1. Introduction
Two-dimensional materials are becoming increasingly important owing to the variety of technological and biomedical applications for which they can be used [
1,
2,
3,
4,
5]. According to the size of the building units, ranging from molecules and macromolecules to nano- and micro-particles, surface patterns can be designed to achieve specific functionalities at different length scales. The formation of well-designed surface structures can often be described by a combination of general ingredients. For instance, the assembly of organic molecules functionalized with multiple carboxyl groups on a highly oriented pyrolytic graphite surface [
6,
7,
8,
9,
10,
11] can be rationalized in terms of the rotational symmetry of the building units, their aspect ratio, the placement of the bonding groups, and possible energy differences between binding groups [
12,
13,
14,
15]. These same general ingredients were proven to be crucial in other molecular-scale processes: by combining the anisotropy of the building blocks with a low bond functionality, it is possible, for instance, to control the emergence of crystal polymorphs in protein systems [
16] or to separate the enantiomers of chiral molecules [
17]. The same approach—non-spherical building units provided with a discrete number of bonding sites—can be also applied at the nanometer scale: DNA origami of different shapes have been programmed to assemble into prescribed two-dimensional tilings by taking advantage of blunt end stacking and hybridization sites, thus providing versatile platforms to engineer optical metamaterials and biomimetic tissues [
18,
19,
20,
21,
22]. At even larger length scales, micrometer non-spherical colloids decorated with attractive spots along their perimeter also form two-dimensional aggregates whose complex geometries can be related to the properties of the constituent units [
23,
24,
25]: close-packed versus porous, surface structures, as well as finite clusters with specific architectures can be designed by tailoring the single particle features [
26,
27,
28,
29,
30]. Colloidal platelets with non-spherical shapes and directional bonding sites—often referred to as patches—constitute an ideal playground for testing and understanding the driving mechanisms of two-dimensional tilings. The self-assembly of patchy colloidal platelets is governed by a competition between shape and bonding anisotropy: on the one hand, non-spherical hard shapes tend to maximize edge-to-edge contacts [
23,
31,
32]; on the other hand, the presence of attractive patches imposes additional constraints on the assembly process, thus leading to a rich aggregation scenario [
24,
25,
26].
We recently considered systems of anisotropic patchy platelets with a small number attractive patches placed in different arrangements along the particle edges, and we rationalized the emerging aggregation behavior in terms of the complex interplay between steric incompatibilities and (un)satisfied bonding geometries [
27,
28,
29]; additionally, by tuning the patch placement on the particle edges, the combination of shape and bond anisotropy was used to drive the assembly from a close-packed arrangement to tunable porous lattices with the same geometric pattern [
27]. We now aim at investigating the effect of the patch size, i.e., of the attractive interaction range of the bonding sites, on the two-dimensional tiling of the same anisotropic patchy colloids.
Within the vast realm of currently available patchy colloids and platelets [
23,
33,
34], the patch size can be tuned in a wide variety of systems by changing the physical or chemical features that characterize the patches, such as the patch surface extension [
35,
36,
37], the patch surface roughness [
38], the length of the DNA strands and ligands grafted onto the particle surface [
24,
39], as well as the pH or the salt concentration of the colloidal dispersion [
40,
41]. The effect of the patch size for spherical particles and colloidal molecules has been investigated in the assembly of finite clusters of various symmetries [
42,
43,
44] and in the spontaneous nucleation of two- and three-dimensional crystals, such as the diamond lattice [
45,
46] and the honeycomb network [
