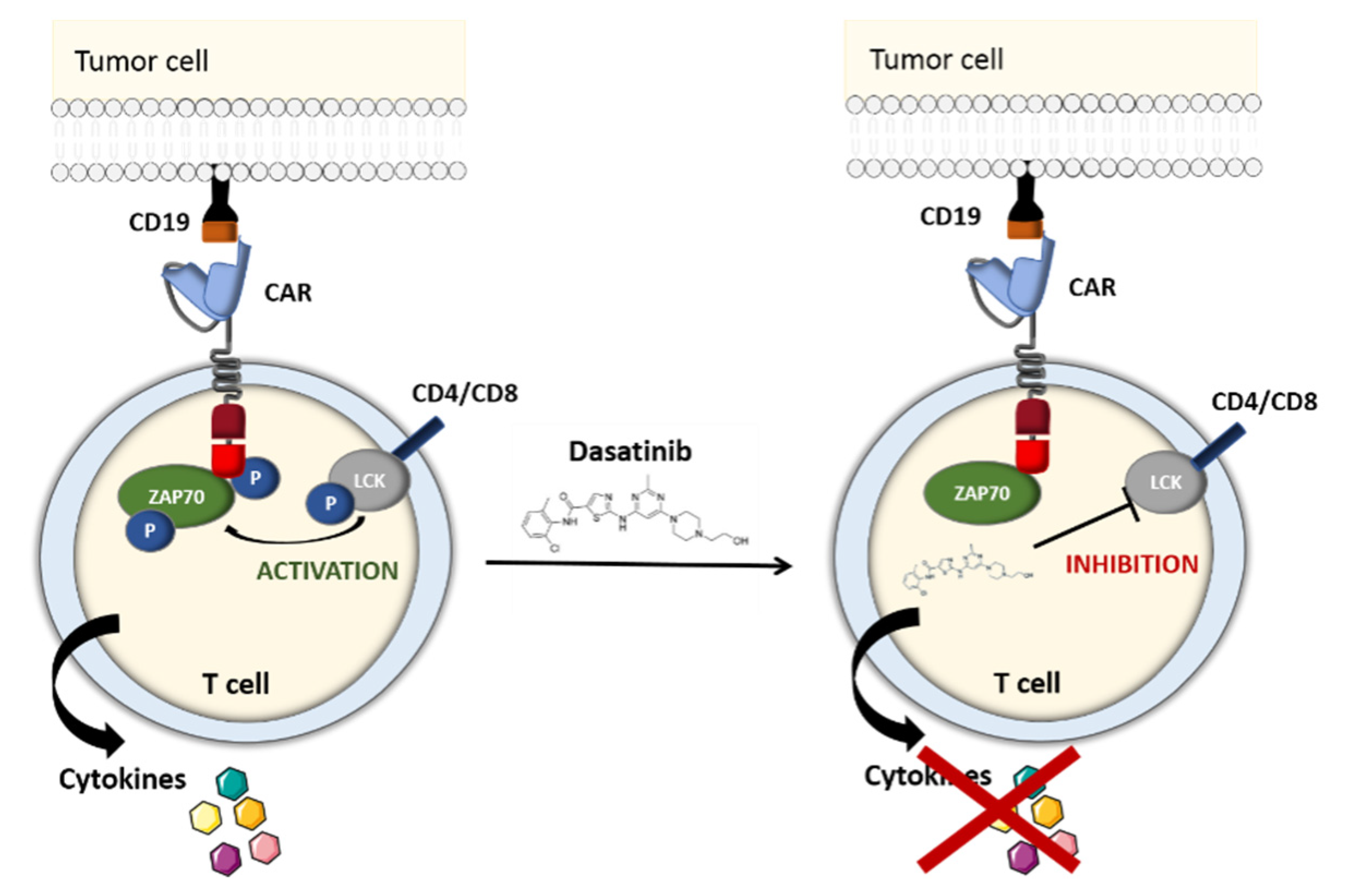
2.1.4. The TKI-Based OFF-Switch
2.2. CAR-T Cells with an ON-Switch
2.2.1. The Tetracycline-On System
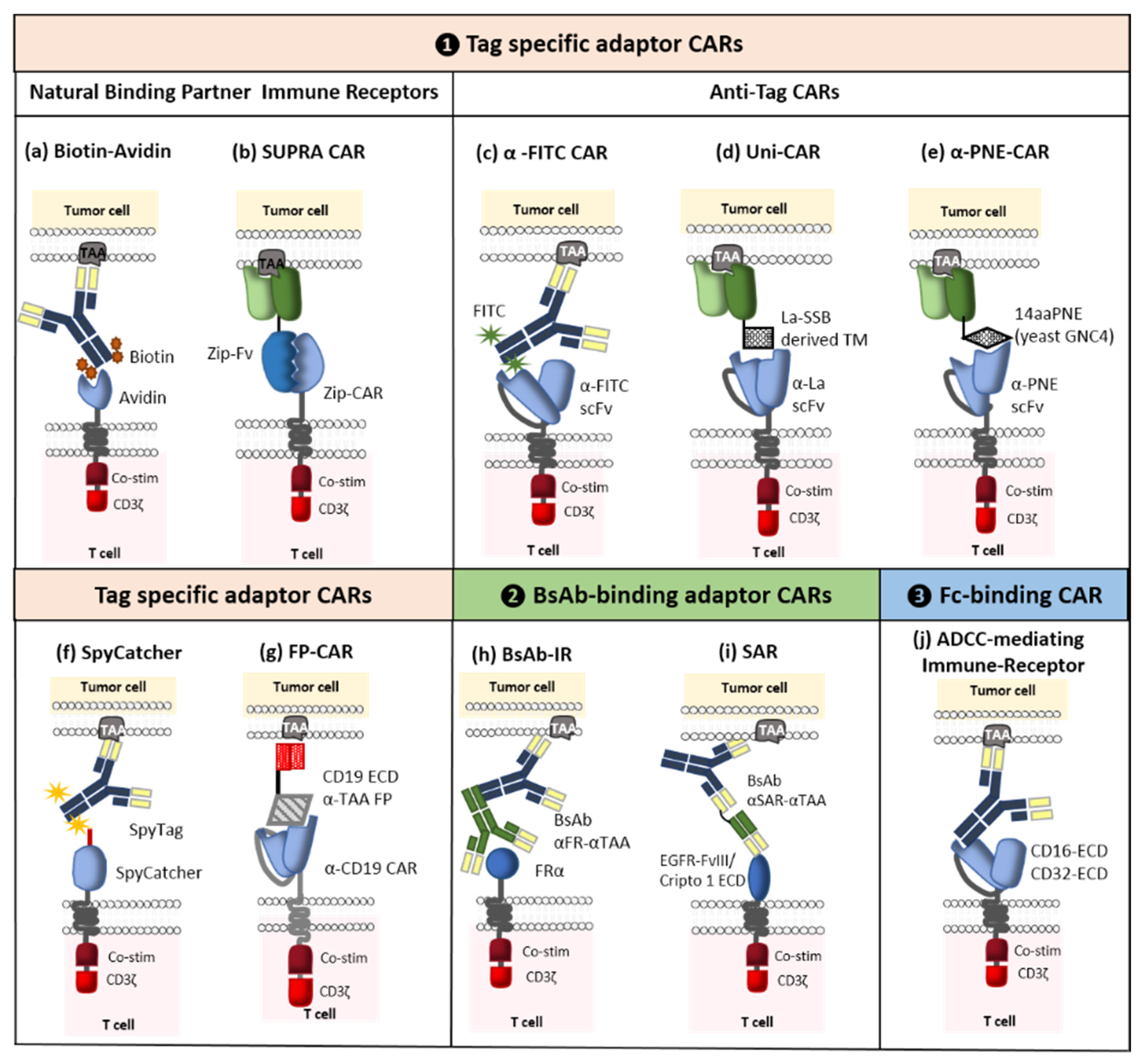
2.2.2. Switchable Adaptor CARs
2.3. Strategies for Eliminating On-Target Off-Tumor Toxicity
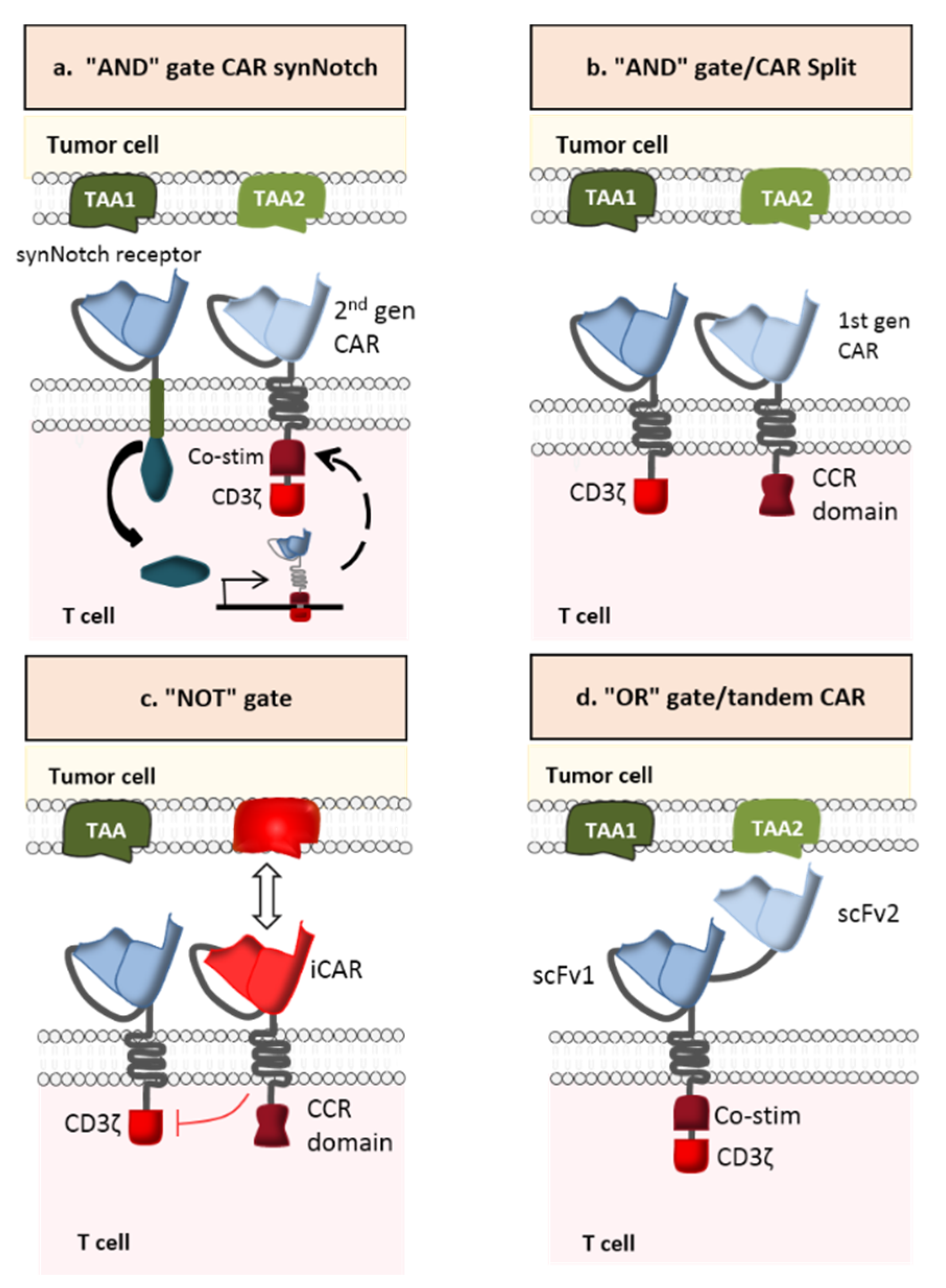
2.3.1. AND-Gate CAR-T Cells
2.3.2. NOT-Gate CAR-T Cells
2.3.3. OR-Gate CAR-T Cells
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- McCarthy, E.F. The Toxins of William B. Coley and the Treatment of Bone and Soft-Tissue Sarcomas. Iowa Orthop. J. 2006, 26, 154–158. [Google Scholar] [PubMed]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495. [Google Scholar] [CrossRef] [PubMed]
- Mach, J.-P. Introduction to monoclonal antibodies. Cancer Immun. 2012, 12, 11. [Google Scholar]
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golay, J.; Andrea, A.E. Combined Anti-Cancer Strategies Based on Anti-Checkpoint Inhibitor Antibodies. Antibodies 2020, 9, 17. [Google Scholar] [CrossRef]
- Irving, B.A.; Weiss, A. The cytoplasmic domain of the T cell receptor ζ chain is sufficient to couple to receptor-associated signal transduction pathways. Cell 1991, 64, 891–901. [Google Scholar] [CrossRef]
- Lee, Y.-H.; Kim, C.H. Evolution of chimeric antigen receptor (CAR) T cell therapy: Current status and future perspectives. Arch. Pharm. Res. 2019, 42, 607–616. [Google Scholar] [CrossRef]
- Letourneur, F.; Klausner, R.D. Activation of T cells by a tyrosine kinase activation domain in the cytoplasmic tail of CD3 epsilon. Science 1992, 255, 79–82. [Google Scholar] [CrossRef]
- Romeo, C.; Amiot, M.; Seed, B. Sequence requirements for induction of cytolysis by the T cell antigen/Fc receptor zeta chain. Cell 1992, 68, 889–897. [Google Scholar] [CrossRef]
- Klampatsa, A.; Haas, A.R.; Moon, E.K.; Albelda, S.M. Chimeric Antigen Receptor (CAR) T Cell Therapy for Malignant Pleural Mesothelioma (MPM). Cancers 2017, 9, 115. [Google Scholar] [CrossRef] [Green Version]
- Marincola, F.M.; Jaffee, E.M.; Hicklin, D.J.; Ferrone, S. Escape of human solid tumors from T-cell recognition: Molecular mechanisms and functional significance. Adv. Immunol. 2000, 74, 181–273. [Google Scholar] [PubMed]
- Garrido, F.; Algarra, I. MHC antigens and tumor escape from immune surveillance. Adv. Cancer Res. 2001, 83, 117–158. [Google Scholar] [PubMed]
- Cabrera, T.; López-Nevot, M.A.; Gaforio, J.J.; Ruiz-Cabello, F.; Garrido, F. Analysis of HLA expression in human tumor tissues. Cancer Immunol. Immunother. 2003, 52, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Barrett, D.M.; Grupp, S.A.; June, C.H. Chimeric Antigen Receptor– and TCR-Modified T Cells Enter Main Street and Wall Street. J. Immunol. 2015, 195, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Fesnak, A.D.; Levine, B.L.; June, C.H. Engineered T Cells: The Promise and Challenges of Cancer Immunotherapy. Nat. Rev. Cancer 2016, 16, 566–581. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Kochenderfer, J.N. Toxicities of chimeric antigen receptor T cells: Recognition and management. Blood 2016, 127, 3321–3330. [Google Scholar] [CrossRef] [Green Version]
- Kulemzin, S.V.; Kuznetsova, V.V.; Mamonkin, M.; Taranin, A.V.; Gorchakov, A.A. CAR T-cell therapy: Balance of efficacy and safety. Mol. Biol. (Mosk.) 2017, 51, 274–287. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Santos, E.; Nikhamin, Y.; Yeh, R.; Matsushita, M.; La Perle, K.; Quintás-Cardama, A.; Larson, S.M.; Sadelain, M. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin. Cancer Res. 2007, 13, 5426–5435. [Google Scholar] [CrossRef] [Green Version]
- Finney, H.M.; Akbar, A.N.; Lawson, A.D.G. Activation of resting human primary T cells with chimeric receptors: Costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J. Immunol. 2004, 172, 104–113. [Google Scholar] [CrossRef]
