Efficacy of Sulforaphane in Neurodegenerative Diseases

Abstract
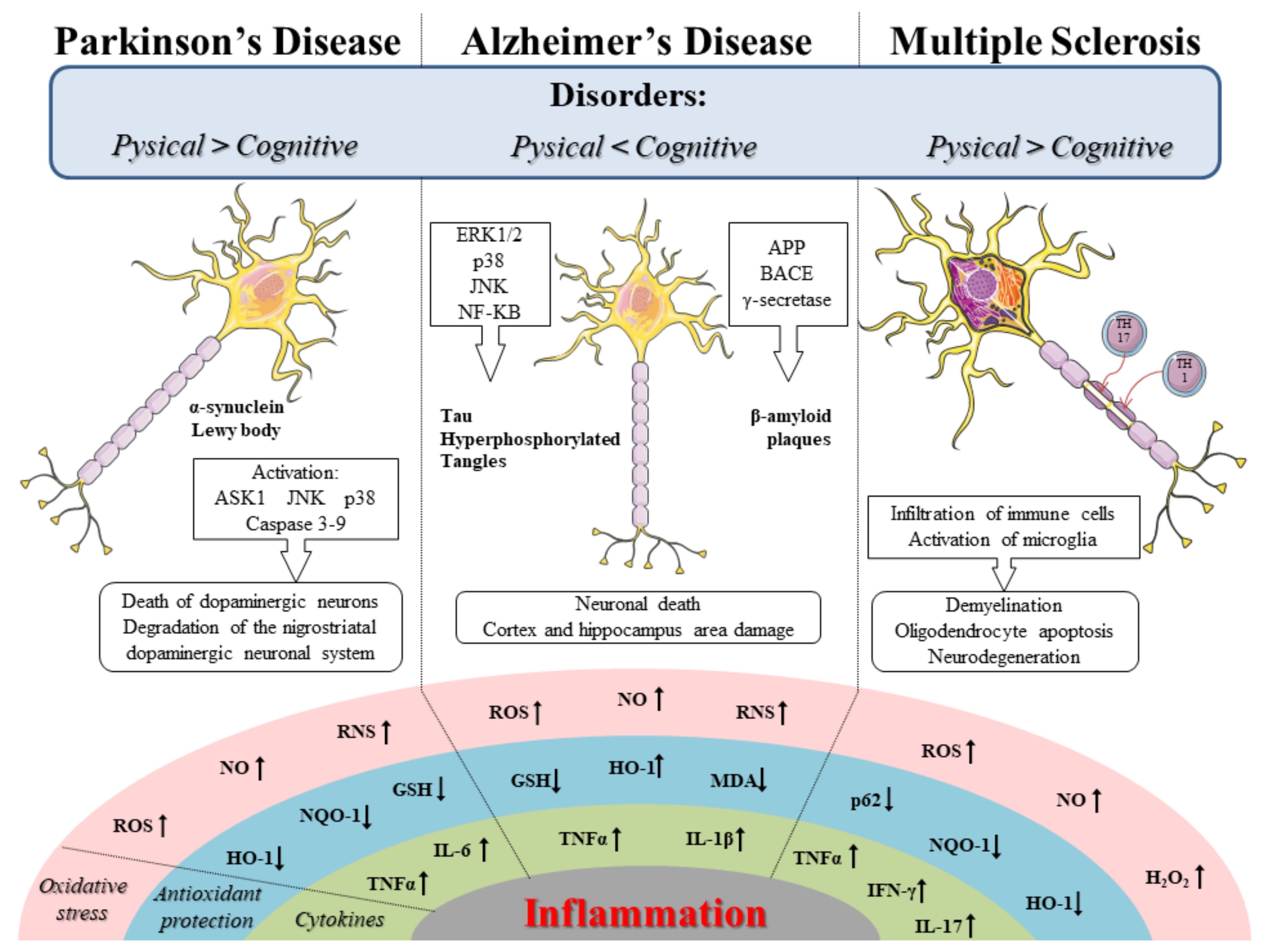
:1. Introduction
2. SFN and Its Molecular Target
3. Effects of Sulforaphane in Experimental Studies of Alzheimer’s Disease
3.1. Effects of Sulforaphane in In Vivo AD Models
3.2. Effects of Sulforaphane in In Vitro AD Models
4. Effects of Sulforaphane in Experimental Studies of Parkinson’s Disease
4.1. Effects of Sulforaphane in In Vitro PD Models
4.2. Effects of Sulforaphane in In Vivo PD Models
5. Effects of Sulforaphane in Experimental Studies of Multiple Sclerosis
5.1. Effects of Sulforaphane in In Vivo MS Models
5.2. Effects of Sulforaphane in In Vitro MS Models
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| SFN | Sulforaphane |
| AD | Alzheimer’s disease |
| PD | Parkinson’s disease |
| MS | multiple sclerosis |
| ESP | epithiospecifier |
| GSH | glutathione |
| Nrf2 | nuclear factor erythroid 2 related factor 2 |
| NQO-1 | nicotinamide adenine dinucleotide phosphate quinone oxidoreductase 1 |
| HO-1 | heme oxygenase 1 |
| ARE | antioxidant response elements |
| Keap1 | Kelch-like ECH associated protein 1 |
| UPS | ubiquitin-proteasome system |
| Maf | musculoaponeurotic fibrosarcoma oncogene homologue |
| ROS | reactive oxygen species reactive |
| RNS | nitrogen species |
| KO | knockout |
| TNF-α | tumor necrosis factor-α |
| IL | interleukin |
| iNOS | inducible nitric oxide synthetase |
| COX-2 | cyclooxygenase-2 |
| MAPK | mitogen-activated protein kinase |
| NF-κB | nuclear factor kappa-B |
| ERK | extracellular signal-regulated kinase |
| JNK | c-Jun N-terminal kinase |
| LC3-II | light chain 3-II |
| ATP | adenosine triphosphate |
| BDNF | brain-derived neurotrophic factor |
| WNT | Wingless type |
| Aβ | beta-amyloid |
| APP | amyloid precursor protein |
| β-secretase1 | BACE-1 |
| MDA | malondialdehyde |
| HSP | heat shock protein |
| CHIP | C-terminus of HSP70-interacting-protein |
| BCL-2 | B cell lymphoma-2 |
| BAX | BCL2-Associated X |
| CNS | central nervous system |
| SOD | superoxide dismutase |
| p75NTR | p75 neurotrophin receptor |
| MPP+ | 1-methyl-4-phenyl pyridine |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| 6-OHDA | 6-hydroxydopamine |
| H2O2 | hydrogen peroxide |
| PI3K/AKT | phosphatidylinositol-3-kinase |
| ASK1/MAP3K5 | apoptosis signal-regulating kinase 1 |
| BBB | blood-brain barrier |
| EAE | experimental autoimmune encephalomyelitis |
| IFN-γ | interferon-γ |
References