47]. In general, the assembly mechanism of these systems is governed by a compromise between selectivity, kinetic accessibility, and thermodynamics. On the one hand, small patches impart a strict directionality to the inter-particle bonds, thus selecting the target structure that is compatible with the fully bonded local arrangement of the building units; on the other hand, the narrower the patches are, the more difficult it is for the particles to rearrange an incorrect aggregate, and the system is likely kinetically trapped. In contrast, large patches introduce a certain degree of flexibility in the bonding mechanism: relaxed bonding restrictions favor local rearrangements—and the formation of the equilibrium structure—but at the same time, bonds become increasingly less selective with the patch size, thus leading to disordered assemblies. It is worth noting that, when patches can form at most one bond, there exists a correspondence between the patch number and the bonding pattern that provides the control over the features of the target crystal or finite cluster. Nonetheless, even when the single bond per patch condition is satisfied, an extreme flexibility of the bonding mechanism can still disfavor the stabilization of ordered phases at any value of the interaction energy [
48].
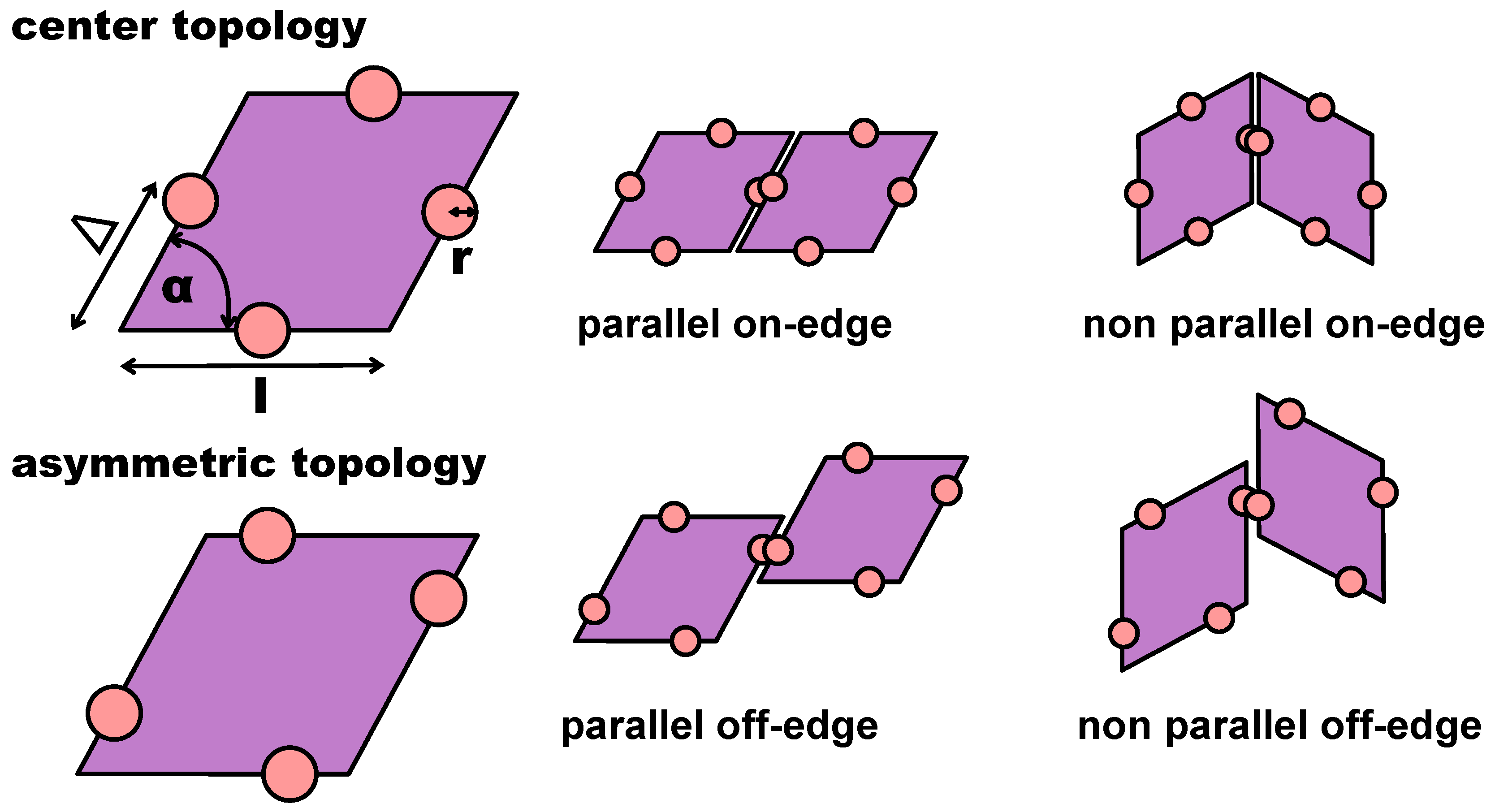
In the present contribution, we consider a system of regular hard patchy rhombi (of edge length
l and opening angle
°) decorated with four identical patches (of size
r placed in different arrangements along the particle perimeter (see
Figure 1). The patch arrangements are determined by a predefined topology and a parameter
, which sets the exact patch positions along the rhombi edges (see again
Figure 1): pairs of patches on adjacent edges are at distance
from their closest vertex, while pairs of patches on opposite edges are described by
; when
, the chosen topology is referred to as asymmetric (note that it was labeled feq-as2 in Reference [
27]), while when
, each patch sits in the center of its respective edge, and the resulting symmetric arrangement is referred to as the center topology. We note that the chosen patch topology is symmetric with respect to
when mirrored along the major axis of the rhombi. Due to their steric incompatibilities, rhombic platelets align with each other in two possible orientations: parallel (p) and non-parallel (np); depending on the patch placement defined by
, these alignments can be realized by edge-to-edge (on-edge) contacts or in staggered (off-edge) configurations (all represented in
Figure 1). If two of these neighboring particles are additionally bonded together via at least one of their patches, they are considered to be members of the same cluster. When all patches are in the center of their respective edge, edge-to-edge—either p or np—configurations are energetically favored, thus leading to a close-packed random (r) tiling: the r-tiling emerges because our patchy rhombi are equally likely to bond in a p- or np-fashion; nonetheless, it is worth noting that, as there are bonding restrictions on particles attaching to already formed clusters, the total number of p- and np-bonds in the resulting r-tiling is not equal [
27]. In contrast, on decreasing
, staggered configurations—again, either p or np—are energetically preferred, thus leading to the formation of pores in the assembling tilings. The emerging porous tilings are characterized by either rhombic pores (p-bonded domains) or by hexagonal and triangular pores (np-bonded domains) [
27]. Additionally, for the chosen patch topology, porous p- and np-clusters grow next to each other within the same sample, but as p- and np-domains are more and more incompatible on decreasing
, a switch to an np-tiling eventually occurs at extreme
-values [
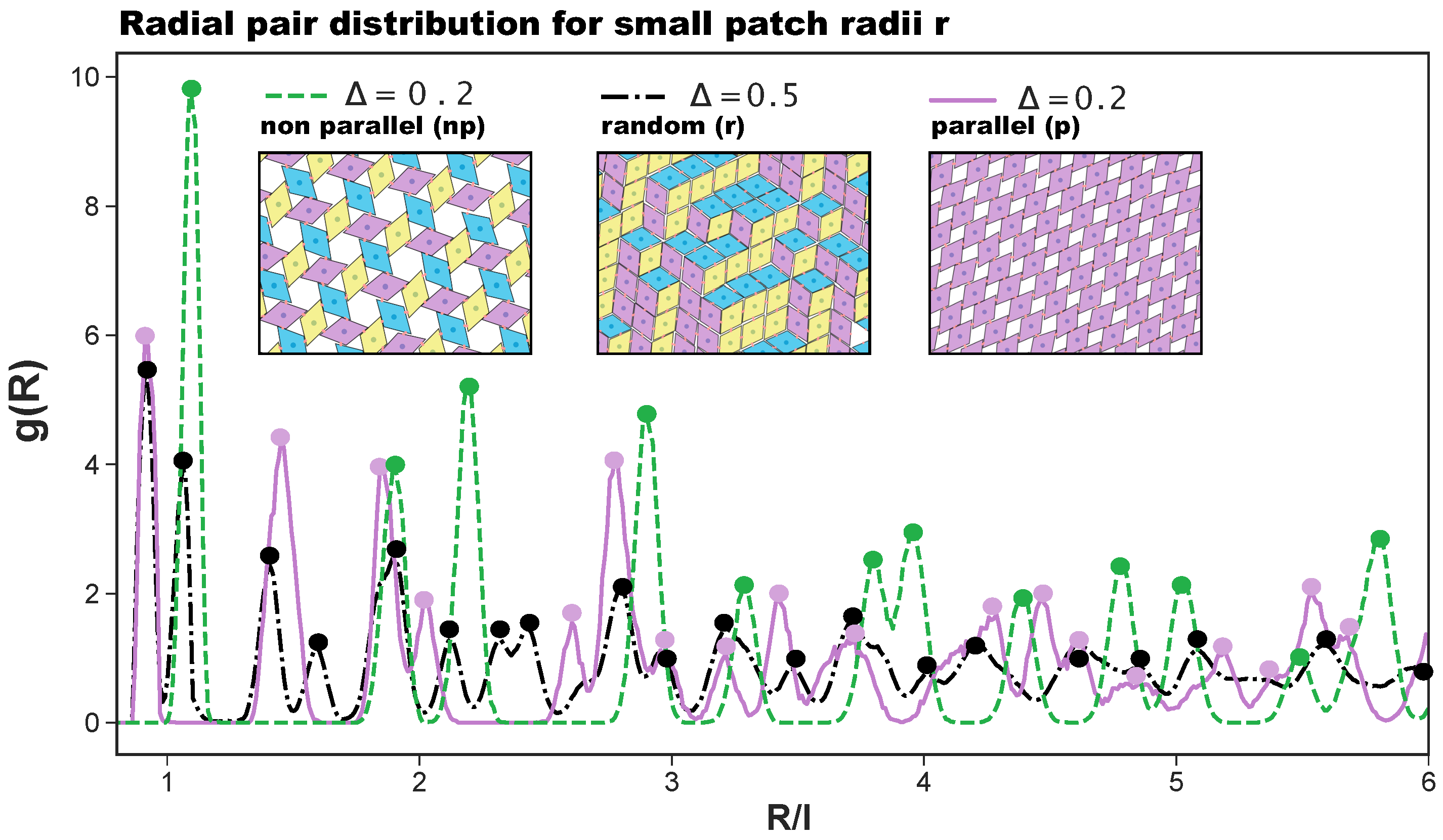
27]. The radial distribution functions that characterize np-, p-, and r-tilings are denoted as
with
R as the center-to-center particle distance and are reported in
Figure 2, together with their corresponding simulation snapshots. We note that, for the asymmetric patch topology with
, the first-neighbor peak of the np-pattern is at
, while the corresponding peak of the p-pattern is at
; in contrast, for the symmetric patch topology, the p-peak is at
and the np-peak at
. Additionally, we note that although the co-occurrence of p- and np-bonding motifs does not create a lattice order, the
of the r-tiling has sharp peaks as typical distances do repeat.
While in Reference [
27], the patch size was set to a few percent of the particle edge length [
12], in the present contribution, we investigate the effect of larger and larger patches on the tiling process. As large patches are less selective, the restrictions induced by the bonding anisotropy are expected to become more relaxed, meaning that the ordered—mostly np—tiling scenario observed at small
r-values might be destroyed on increasing the patch size. Nonetheless, shape anisotropy still plays a role in the assembly process, thus possibly leading to a complex interplay between flexible bonds and non-spherical particle shapes. Such an interplay has to be rationalized in view of emerging poly-bonds: patches, as well as particles are expected to bond in multiple ways as soon as the patches allow for a high flexibility in both bond movements and bonding restrictions.