- Imai, C.; Mihara, K.; Andreansky, M.; Nicholson, I.C.; Pui, C.-H.; Geiger, T.L.; Campana, D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 2004, 18, 676–684. [Google Scholar] [CrossRef] [Green Version]
- Song, D.-G.; Ye, Q.; Poussin, M.; Harms, G.M.; Figini, M.; Powell, D.J. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood 2012, 119, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.J.; Oertle, J.; Warren, D.; Prato, D. Chimeric antigen receptor (CAR) T cell therapy for malignant cancers: Summary and perspective. J. Cell. Immunother. 2016, 2, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhao, Y. Increasing the safety and efficacy of chimeric antigen receptor T cell therapy. Protein Cell 2017, 8, 573–589. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Teachey, D.T.; Porter, D.L.; Grupp, S.A. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood 2015, 125, 4017–4023. [Google Scholar] [CrossRef] [Green Version]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Fillat, C.; Carrió, M.; Cascante, A.; Sangro, B. Suicide gene therapy mediated by the Herpes Simplex virus thymidine kinase gene/Ganciclovir system: Fifteen years of application. Curr. Gene Ther. 2003, 3, 13–26. [Google Scholar] [CrossRef]
- Ciceri, F.; Bonini, C.; Marktel, S.; Zappone, E.; Servida, P.; Bernardi, M.; Pescarollo, A.; Bondanza, A.; Peccatori, J.; Rossini, S.; et al. Antitumor effects of HSV-TK-engineered donor lymphocytes after allogeneic stem-cell transplantation. Blood 2007, 109, 4698–4707. [Google Scholar] [CrossRef]
- Greco, R.; Oliveira, G.; Stanghellini, M.T.L.; Vago, L.; Bondanza, A.; Peccatori, J.; Cieri, N.; Marktel, S.; Mastaglio, S.; Bordignon, C.; et al. Improving the safety of cell therapy with the TK-suicide gene. Front. Pharmacol. 2015, 6. [Google Scholar] [CrossRef]
- Beltinger, C.; Fulda, S.; Kammertoens, T.; Meyer, E.; Uckert, W.; Debatin, K.M. Herpes simplex virus thymidine kinase/ganciclovir-induced apoptosis involves ligand-independent death receptor aggregation and activation of caspases. Proc. Natl. Acad. Sci. USA 1999, 96, 8699–8704. [Google Scholar] [CrossRef] [Green Version]
- Traversari, C.; Marktel, S.; Magnani, Z.; Mangia, P.; Russo, V.; Ciceri, F.; Bonini, C.; Bordignon, C. The potential immunogenicity of the TK suicide gene does not prevent full clinical benefit associated with the use of TK-transduced donor lymphocytes in HSCT for hematologic malignancies. Blood 2007, 109, 4708–4715. [Google Scholar] [CrossRef] [Green Version]
- Tiberghien, P.; Ferrand, C.; Lioure, B.; Milpied, N.; Angonin, R.; Deconinck, E.; Certoux, J.M.; Robinet, E.; Saas, P.; Petracca, B.; et al. Administration of herpes simplex-thymidine kinase-expressing donor T cells with a T-cell-depleted allogeneic marrow graft. Blood 2001, 97, 63–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciceri, F.; Bonini, C.; Stanghellini, M.T.L.; Bondanza, A.; Traversari, C.; Salomoni, M.; Turchetto, L.; Colombi, S.; Bernardi, M.; Peccatori, J.; et al. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): A non-randomised phase I-II study. Lancet Oncol. 2009, 10, 489–500. [Google Scholar] [CrossRef]
- Straathof, K.C.; Pulè, M.A.; Yotnda, P.; Dotti, G.; Vanin, E.F.; Brenner, M.K.; Heslop, H.E.; Spencer, D.M.; Rooney, C.M. An inducible caspase 9 safety switch for T-cell therapy. Blood 2005, 105, 4247–4254. [Google Scholar] [CrossRef] [PubMed]
- Di Stasi, A.; Tey, S.-K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoyos, V.; Savoldo, B.; Quintarelli, C.; Mahendravada, A.; Zhang, M.; Vera, J.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Dotti, G. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia 2010, 24, 1160–1170. [Google Scholar] [CrossRef] [Green Version]
- Budde, L.E.; Berger, C.; Lin, Y.; Wang, J.; Lin, X.; Frayo, S.E.; Brouns, S.A.; Spencer, D.M.; Till, B.G.; Jensen, M.C.; et al. Combining a CD20 Chimeric Antigen Receptor and an Inducible Caspase 9 Suicide Switch to Improve the Efficacy and Safety of T Cell Adoptive Immunotherapy for Lymphoma. PLoS ONE 2013, 8, e82742. [Google Scholar] [CrossRef] [Green Version]
- Diaconu, I.; Ballard, B.; Zhang, M.; Chen, Y.; West, J.; Dotti, G.; Savoldo, B. Inducible Caspase-9 Selectively Modulates the Toxicities of CD19-Specific Chimeric Antigen Receptor-Modified T Cells. Mol. Ther. 2017, 25, 580–592. [Google Scholar] [CrossRef] [Green Version]
- Minagawa, K.; Jamil, M.O.; Al-Obaidi, M.; Pereboeva, L.; Salzman, D.; Erba, H.P.; Lamb, L.S.; Bhatia, R.; Mineishi, S.; Di Stasi, A. In Vitro Pre-Clinical Validation of Suicide Gene Modified Anti-CD33 Redirected Chimeric Antigen Receptor T-Cells for Acute Myeloid Leukemia. PLoS ONE 2016, 11, e0166891. [Google Scholar] [CrossRef] [Green Version]
- Warda, W.; Larosa, F.; Neto Da Rocha, M.; Trad, R.; Deconinck, E.; Fajloun, Z.; Faure, C.; Caillot, D.; Moldovan, M.; Valmary-Degano, S.; et al. CML Hematopoietic Stem Cells Expressing IL1RAP Can Be Targeted by Chimeric Antigen Receptor-Engineered T Cells. Cancer Res. 2019, 79, 663–675. [Google Scholar] [CrossRef] [Green Version]
- Griffioen, M.; van Egmond, E.H.M.; Kester, M.G.D.; Willemze, R.; Falkenburg, J.H.F.; Heemskerk, M.H.M. Retroviral transfer of human CD20 as a suicide gene for adoptive T-cell therapy. Haematologica 2009, 94, 1316–1320. [Google Scholar] [CrossRef] [Green Version]
- Vogler, I.; Newrzela, S.; Hartmann, S.; Schneider, N.; von Laer, D.; Koehl, U.; Grez, M. An Improved Bicistronic CD20/tCD34 Vector for Efficient Purification and In Vivo Depletion of Gene-Modified T Cells for Adoptive Immunotherapy. Mol. Ther. 2010, 18, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chang, W.-C.; Wong, C.W.; Colcher, D.; Sherman, M.; Ostberg, J.R.; Forman, S.J.; Riddell, S.R.; Jensen, M.C. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood 2011, 118, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Paszkiewicz, P.J.; Fräßle, S.P.; Srivastava, S.; Sommermeyer, D.; Hudecek, M.; Drexler, I.; Sadelain, M.; Liu, L.; Jensen, M.C.; Riddell, S.R.; et al. Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J. Clin. Investig. 2016, 126, 4262–4272. [Google Scholar] [CrossRef] [PubMed]
- Kieback, E.; Charo, J.; Sommermeyer, D.; Blankenstein, T.; Uckert, W. A safeguard eliminates T cell receptor gene-modified autoreactive T cells after adoptive transfer. Proc. Natl. Acad. Sci. USA 2008, 105, 623–628. [Google Scholar] [CrossRef] [Green Version]
- Introna, M.; Barbui, A.M.; Bambacioni, F.; Casati, C.; Gaipa, G.; Borleri, G.; Bernasconi, S.; Barbui, T.; Golay, J.; Biondi, A.; et al. Genetic modification of human T cells with CD20: A strategy to purify and lyse transduced cells with anti-CD20 antibodies. Hum. Gene Ther. 2000, 11, 611–620. [Google Scholar] [CrossRef]
- Serafini, M.; Manganini, M.; Borleri, G.; Bonamino, M.; Imberti, L.; Biondi, A.; Golay, J.; Rambaldi, A.; Introna, M. Characterization of CD20-transduced T lymphocytes as an alternative suicide gene therapy approach for the treatment of graft-versus-host disease. Hum. Gene Ther. 2004, 15, 63–76. [Google Scholar] [CrossRef]
- Philip, B.; Kokalaki, E.; Mekkaoui, L.; Thomas, S.; Straathof, K.; Flutter, B.; Marin, V.; Marafioti, T.; Chakraverty, R.; Linch, D.; et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood 2014, 124, 1277–1287. [Google Scholar] [CrossRef]
- Koneru, M.; Purdon, T.J.; Spriggs, D.; Koneru, S.; Brentjens, R.J. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology 2015, 4, e994446. [Google Scholar] [CrossRef] [Green Version]