2. Results
Our patchy rhombi model—first introduced in Reference [
27]—is detailed in
Section 4 with the particle parameters reported therein. To simulate the two-dimensional assembly process of these building units on a non-interacting substrate, we perform grand canonical Monte Carlo simulations; the details of the simulations are reported in
Section 4 together with the simulation parameters.
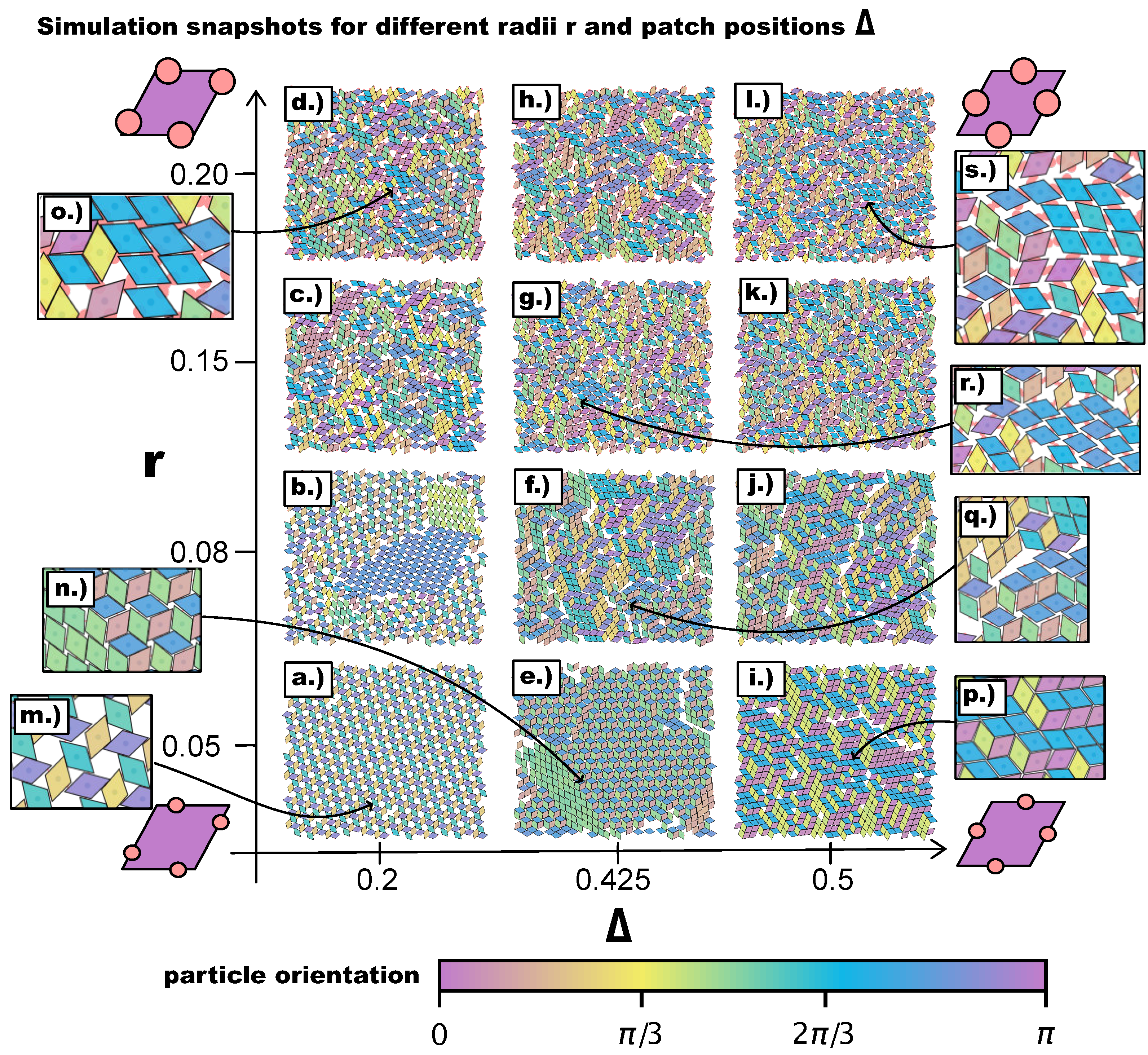
We begin our discussion with a qualitative, visual analysis of the typical morphologies observed in the emerging tilings on increasing the patch size
r at different
-values.
Figure 3 provides an overview of simulation snapshots at selected
-values. The multiple colors of the particles, indicating their orientations, help visualize differently ordered domains. At
, we obtain the same tilings as observed in Reference [
27], i.e., a porous np-tiling for the most asymmetric topology (see
Figure 3a,m) and an r-tiling for the center topology (see
Figure 3i,p). In between these two regimes, we observe a coexistence of p- and np-domains (see
Figure 3e,n). We note that, on reducing the asymmetry of the topology, the hexagonal/triangular, as well as rhombic pores shrink, leading from open to close-packed tilings. Moreover, the more the patches move towards the center topology, the more commensurable the p- and the np-tiling become, and the higher the probability to find p- and np-domains within the same system. Interestingly, at
, we observe coexisting p- and np-domains already at
(see
Figure 3b), as an effect of the larger patch radius that enhances commensurability and promotes p-bonding—as detailed in the following. On reducing the asymmetry of the topology, p- and np-domains can still be identified: at
, they form a mixed bonding motif (see
Figure 3f,q) that develops into an r-tiling at
(see
Figure 3j). A different picture presents itself as the radius is further increased to
and then to
. In many of these domains, two new phenomena appear (see the discussion of
Figure 4): (1) pairs of particles can be bonded to each other via more than one patch (they are thus referred to as pairs of poly-bonded particles), and (2) a patch can form a bond with more than one patch (it is thus referred to as a poly-bonded patch); in the discussion of
Figure 5, we describe the impact of these bonding scenarios on the tilings in detail. In general, the poly-bond scenarios seem to favor a p-alignment between the particles (see
Figure 3o,r,s), as well as the emergence of mixed bond types: as more orientations are possible, more bonded motifs are allowed (see
Figure 3c,d,g,h,l).