- Chung, H.K.; Jacobs, C.L.; Huo, Y.; Yang, J.; Krumm, S.A.; Plemper, R.K.; Tsien, R.Y.; Lin, M.Z. Tunable and reversible drug control of protein production via a self-excising degron. Nat. Chem. Biol. 2015, 11, 713–720. [Google Scholar] [CrossRef]
- Juillerat, A.; Tkach, D.; Busser, B.W.; Temburni, S.; Valton, J.; Duclert, A.; Poirot, L.; Depil, S.; Duchateau, P. Modulation of chimeric antigen receptor surface expression by a small molecule switch. BMC Biotechnol. 2019, 19. [Google Scholar] [CrossRef] [Green Version]
- Valton, J.; Guyot, V.; Boldajipour, B.; Sommer, C.; Pertel, T.; Juillerat, A.; Duclert, A.; Sasu, B.J.; Duchateau, P.; Poirot, L. A Versatile Safeguard for Chimeric Antigen Receptor T-Cell Immunotherapies. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Casucci, M.; Bondanza, A. Suicide Gene Therapy to Increase the Safety of Chimeric Antigen Receptor-Redirected T Lymphocytes. J. Cancer 2011, 2, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Khoury, H.J.; Guilhot, F.; Hughes, T.P.; Kim, D.-W.; Cortes, J.E. Dasatinib treatment for Philadelphia chromosome-positive leukemias: Practical considerations. Cancer 2009, 115, 1381–1394. [Google Scholar] [CrossRef] [PubMed]
- Conchon, M.; Freitas, C.M.B.d.M.; Rego, M.A.d.C.; Braga Junior, J.W.R. Dasatinib-clinical trials and management of adverse events in imatinib resistant/intolerant chronic myeloid leukemia. Rev. Bras. Hematol. Hemoter 2011, 33, 131–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mestermann, K.; Giavridis, T.; Weber, J.; Rydzek, J.; Frenz, S.; Nerreter, T.; Mades, A.; Sadelain, M.; Einsele, H.; Hudecek, M. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Rydzek, J.; Nerreter, T.; Peng, H.; Jutz, S.; Leitner, J.; Steinberger, P.; Einsele, H.; Rader, C.; Hudecek, M. Chimeric Antigen Receptor Library Screening Using a Novel NF-κB/NFAT Reporter Cell Platform. Mol. Ther. 2019, 27, 287–299. [Google Scholar] [CrossRef] [Green Version]
- Giavridis, T.; van der Stegen, S.J.C.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018, 24, 731–738. [Google Scholar] [CrossRef]
- Wu, B.X.; Song, N.-J.; Riesenberg, B.P.; Li, Z. Development of molecular and pharmacological switches for chimeric antigen receptor T cells. Exp. Hematol. Oncol. 2019, 8, 27. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-Y.; Roybal, K.T.; Puchner, E.M.; Onuffer, J.; Lim, W.A. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science 2015, 350, aab4077. [Google Scholar] [CrossRef] [Green Version]
- Loew, R.; Heinz, N.; Hampf, M.; Bujard, H.; Gossen, M. Improved Tet-responsive promoters with minimized background expression. BMC Biotechnol. 2010, 10, 81. [Google Scholar] [CrossRef] [Green Version]
- Gossen, M.; Bujard, H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. USA 1992, 89, 5547–5551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urlinger, S.; Baron, U.; Thellmann, M.; Hasan, M.T.; Bujard, H.; Hillen, W. Exploring the sequence space for tetracycline-dependent transcriptional activators: Novel mutations yield expanded range and sensitivity. Proc. Natl. Acad. Sci. USA 2000, 97, 7963–7968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinz, N.; Schambach, A.; Galla, M.; Maetzig, T.; Baum, C.; Loew, R.; Schiedlmeier, B. Retroviral and transposon-based tet-regulated all-in-one vectors with reduced background expression and improved dynamic range. Hum. Gene Ther. 2011, 22, 166–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.-Y.; Wei, D.; Liu, Z.-K.; Yong, Y.-L.; Wei, W.; Zhang, Z.-Y.; Lv, J.-J.; Zhang, Z.; Chen, Z.-N.; Bian, H. Doxycycline Inducible Chimeric Antigen Receptor T Cells Targeting CD147 for Hepatocellular Carcinoma Therapy. Front. Cell Dev. Biol. 2019, 7. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.; He, D.; Li, C.; Wang, H.; Yang, G. Development of Inducible CD19-CAR T Cells with a Tet-On System for Controlled Activity and Enhanced Clinical Safety. Int J. Mol. Sci 2018, 19, 3455. [Google Scholar] [CrossRef] [Green Version]