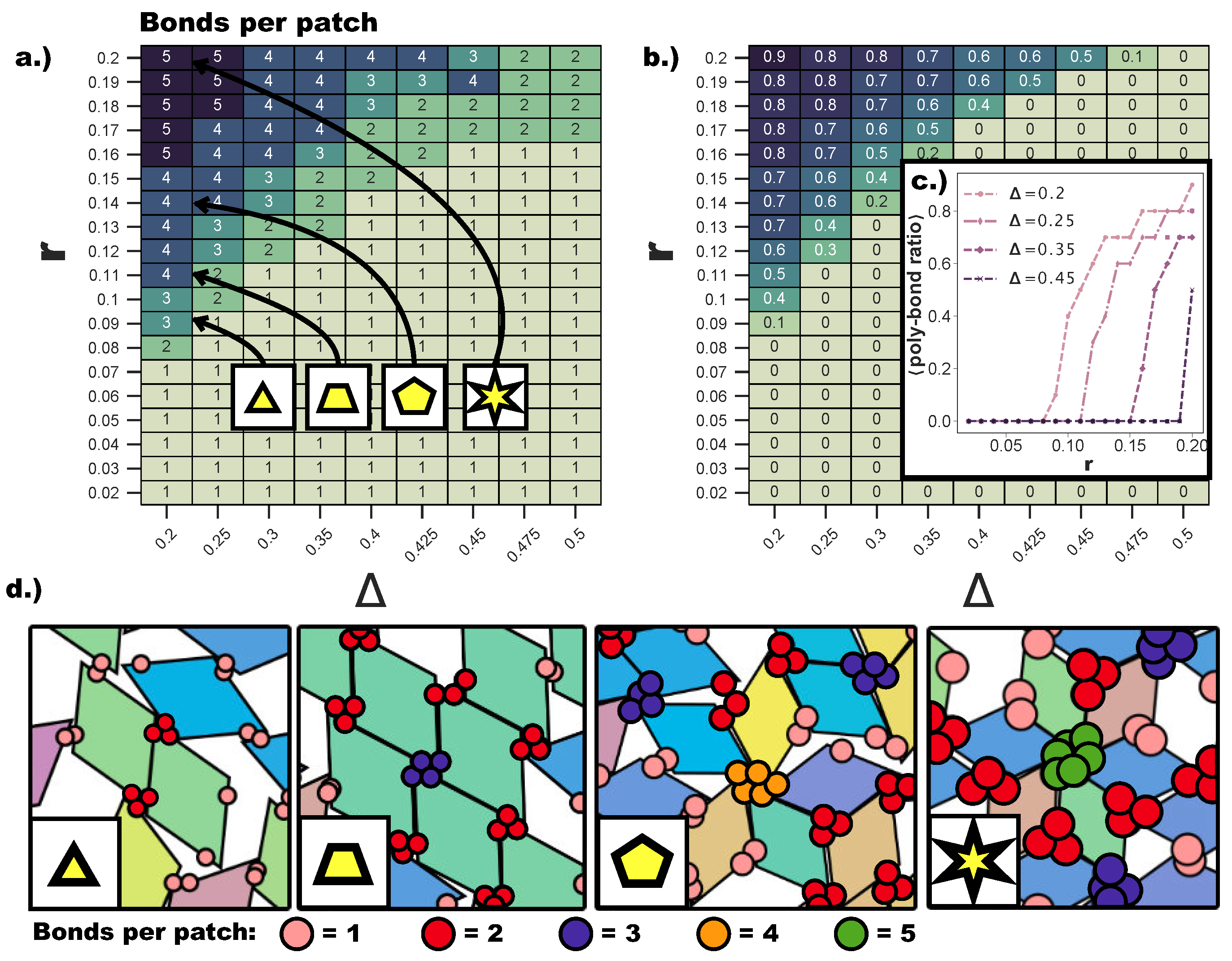
Even though both poly-bond scenarios have an impact on the tiling process, we first focus on the effects related to the emergence of bonding configurations where a single patch can bond to two or more patches at the same time. Due to the anisotropy of our building blocks, the interplay between the patch size and the patch position is highly non-trivial: the whole
parameter space must be spanned to detect where the single bond per patch condition is no longer guaranteed.
Figure 4a displays a heat map of the maximum number of bonds per patch over the whole range of (
)-values. Clearly, bigger patches favor the formation of many bonds per patch, but we find that the first occurrence of poly-bonds per patch is strongly dependent also on
. While at
, the first poly-bonds per patch occur already at
, at
, the first poly-bonds per patch appear only at
. This can be rationalized by taking into account the effect of the particle shape: for extremely asymmetric patch topologies, the patches are closer to the corners of the rhombi, thus making themselves accessible to more than one patch, if the patch size is large enough; in contrast, the closer the patch position is to the center, the more the rhombi edges shield the patch and the larger
r needs to be to allow for poly-bonds. We observe that the critical patch size for which the first poly-bonds start to appear scales with
. We also observe that, as soon as the geometric factors allow for the formation of multiple bonds per patch, the fraction of patches that form poly-bonds in the system grows sharply for any further increase of
r.
Figure 4b shows the average fraction of patches that formed poly-bonds as a heat map in the
-plane, while
Figure 4c shows the average poly-bond per patch ratio as a function of
r at selected
-values. For
, the average fraction of poly-bonded patches goes up to 0.9 at the maximum
r-value considered here, while at the same
r, the fraction of poly-bonds per patch at
is about 0.5. It is crucial to note that the maximum number of patches that are allowed to bond at the same site has a drastic effect on the particle arrangement, as shown in
Figure 4d, where snapshots of typical patch configurations at
and different
r-values are displayed. When patches can bond with up to two/three other patches, a p-bond scenario is promoted (see the first and the second panel of
Figure 4d), while patches are able to bond with four/five patches only in five- and six-star configurations, which are np-bond motifs (see the third and the fourth panel of
Figure 4d). The bonding motifs favored by the described poly-bonds per patch impact the ordering of the self-assembled tilings heavily, as described in the following.
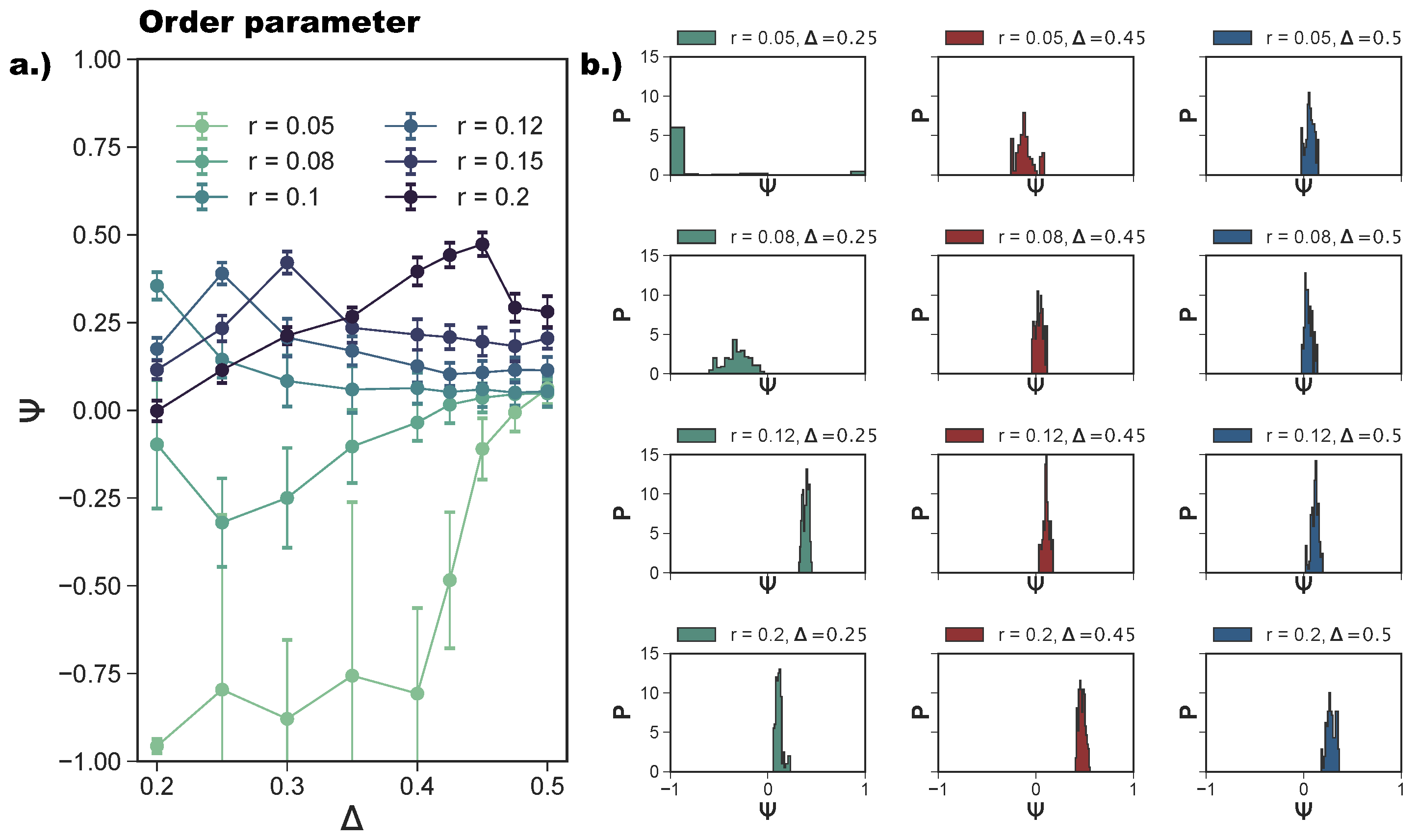
We quantify the ordering of a tiling via a suitably designed order parameter
(see
Section 4) that takes values between
(indicating that all neighbors of all particles are oriented in an np-fashion) and 1 (indicating that all neighbors of all particles are oriented in a p-fashion); a perfect random tiling corresponds to
. We note that
can fluctuate around zero for different reasons: if the system is in a fluid phase, if the system forms a perfect random tiling, or if domains with different bonding types develop during the assembly process. To differentiate between these cases and better characterize the spatial arrangements of the different tilings, we also calculate the radial distribution function,
, where
R is the center-to-center distance between two rhombi.