- Drent, E.; Poels, R.; Mulders, M.J.; van de Donk, N.W.C.J.; Themeli, M.; Lokhorst, H.M.; Mutis, T. Feasibility of controlling CD38-CAR T cell activity with a Tet-on inducible CAR design. PLoS ONE 2018, 13, e0197349. [Google Scholar] [CrossRef]
- González-Zorn, B.; Escudero, J.A. Ecology of antimicrobial resistance: Humans, animals, food and environment. Int. Microbiol. 2012, 15, 101–109. [Google Scholar]
- Clémenceau, B.; Congy-Jolivet, N.; Gallot, G.; Vivien, R.; Gaschet, J.; Thibault, G.; Vié, H. Antibody-dependent cellular cytotoxicity (ADCC) is mediated by genetically modified antigen-specific human T lymphocytes. Blood 2006, 107, 4669–4677. [Google Scholar] [CrossRef] [Green Version]
- Arndt, C.; Fasslrinner, F.; Loureiro, L.R.; Koristka, S.; Feldmann, A.; Bachmann, M. Adaptor CAR Platforms—Next Generation of T Cell-Based Cancer Immunotherapy. Cancers 2020, 12, 1302. [Google Scholar] [CrossRef]
- Tanaka, H.; Fujiwara, H.; Ochi, F.; Tanimoto, K.; Casey, N.; Okamoto, S.; Mineno, J.; Kuzushima, K.; Shiku, H.; Sugiyama, T.; et al. Development of Engineered T Cells Expressing a Chimeric CD16-CD3ζ Receptor to Improve the Clinical Efficacy of Mogamulizumab Therapy Against Adult T-Cell Leukemia. Clin. Cancer Res. 2016, 22, 4405–4416. [Google Scholar] [CrossRef] [Green Version]
- Bridgeman, J.S.; Hawkins, R.E.; Hombach, A.A.; Abken, H.; Gilham, D.E. Building better chimeric antigen receptors for adoptive T cell therapy. Curr. Gene Ther. 2010, 10, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Kudo, K.; Imai, C.; Lorenzini, P.; Kamiya, T.; Kono, K.; Davidoff, A.M.; Chng, W.J.; Campana, D. T Lymphocytes Expressing a CD16 Signaling Receptor Exert Antibody-Dependent Cancer Cell Killing. Cancer Res. 2014, 74, 93–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rataj, F.; Jacobi, S.J.; Stoiber, S.; Asang, F.; Ogonek, J.; Tokarew, N.; Cadilha, B.L.; van Puijenbroek, E.; Heise, C.; Duewell, P.; et al. High-affinity CD16-polymorphism and Fc-engineered antibodies enable activity of CD16-chimeric antigen receptor-modified T cells for cancer therapy. Br. J. Cancer 2019, 120, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Caratelli, S.; Arriga, R.; Sconocchia, T.; Ottaviani, A.; Lanzilli, G.; Pastore, D.; Cenciarelli, C.; Venditti, A.; Del Principe, M.I.; Lauro, D.; et al. In vitro elimination of epidermal growth factor receptor-overexpressing cancer cells by CD32A-chimeric receptor T cells in combination with cetuximab or panitumumab. Int. J. Cancer 2020, 146, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Minutolo, N.G.; Hollander, E.E.; Powell, D.J.J. The Emergence of Universal Immune Receptor T Cell Therapy for Cancer. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- FDA Places Clinical Hold on Unum Therapeutics Phase 1 Trial Evaluating ACTR087 for Relapsed/Refractory CD20+ B Cell Non-Hodgkin Lymphoma. Available online: https://www.trialsitenews.com/fda-places-clinical-hold-on-unum-therapeutics-phase-1-trial-evaluating-actr087-for-relapsed-refractory-cd20-b-cell-non-hodgkin-lymphoma/ (accessed on 11 June 2020).
- Unum Therapeutics Inc. Unum Therapeutics Provides Updates to Its Phase 1 Trial of ACTR707 for HER2+ Solid Tumor Cancers. Available online: http://www.globenewswire.com/news-release/2020/01/29/1976692/0/en/Unum-Therapeutics-Provides-Updates-to-its-Phase-1-Trial-of-ACTR707-for-HER2-Solid-Tumor-Cancers.html (accessed on 11 June 2020).