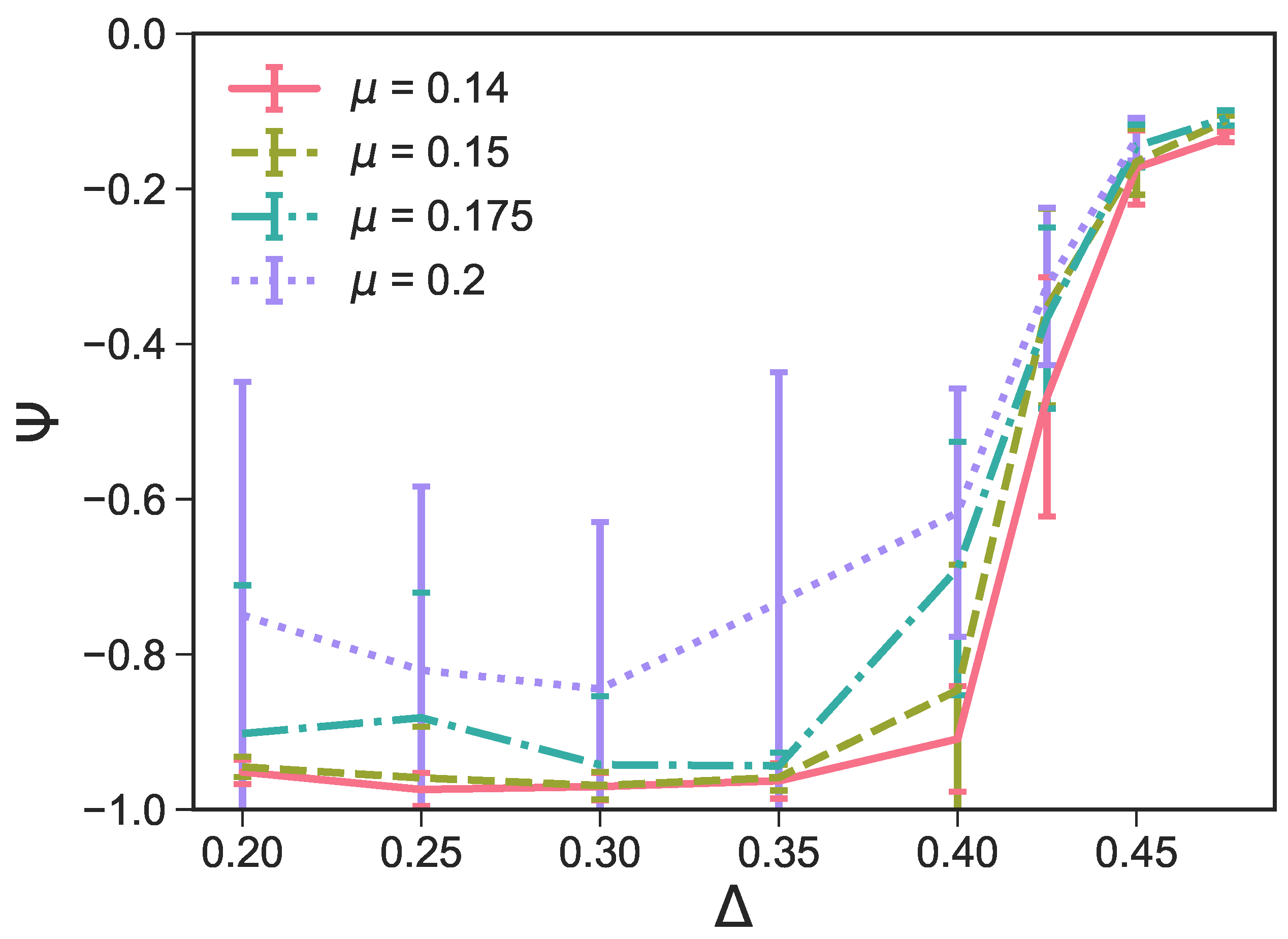
In
Figure 5a, we report the average
as a function of
at different
r-values. For
, we confirm—as suggested by the snapshot analysis and previous results [
27]—that the more asymmetric the topology is, the more the np-tiling is favored: for
,
. It is worth noting that, for
, and
, parallel simulation runs can either form a porous np-tiling with
or a porous p-tiling with
: the latter appears less often than the former, as highlighted in
Figure 5b, where histograms for
are reported (see the green histogram corresponding to
and
); this behavior results in a bimodal distribution and explains the large error bars on
for these systems. When
is increased beyond
,
grows monotonically towards zero, because p- and np-tilings become more commensurable (see the red histogram corresponding to
and
together with
Figure 3e) until the random tiling emerges (see the red histogram corresponding to
and
together with
Figure 3i). On increasing the patch size beyond our reference value
, the enhanced angular freedom in bonding leads to the formation of more mixed configurations either as coexisting p- and np-clusters (see
Figure 3) or as mixed bond types (see
Figure 4d). At
, p-domains are promoted with respect to np-ones, as highlighted by the shift of
towards zero at all
-values (see
Figure 3b and the corresponding green histogram in
Figure 5b). This behavior can be associated with the higher commensurability of the porous p- and np-tilings—due to the larger patches—promoting the parallel growth of these two types of domains in the same sample (see
Figure 3b). On further increasing
r, the interplay between the patch size and the patch position becomes quite complex: the specific trends of
as a function of
at selected
r-values (see
Figure 5a) can be related to the presence of different poly-bonds per patch in the system. At
, p-domains are largely promoted for the most asymmetric patch topology. This phenomenon can be understood by taking into account the occurrence of poly-bonds per patch (see
Figure 4b): at
, a fraction of
of all patches bond with up to three patches, while at larger
-values, poly-bonds are still negligible, and
approaches zero quickly. The visual analysis of particle arrangements when a patch can bond to two other patches (see the first and second panels of
Figure 4d) suggests that this bonding scenario promotes the p-alignment between the rhombi. The same bonding scenario can be observed at
: p-domains are largely promoted up to
, where a fraction of
of all patches bond with two or three patches (see again
Figure 4a,b), while
at larger
-values where poly-bonds per patch are not present in a significant amount. In general, when
,
develops a maximum, and the corresponding
-value increases on increasing
r: for the largest patches, i.e., for
r = 0.2,
grows monotonically from zero at
= 0.2 to almost
at
= 0.45, signaling a tendency to form p-domains (see the red histogram corresponding to
and
in
Figure 5b); as soon as
,
drops again towards zero. This peculiar behavior stems from the occurrence of different poly-bonds per patch in the system: when the poly-bonds per patch are up to two/three, p-bonds are promoted (first and second panels of
Figure 4d), while when the poly-bonds per patch are up to four/five, np-bonds are favored again within the five-star and six-star motifs (third and fourth panels of
Figure 4d). When the patch topology is such that the single bond per particle condition is recovered at the given patch size
r,
tends towards zero.
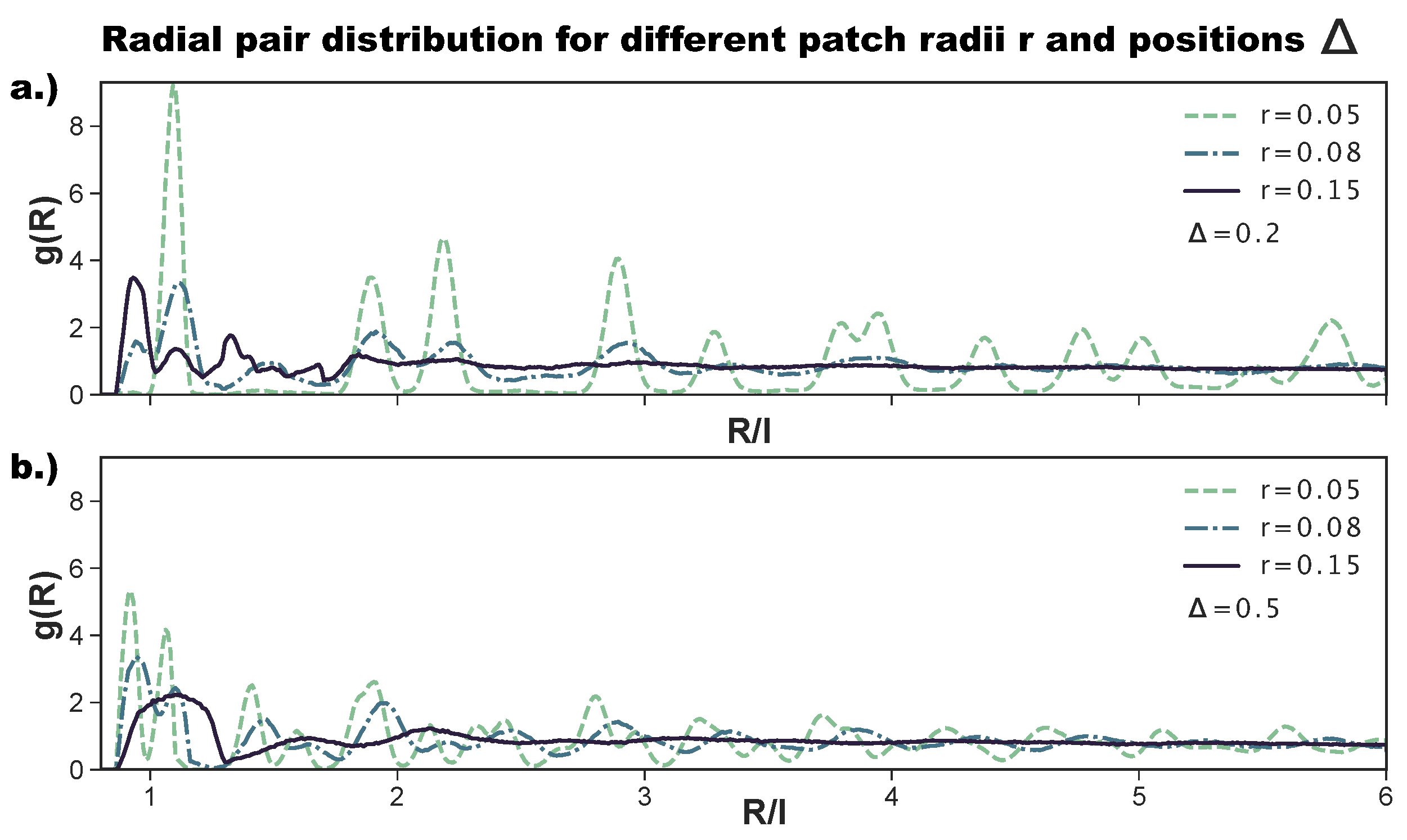
As anticipated above, to better characterize the change in the ordering of the tilings on increasing the patch size, we also consider the radial distribution function. While the ordered tilings are expected to show significant peaks and troughs, non-crystalline structures are expected to show a less pronounced behavior and quickly converge to one.
Figure 6a shows the
for the most asymmetric patch topology at selected
r-values. We observe that, for
,
has the sharp peaks of an ordered hexagonal tiling, corresponding to an np-pattern (see
Figure 2 as a reference). Already at
, the peaks are significantly less pronounced; at short inter-particle distances, we observe the appearance of the peak related to p-bonded particles (see again
Figure 2 as a reference), while at large
-values, the peaks vanish, indicating a lower long range order. At
, all peaks but those indicating the nearest neighbors have vanished, indicating a complete loss of long range order. We note that the p-peak at small inter-particle distances is in this case more pronounced than the np-peak, confirming the predominance of p- over np-bonds at this
-combination. For the central topology—reported in
Figure 6b—we observe sharp peaks for
, which correspond to an r-tiling (see
Figure 2 as a reference). Again, for
,
has less pronounced peaks, and at
, the only well-pronounced peak of the
is the one indicating the first neighbor shell; we note that this peak is relatively broad, as the many different bonding motifs (see
Figure 4d) imply the existence of many possible distances for the first neighbors. In summary, for both patch topologies, large patch radii, i.e., high bond flexibilities, destroy the order of the tilings, as at the largest bond flexibility, edge-to-edge alignments are no longer crucial for the bond formation.