- Urbanska, K.; Lanitis, E.; Poussin, M.; Lynn, R.C.; Gavin, B.P.; Kelderman, S.; Yu, J.; Scholler, N.; Powell, D.J. A Universal Strategy for Adoptive Immunotherapy of Cancer through Use of a Novel T-cell Antigen Receptor. Cancer Res. 2012, 72, 1844–1852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamada, K.; Geng, D.; Sakoda, Y.; Bansal, N.; Srivastava, R.; Li, Z.; Davila, E. Redirecting gene-modified T cells toward various cancer types using tagged antibodies. Clin. Cancer Res. 2012, 18, 6436–6445. [Google Scholar] [CrossRef] [Green Version]
- Cartellieri, M.; Loff, S.; von Bonin, M.; Bejestani, E.P.; Ehninger, A.; Feldmann, A.; Koristka, S.; Arndt, C.; Ehninger, G.; Bachmann, M.P. Unicar: A Novel Modular Retargeting Platform Technology for CAR T Cells. Blood 2015, 126, 5549. [Google Scholar] [CrossRef]
- Rodgers, D.T.; Mazagova, M.; Hampton, E.N.; Cao, Y.; Ramadoss, N.S.; Hardy, I.R.; Schulman, A.; Du, J.; Wang, F.; Singer, O.; et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc. Natl. Acad. Sci. USA 2016, 113, E459–E468. [Google Scholar] [CrossRef] [Green Version]
- Raj, D.; Yang, M.-H.; Rodgers, D.; Hampton, E.N.; Begum, J.; Mustafa, A.; Lorizio, D.; Garces, I.; Propper, D.; Kench, J.G.; et al. Switchable CAR-T cells mediate remission in metastatic pancreatic ductal adenocarcinoma. Gut 2019, 68, 1052–1064. [Google Scholar] [CrossRef] [Green Version]
- Viaud, S.; Ma, J.S.Y.; Hardy, I.R.; Hampton, E.N.; Benish, B.; Sherwood, L.; Nunez, V.; Ackerman, C.J.; Khialeeva, E.; Weglarz, M.; et al. Switchable control over in vivo CAR T expansion, B cell depletion, and induction of memory. Proc. Natl. Acad. Sci. USA 2018, 115, E10898–E10906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, J.H.; Collins, J.J.; Wong, W.W. Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell 2018, 173, 1426–1438.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minutolo, N.G.; Sharma, P.; Poussin, M.; Shaw, L.C.; Brown, D.P.; Hollander, E.E.; Smole, A.; Rodriguez-Garcia, A.; Hui, J.Z.; Zappala, F.; et al. Quantitative Control of Gene-Engineered T-Cell Activity through the Covalent Attachment of Targeting Ligands to a Universal Immune Receptor. J. Am. Chem. Soc. 2020, 142, 6554–6568. [Google Scholar] [CrossRef]
- AbbVie & Scripps-based Calibr Moves Novel ‘Switchable’ CAR-T Technology to Phase I Clinical Trial. Available online: https://www.trialsitenews.com/abbvie-scripps-based-calibr-moves-novel-switchable-car-t-technology-to-phase-i-clinical-trial/ (accessed on 13 June 2020).
- Stamova, S.; Koristka, S.; Keil, J.; Arndt, C.; Feldmann, A.; Michalk, I.; Bartsch, H.; Bippes, C.C.; Schmitz, M.; Cartellieri, M.; et al. Cancer Immunotherapy by Retargeting of Immune Effector Cells via Recombinant Bispecific Antibody Constructs. Antibodies 2012, 1, 172–198. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.G.; Marks, I.; Srinivasarao, M.; Kanduluru, A.K.; Mahalingam, S.M.; Liu, X.; Chu, H.; Low, P.S. Use of a Single CAR T Cell and Several Bispecific Adapters Facilitates Eradication of Multiple Antigenically Different Solid Tumors. Cancer Res. 2019, 79, 387–396. [Google Scholar] [CrossRef] [Green Version]
- Urbanska, K.; Lynn, R.C.; Stashwick, C.; Thakur, A.; Lum, L.G.; Powell, D.J. Targeted cancer immunotherapy via combination of designer bispecific antibody and novel gene-engineered T cells. J. Transl. Med. 2014, 12, 347. [Google Scholar] [CrossRef] [Green Version]
- Karches, C.H.; Benmebarek, M.-R.; Schmidbauer, M.L.; Kurzay, M.; Klaus, R.; Geiger, M.; Rataj, F.; Cadilha, B.L.; Lesch, S.; Heise, C.; et al. Bispecific Antibodies Enable Synthetic Agonistic Receptor-Transduced T Cells for Tumor Immunotherapy. Clin. Cancer Res. 2019, 25, 5890–5900. [Google Scholar] [CrossRef] [Green Version]
- Ambrose, C.; Su, L.; Wu, L.; Lobb, R.R.; Rennert, P.D. Abstract 3768: CAR T cells specific for CD19 can be redirected to kill CD19 negative tumors. Cancer Res. 2017, 77, 3768. [Google Scholar]
- Rennert, P.; Su, L.; Dufort, F.; Birt, A.; Sanford, T.; Wu, L.; Ambrose, C.; Lobb, R. A Novel CD19-Anti-CD20 Bridging Protein Prevents and Reverses CD19-Negative Relapse from CAR19 T Cell Treatment In Vivo. Blood 2019, 134, 252. [Google Scholar] [CrossRef]
- Wei, J.; Han, X.; Bo, J.; Han, W. Target selection for CAR-T therapy. J. Hematol. Oncol. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Wang, Y.; Wei, J.; Han, W. Multi-antigen-targeted chimeric antigen receptor T cells for cancer therapy. J. Hematol. Oncol. 2019, 12, 128. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200. [Google Scholar] [CrossRef] [PubMed]
- Kloss, C.C.; Condomines, M.; Cartellieri, M.; Bachmann, M.; Sadelain, M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol. 2013, 31, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Wilkie, S.; van Schalkwyk, M.C.I.; Hobbs, S.; Davies, D.M.; van der Stegen, S.J.C.; Pereira, A.C.P.; Burbridge, S.E.; Box, C.; Eccles, S.A.; Maher, J. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J. Clin. Immunol. 2012, 32, 1059–1070. [Google Scholar] [CrossRef]
- Feldmann, A.; Arndt, C.; Bergmann, R.; Loff, S.; Cartellieri, M.; Bachmann, D.; Aliperta, R.; Hetzenecker, M.; Ludwig, F.; Albert, S.; et al. Retargeting of T lymphocytes to PSCA- or PSMA positive prostate cancer cells using the novel modular chimeric antigen receptor platform technology “UniCAR”. Oncotarget 2017, 8, 31368–31385. [Google Scholar] [CrossRef] [Green Version]
- Lam, J.S.; Yamashiro, J.; Shintaku, I.P.; Vessella, R.L.; Jenkins, R.B.; Horvath, S.; Said, J.W.; Reiter, R.E. Prostate stem cell antigen is overexpressed in prostate cancer metastases. Clin. Cancer Res. 2005, 11, 2591–2596. [Google Scholar] [CrossRef] [Green Version]