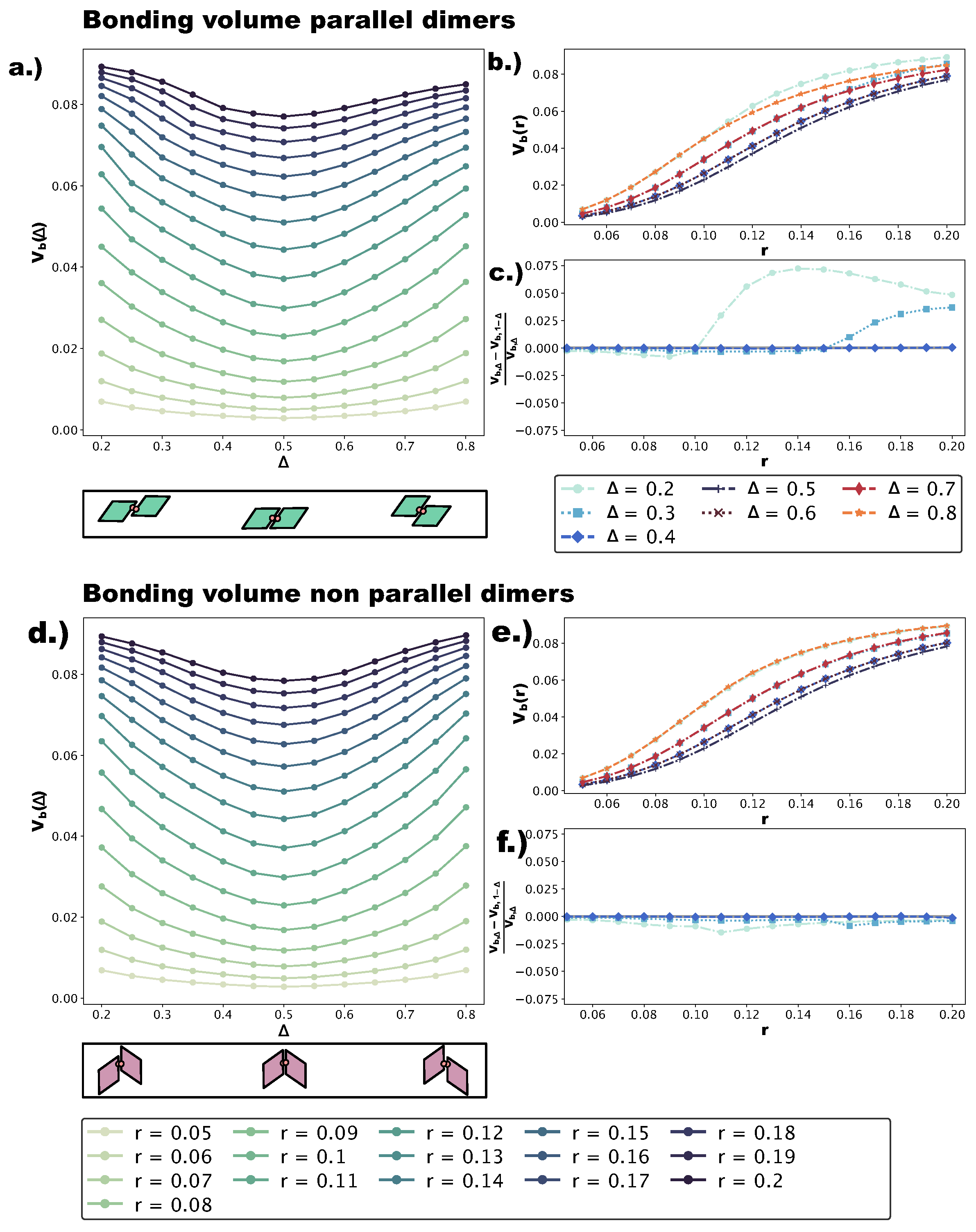
We quantify the change in bond flexibility with increasing
r at different values of
with the bonding volume
, i.e., the volume in configuration space occupied by all possible relative positions and orientations a particle can assume while staying bonded to its neighbor.
Figure 7a,d shows
at different
r-values as a function of
for p- and np-bonds, respectively. Sketches of the dimers used in the calculations (see
Section 4) are reported below the corresponding panels; note that we numerically evaluate
for
, while in the self-assembly simulations, we consider
. This is due to the fact that, while the asymmetric and center topologies for four patches are mirrored with respect to
, the parallel asymmetric topology for one patch is not: for
, bonded patches are closer to the small angles; for
, bonded patches are closer to the big angles. Of course, in the four-patch systems, both bonding types are observed. We find that, for both p- and np-bonds,
is minimal at
and increases monotonically towards
and
. This can be understood intuitively by considering that the more off-center the patches are placed, the more the bonded particles are allowed to wiggle. On increasing
r,
grows at all
-values, with the bonding volume remaining the highest at
and
(corresponding to the most asymmetric patch topologies) and the lowest at
(central patch topology). To better characterize the growth of
with
r at all
-values, we represent
in
Figure 7b,e as a function of
r at different
-values for the p- and np-dimers, respectively. For both p- and np-bonds, the increase in
is the steepest at intermediate
r-values, roughly between
and
, while it is flatter at small (
) and large (
)
r-values. Both the growth trend and the absolute growth in
are similar for p- and np-bonds. In contrast,
is asymmetric with respect to
for p-bonds, whereas it is symmetric for np-bonds. These conclusions can be already drawn from
Figure 7a,d and are quantified in
Figure 7c,f, where we display
at selected
-values. We observe that for p-bonds, the asymmetry grows with
r to the point that, at
,
is approximately
larger than
at
for large
r-values, while for np-bonds, we do not observe a significant asymmetry (see
Figure 7f). We note that, as the effective interaction energy of a single patch is proportional to
, a higher bonding volume implies a higher effective energy, favoring the parallel growth of multiple domains and the formation of grain boundaries. Furthermore, a higher bonding volume contributes to the emergence of poly-bonds especially at extreme
-values. The reason for this is geometrical: off-center patch positions are more accessible, and as soon as—for the given patch topology—the bonding volume is large enough, more than one patch can bond at the same site.
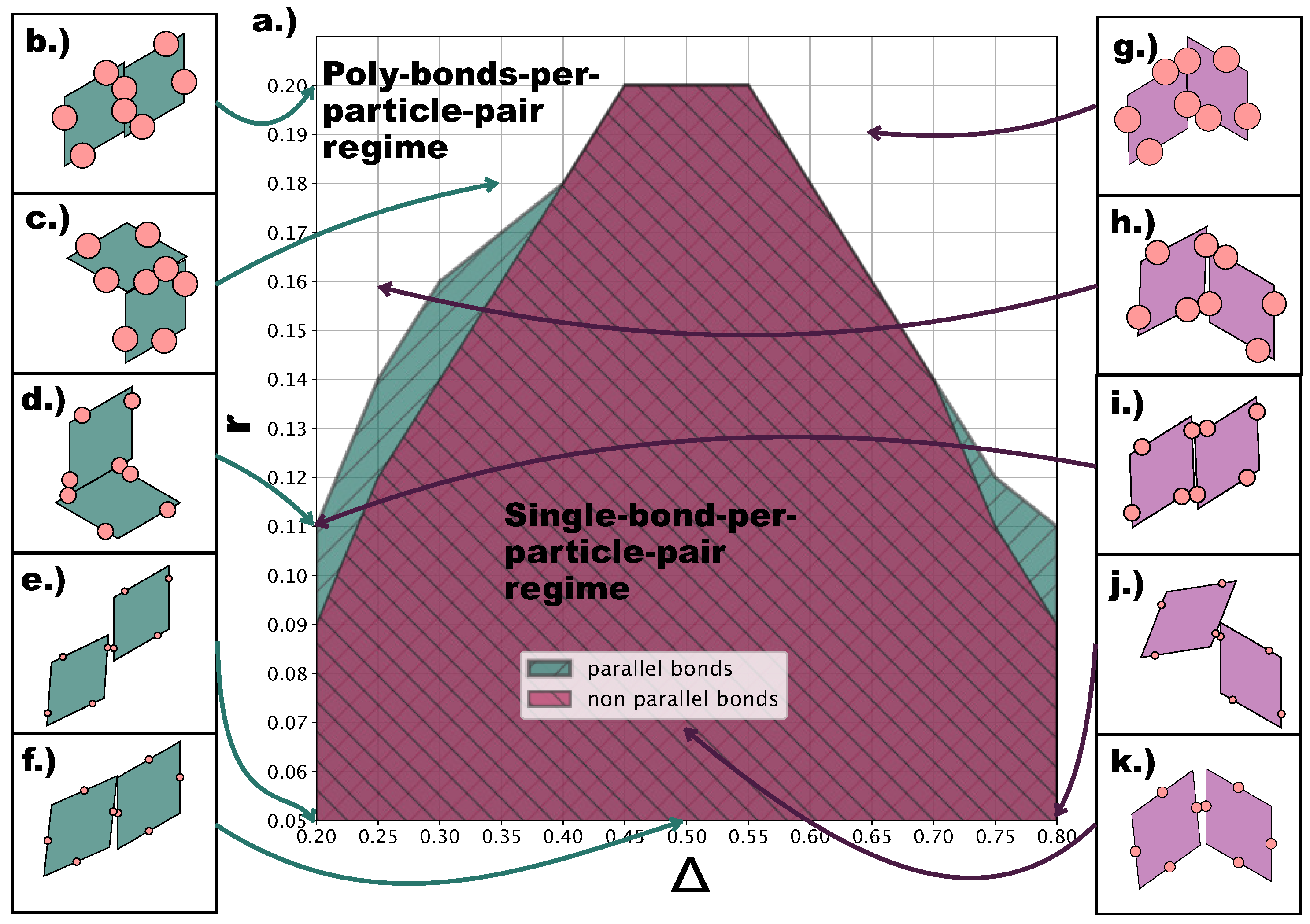
At this point, it is important to describe the effect of the emerging poly-bonds per particle pair as they impact both the bond flexibility and the ordering of the tilings.