- Silver, D.A.; Pellicer, I.; Fair, W.R.; Heston, W.D.; Cordon-Cardo, C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin. Cancer Res. 1997, 3, 81–85. [Google Scholar]
- Lanitis, E.; Poussin, M.; Klattenhoff, A.W.; Song, D.; Sandaltzopoulos, R.; June, C.H.; Powell, D.J. Chimeric Antigen Receptor T Cells with Dissociated Signaling Domains Exhibit Focused Antitumor Activity with Reduced Potential for Toxicity In Vivo. Cancer Immunol. Res. 2013, 1, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Roybal, K.T.; Williams, J.Z.; Morsut, L.; Rupp, L.J.; Kolinko, I.; Choe, J.H.; Walker, W.J.; McNally, K.A.; Lim, W.A. Engineering T Cells with Customized Therapeutic Response Programs Using Synthetic Notch Receptors. Cell 2016, 167, 419–432.e16. [Google Scholar] [CrossRef] [Green Version]
- Jaspers, J.E.; Brentjens, R.J. Development of CAR T cells designed to improve antitumor efficacy and safety. Pharmacol. Ther. 2017, 178, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S.; Salter, A.I.; Liggitt, D.; Yechan-Gunja, S.; Sarvothama, M.; Cooper, K.; Smythe, K.S.; Dudakov, J.A.; Pierce, R.H.; Rader, C.; et al. Logic-Gated ROR1 Chimeric Antigen Receptor Expression Rescues T Cell-Mediated Toxicity to Normal Tissues and Enables Selective Tumor Targeting. Cancer Cell 2019, 35, 489–503.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roybal, K.T.; Rupp, L.J.; Morsut, L.; Walker, W.J.; McNally, K.A.; Park, J.S.; Lim, W.A. Precision Tumor Recognition by T Cells with Combinatorial Antigen-Sensing Circuits. Cell 2016, 164, 770–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukumaran, S.; Watanabe, N.; Bajgain, P.; Raja, K.; Mohammed, S.; Fisher, W.E.; Brenner, M.K.; Leen, A.M.; Vera, J.F. Enhancing the Potency and Specificity of Engineered T Cells for Cancer Treatment. Cancer Discov. 2018, 8, 972–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedorov, V.D.; Themeli, M.; Sadelain, M. PD-1– and CTLA-4–Based Inhibitory Chimeric Antigen Receptors (iCARs) Divert Off-Target Immunotherapy Responses. Sci. Transl. Med. 2013, 5, 215ra172. [Google Scholar] [CrossRef] [Green Version]
- Hegde, M.; Corder, A.; Chow, K.K.; Mukherjee, M.; Ashoori, A.; Kew, Y.; Zhang, Y.J.; Baskin, D.S.; Merchant, F.A.; Brawley, V.S.; et al. Combinational Targeting Offsets Antigen Escape and Enhances Effector Functions of Adoptively Transferred T Cells in Glioblastoma. Mol. Ther. 2013, 21, 2087–2101. [Google Scholar] [CrossRef] [Green Version]
- Ruella, M.; Barrett, D.M.; Kenderian, S.S.; Shestova, O.; Hofmann, T.J.; Perazzelli, J.; Klichinsky, M.; Aikawa, V.; Nazimuddin, F.; Kozlowski, M.; et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J. Clin. Investig. 2016, 126, 3814–3826. [Google Scholar] [CrossRef] [Green Version]
- Feng, K.; Guo, Y.; Liu, Y.; Dai, H.; Wang, Y.; Lv, H.; Huang, J.; Yang, Q.; Han, W. Cocktail treatment with EGFR-specific and CD133-specific chimeric antigen receptor-modified T cells in a patient with advanced cholangiocarcinoma. J. Hematol. Oncol. 2017, 10. [Google Scholar] [CrossRef] [Green Version]
- Zah, E.; Lin, M.-Y.; Silva-Benedict, A.; Jensen, M.C.; Chen, Y.Y. T cells expressing CD19/CD20 bi-specific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol. Res. 2016, 4, 498–508. [Google Scholar] [CrossRef] [Green Version]
- Grada, Z.; Hegde, M.; Byrd, T.; Shaffer, D.R.; Ghazi, A.; Brawley, V.S.; Corder, A.; Schönfeld, K.; Koch, J.; Dotti, G.; et al. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol. Ther. Nucleic Acids 2013, 2, e105. [Google Scholar] [CrossRef]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.H.; et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J. Clin. Investig. 2016, 126, 3036–3052. [Google Scholar] [CrossRef] [Green Version]
- De Munter, S.; Ingels, J.; Goetgeluk, G.; Bonte, S.; Pille, M.; Weening, K.; Kerre, T.; Abken, H.; Vandekerckhove, B. Nanobody Based Dual Specific CARs. Int. J. Mol. Sci. 2018, 19, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, H.; Wang, Z.; Wang, Y.; Liu, Y.; Dai, H.; Tong, C.; Guo, Y.; Guo, B.; Ti, D.; Han, X.; et al. Haploidentical CD19/CD22 bispecific CAR-T cells induced MRD-negative remission in a patient with relapsed and refractory adult B-ALL after haploidentical hematopoietic stem cell transplantation. J. Hematol. Oncol. 2019, 12, 57. [Google Scholar] [CrossRef] [PubMed]
- Bielamowicz, K.; Fousek, K.; Byrd, T.T.; Samaha, H.; Mukherjee, M.; Aware, N.; Wu, M.-F.; Orange, J.S.; Sumazin, P.; Man, T.-K.; et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol. 2018, 20, 506–518. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Abbreviation | Referring to | Abbreviation | Referring to |
|---|---|---|---|
| ADCC | Antibody-dependent cellular cytotoxicity | HSV-TK | Herpes simplex virus thymidine kinase gene |
| ALL | Acute lymphoblastic leukemia | iCAR | Inhibitory CAR |
| AML | Acute myelogenous leukemia | iCasp9 | Inducible caspase-9 |
| ASN | Asunaprevir | IFNγ | Interferon gamma |
| BBIR | Biotin-binding immune receptor | IgG | Immunoglobulin G |
| BMT | Bone marrow transplantation | IL-2 | Interleukin-2 |
| BPDCN | Blastic plasmacytoid dendritic cell neoplasm | ITAM | Immunoreceptor tyrosine-based motif |
| BsAb | Bispecific antibody | mAb | Monoclonal antibody |
| BsAb-IR | BsAb immune receptor | MSLN | Mesothelin |
| CAR-T cell | Chimeric antigen receptor T cell | MHC | Major histocompatibility complex |
| CD | Cluster of differentiation | NHL | Non-Hodgkin’s lymphoma |
| CID | Chemical induction of dimerization | NK | Natural killer |
| CLL | Chronic lymphoblastic leukemia | PD-1 | Programmed cell death-1 |
| CNS | Central nervous system | PNE | Peptide neoepitope |
| CRS | Cytokine release syndrome | PSCA | Prostate stem cell antigen |
| CTLA-4 | Cytotoxic T-lymphocyte-associated protein 4 | PSMA | Prostate-specific membrane antigen |
| rtTA | Reverse Tet transactivator | ||
| DIPG | Diffuse intrinsic pontine glioma | SAR | Synthetic agonistic receptor |
| DLBCL | Diffuse large B cell lymphoma | SWIFF-CAR | Switch-off CAR |
| DMG | Diffuse midline glioma | SMASh | Small-molecule-assisted shutoff |
| Dox | Doxycycline | SUPRA | Split, universal, and programmable |
| ECD | Extracellular domain | SynNotch | Synthetic Notch |
| EGFR | Epidermal growth factor receptor | TAA | Tumor-associated antigen |
| Fab | Fragment antigen binding | TCR | T cell receptor |
| FDA | Food and Drug Administration | Tet | Tetracycline |
| FITC | Fluorescein isothiocyanate | TKI | Tyrosine kinase inhibitor |
| GBM | Glioblastoma | TM | Transmembrane |
| GCV | Ganciclovir | TNF-α | Tumor necrosis factor alfa |
| GMP | Good manufacturing practice | TRUCK | T cells redirected for universal cytokine-mediated killing |
| GVHD | Graft-versus-host disease | TSA | Tumor-specific antigen |
| HER2 | Human epidermal growth factor receptor-2 | ZAP70 | Zeta-chain-associated protein kinase 70 |
| CAR-T Cells | Cancer Type | Time after Suicide Induction | Percentage of Eradicated CAR-T Cells | Reference |
|---|---|---|---|---|
| iCasp9-IL15-CD19CAR | B-cell malignancies | 24 h | >95.0% | [35] |
| iCasp9-CD20CAR-Δ19 | B-cell malignancies | 24 h 72 h | 90.0% 98.0% | [36] |
| iCasp9-CD20CAR-ΔNCFR | B-cell malignancies | 48 h | >99.0% | [37] |
| iCasp9-CD33CAR-Δ19 | AML | N/A | 76.4% | [38] |
| iCasp9-IL1RAPCAR-Δ19 | CML | N/A | 87.0% | [39] |
| Target | Cancer Types | NCT Number |
|---|---|---|
| GD2 | DIPG and DMG | NCT04196413 |
| Relapsed/refractory neuroblastoma | NCT01822652 NCT03721068 NCT03373097 | |
| Refractory/metastatic GD2-positive sarcoma or neuroblastoma | NCT01953900 NCT02107963 | |
| Solid cancers | NCT02992210 | |
| CD19 | Relapsed/refractory ALL | NCT03016377 NCT03594162 |
| Relapsed/refractory B-cell lymphoma | NCT03696784 NCT03579927 | |
| CD19 & CD22 | Relapsed/refractory B-cell lymphoma | NCT03098355 |
| CD30 | Relapsed/refractory CD30-positive lymphomas | NCT02274584 |
| Mesothelin | Solid cancers | NCT02414269 |
| Advanced breast cancer | NCT02792114 |
| Target | Cancer Types | NCT Number |
|---|---|---|
| CD19 | CLL, NHL and ALL | NCT01865617 NCT03103971 NCT01815749 NCT03085173 NCT02146924 NCT02051257 NCT03579888 NCT02028455 NCT02746952 |
| EGFR | Recurrent/refractory solid tumors | NCT03618381 |
| Recurrent or refractory pediatric CNS tumors | NCT03638167 | |
| HER2 | HER2-positive recurrent/refractory pediatric CNS tumors | NCT03500991 |
| BCMA | MM | NCT03070327 |
| CD22 | CD22+ leukemia and lymphoma | NCT03244306 |
| CD123 | CD123+ relapsed/refractory AML or persistent/recurrent BPDCN | NCT03114670 NCT02159495 |
| CD171 | Recurrent/refractory neuroblastoma or ganglioneuroblastoma | NCT02311621 |
| PD-1 | Recurrent glioblastoma multiforme | NCT02937844 |
| CAR Adaptor | Adaptor Molecule | Cancer Type | NCT Number |
|---|---|---|---|
| CD16-BB/ζ (ACTR087) | Rituximab | Refractory or relapsed CD20-positive B cell lymphoma | NCT02776813 |
| CD16-28 (ACTR707) | NCT03189836 | ||
| ACTR087 ACTR707 | Trastuzumab | HER2-positive advanced solid tumor cancers | NCT03680560 |
| ACTR087 ACTR707 | Rituximab Trastuzumab SEA-BCMA | B cell lymphoma MM HER2-positive solid tumor cancers | NCT02840110 |
| ACTR087 | SEA-BCMA | Relapsed or refractory MM | NCT03266692 |
| CAR-T Cell Type | Target | Cancer Types | Reference |
|---|---|---|---|
| Dual CAR-T cell | PSMA/PSCA | Prostate cancer | [96] |
| FRa/MSLN | Ovarian cancer | [101] | |
| CD19/GFP | Experimental cancer | [105] | |
| CD19/MSLN | Experimental cancer | [105] | |
| Trivalent CAR-T cell | PSCA/TGFβ/IL4 | Pancreatic cancer | [106] |
| CAR-T Cell Type | Target | Cancer Type | Reference |
|---|---|---|---|
| Pooled CAR-T cell | HER2/IL-13Rα2 | GMB | [108] |
| CD19/CD123 | B-ALL | [109] | |
| CD19/CD20 | B-ALL | [111] | |
| EGFR/CD133 | Cholangiocarcinoma | [110] | |
| Dual CAR-T cell | CD19/CD123 | B-ALL | [109] |
| HER2/IL-13Rα2 | GMB | [108] | |
| Tandem CAR-T cell | CD19/CD20 | B cell malignancies | [111] |
| HER2/IL-13Rα2 | GMB | [108] | |
| CD20/HER2 | Experimental Cancer | [114] | |
| CD19/HER2 | Experimental Cancer | [112] | |
| Trivalent CAR-T cell | HER2/IL13Rα2/EphA2 | GMB | [116] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrea, A.E.; Chiron, A.; Bessoles, S.; Hacein-Bey-Abina, S. Engineering Next-Generation CAR-T Cells for Better Toxicity Management. Int. J. Mol. Sci. 2020, 21, 8620. https://doi.org/10.3390/ijms21228620
Andrea AE, Chiron A, Bessoles S, Hacein-Bey-Abina S. Engineering Next-Generation CAR-T Cells for Better Toxicity Management. International Journal of Molecular Sciences. 2020; 21(22):8620. https://doi.org/10.3390/ijms21228620
Chicago/Turabian StyleAndrea, Alain E., Andrada Chiron, Stéphanie Bessoles, and Salima Hacein-Bey-Abina. 2020. "Engineering Next-Generation CAR-T Cells for Better Toxicity Management" International Journal of Molecular Sciences 21, no. 22: 8620. https://doi.org/10.3390/ijms21228620
APA StyleAndrea, A. E., Chiron, A., Bessoles, S., & Hacein-Bey-Abina, S. (2020). Engineering Next-Generation CAR-T Cells for Better Toxicity Management. International Journal of Molecular Sciences, 21(22), 8620. https://doi.org/10.3390/ijms21228620