Figure 8a shows for which
-values p- (turquoise) and np- (burgundy) poly-bonds can occur, where p and np refers to the initial bonding state of the particle pair at the start of a Monte Carlo simulation (see
Section 4). It is important to stress that—at a given
-combination—poly-bonded particle pairs can be both p- and np-bonded, and by virtue of this, the orientational alignment of the particles can change during the simulation with respect to its starting alignment (see
Figure 8b–k). As p- and np-configurations can interchange during the simulation, we refer to the single-bond per particle pair scenario as the one resulting from the overlap between the two shaded areas in
Figure 8a. The figure suggests that poly-bonds per particle pair form because of purely geometric factors: patches in a more off-center position favor the formation of poly-bonds per particle pair at smaller
r-values with respect to patches in more centered positions. For the studied assemblies, poly-bonds per particle pair often co-occur with poly-bonds per patch: this is visualized by the snapshots labeled with the triangle and the trapezoid in
Figure 4d, where poly-bonds per particle pair of the type depicted in
Figure 8i co-occur with poly-bonds per patch. Thus, in the
-dependent regime of
r-values where the single-bond per particle pair condition is no longer guaranteed, the formation of multiple bonds between pairs of particles promotes the emergence of p-domains. Nonetheless, it should be noted that, at any value of
, poly-bonds per particle pair only appear at a value of
r larger than the
r-value corresponding to the sharp rise in p-bonds, suggesting that the previously observed predominance of p-patterns on increasing the patch size is initiated by the poly-bonds per patch.
As anticipated, poly-bonds per particle pair have a very strong effect on the bond flexibility: when two particles bond together twice—and this happens on increasing
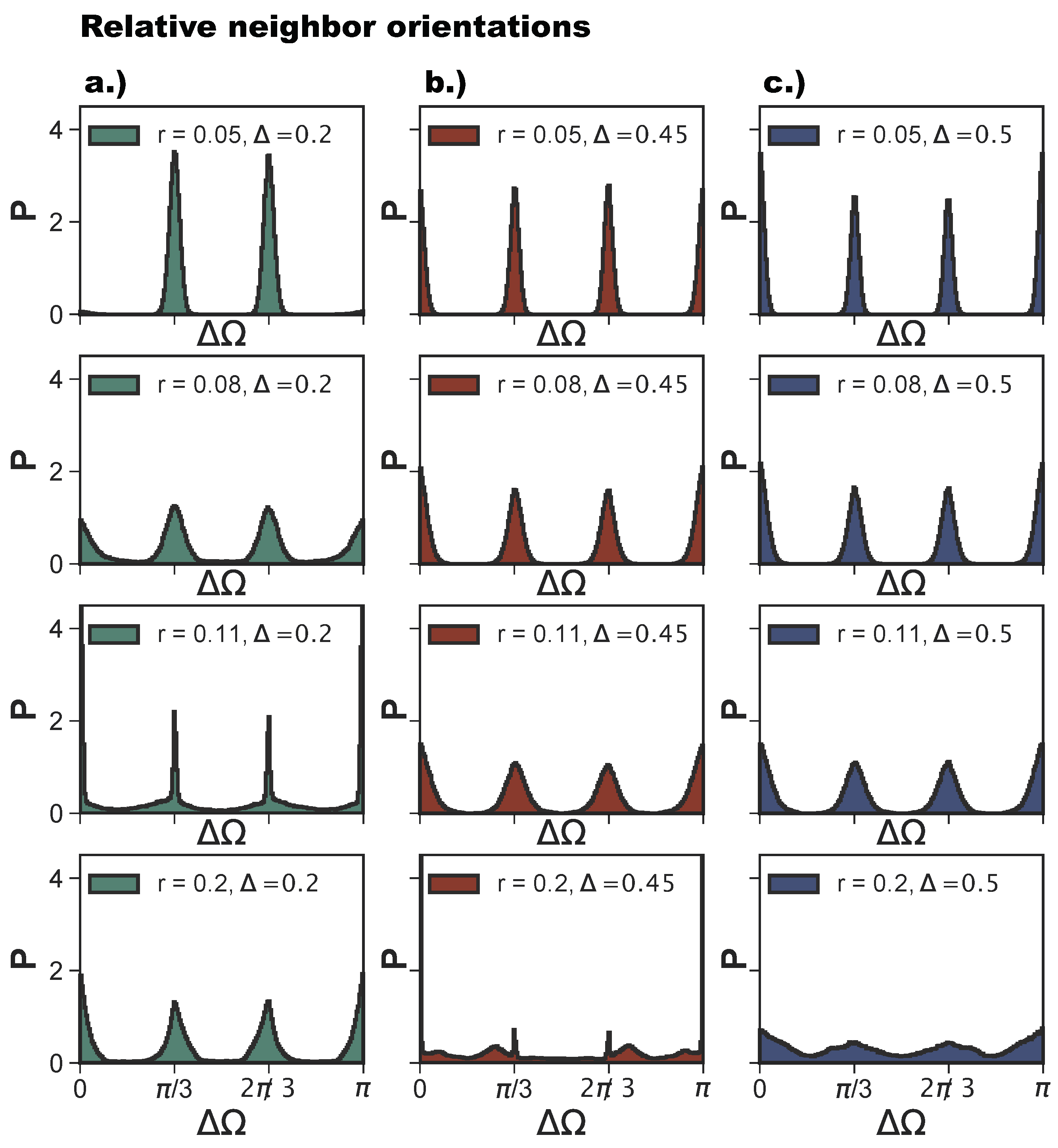
r—the growing bond flexibility is reduced by the double bond between the particles. The reduction of the bond flexibility as soon as poly-bonds per particle pair occur is shown by the histograms of the relative particle orientations,
, defined in
Section 4. For
(see
Figure 9a),
for
shows the typical narrow peaks at
and
of an exclusively np-tiling; as
r increases to
, the np-peaks become wider, and the peaks corresponding to the emerging p-bonds appear at
and zero. At the same time, all peaks become broader as a consequence of the higher bonding flexibility. As
r increases further into the poly-bond per particle pair regime, i.e., at
, we observe a narrowing of the peaks, which indicates a loss of orientational flexibility for a large proportion of bonds in the systems. Note that now the peaks at
and zero are more pronounced than those at
and
as there are more p-bonds. The most typical configuration is shown in
Figure 8i. With a further increase of
r to
, the peaks widen again, but remain more pronounced than those at
r-values corresponding to the single-bond per particle pair regime (at
). For
(see
Figure 9b), we observe the same trend, with the narrowing of the peaks occurring at
, which is the patch radius at which the poly-bonds per particle pair first occur at this
-value. For
and
, we observe four sharp peaks, indicating the random tiling phase. As
r increases, these peaks become broader. At
, the peaks are still visible despite the high orientational flexibility of the bonds. The near absence of poly-bonds per particle pair (see
Figure 8a) allows for orientations outside the peak positions.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}