- Panjwani, A.A.; Liu, H.; Fahey, J.W. Crucifers and related vegetables and supplements for neurologic disorders: What is the evidence? Curr. Opin. Clin. Nutr. 2018, 21, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Vanduchova, A.; Anzenbacher, P.; Anzenbacherova, E. Isothiocyanate from Broccoli, Sulforaphane, and Its Properties. J. Med. Food 2019, 22, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Perocco, P.; Bronzetti, G.; Canistro, D.; Valgimigli, L.; Sapone, A.; Affatato, A.; Pedulli, G.F.; Pozzetti, L.; Broccoli, M.; Iori, R.; et al. Glucoraphanin, the bioprecursor of the widely extolled chemopreventive agent sulforaphane found in broccoli, induces Phase-I xenobiotic metabolizing enzymes and increases free radical generation in rat liver. Mutat. Res. Fundam. Mol. Mech. Mutagenes. 2006, 595, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Ruhee, R.T.; Suzuki, K. The Integrative Role of Sulforaphane in Preventing Inflammation, Oxidative Stress and Fatigue: A Review of a Potential Protective Phytochemical. Antioxidants 2020, 9, 521. [Google Scholar] [CrossRef]
- Clarke, J.D.; Dashwood, R.H.; Ho, E. Multi-targeted prevention of cancer by sulforaphane. Cancer Lett. 2008, 269, 291–304. [Google Scholar] [CrossRef] [Green Version]
- Manach, C.; Scalbert, A.; Morand, C.; Remesy, C.; Jimenez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef] [Green Version]
- Hu, R.; Hebbar, V.; Kim, B.R.; Chen, C.; Winnik, B.; Buckley, B.; Soteropoulos, P.; Tolias, P.; Hart, R.P.; Kong, A.N. In vivo pharmacokinetics and regulation of gene expression profiles by isothiocyanate sulforaphane in the rat. J. Pharmacol. Exp. Ther. 2004, 310, 263–271. [Google Scholar] [CrossRef] [Green Version]
- Uddin, M.S.; Mamun, A.A.; Jakaria, M.; Thangapandiyan, S.; Ahmad, J.; Rahman, M.A.; Mathew, B.; Abdel-Daim, M.M.; Aleya, L. Emerging promise of sulforaphane-mediated Nrf2 signaling cascade against neurological disorders. Sci. Total Environ. 2020, 707, 135624. [Google Scholar] [CrossRef]
- Zhang, Y.S.; Callaway, E.C. High cellular accumulation of sulphoraphane, a dietary anticarcinogen, is followed by rapid transporter-mediated export as a glutathione conjugate. Biochem. J. 2002, 364, 301–307. [Google Scholar] [CrossRef] [Green Version]
- Eggler, A.L.; Gay, K.A.; Mesecar, A.D. Molecular mechanisms of natural products in chemoprevention: Induction of cytoprotective enzymes by Nrf2. Mol. Nutr. Food Res. 2008, 52 (Suppl. 1), S84–S94. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, S.; Jaiswal, A.K. Functional characterization and role of INrf2 in antioxidant response element-mediated expression and antioxidant induction of NAD(P)H:quinone oxidoreductase1 gene. Oncogene 2001, 20, 3906–3917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “Tethering” mechanism—A two-site interaction model for the Nrf2-Keap1 complex. J. Biol. Chem. 2006, 281, 24756–24768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; An, C.; Gao, Y.; Leak, R.K.; Chen, J.; Zhang, F. Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog. Neurobiol. 2013, 100, 30–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osburn, W.O.; Wakabayashi, N.; Misra, V.; Nilles, T.; Biswal, S.; Trush, M.A.; Kensler, T.W. Nrf2 regulates an adaptive response protecting against oxidative damage following diquat-mediated formation of superoxide anion. Arch. Biochem. Biophys. 2006, 454, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, A.; Kang, M.I.; Watai, Y.; Tong, K.I.; Shibata, T.; Uchida, K.; Yamamoto, M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol. Cell. Biol. 2006, 26, 221–229. [Google Scholar] [CrossRef] [Green Version]
- Itoh, K.; Tong, K.I.; Yamamoto, M. Molecular mechanism activating Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic. Biol. Med. 2004, 36, 1208–1213. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2 small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Kensler, T.W.; Egner, P.A.; Agyeman, A.S.; Visvanathan, K.; Groopman, J.D.; Chen, J.G.; Chen, T.Y.; Fahey, J.W.; Talalay, P. Keap1-nrf2 signaling: A target for cancer prevention by sulforaphane. Top. Curr. Chem. 2013, 329, 163–177. [Google Scholar] [CrossRef] [Green Version]
- Bergstrom, P.; Andersson, H.C.; Gao, Y.; Karlsson, J.O.; Nodin, C.; Anderson, M.F.; Nilsson, M.; Hammarsten, O. Repeated transient sulforaphane stimulation in astrocytes leads to prolonged Nrf2-mediated gene expression and protection from superoxide-induced damage. Neuropharmacology 2011, 60, 343–353. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, K.; Kume, T.; Muto, C.; Takada-Takatori, Y.; Izumi, Y.; Sugimoto, H.; Akaike, A. Glutathione Biosynthesis via Activation of the Nuclear Factor E2-Related Factor 2 (Nrf2)—Antioxidant-Response Element (ARE) Pathway Is Essential for Neuroprotective Effects of Sulforaphane and 6-(Methylsulfinyl) Hexyl Isothiocyanate. J. Pharm. Sci. 2011, 115, 320–328. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.; Yan, W.; Chen, S.; Sun, C.R.; Zhang, J.M. The role of Nrf2 signaling in the regulation of antioxidants and detoxifying enzymes after traumatic brain injury in rats and mice. Acta Pharmacol. Sin. 2010, 31, 1421–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beal, M.F. Therapeutic approaches to mitochondrial dysfunction in Parkinson’s disease. Parkinsonism Relat. Disord. 2009, 15 (Suppl. 3), S189–S194. [Google Scholar] [CrossRef]
- Klomparens, E.A.; Ding, Y. The neuroprotective mechanisms and effects of sulforaphane. Brain Circ. 2019, 5, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Innamorato, N.G.; Rojo, A.I.; Garcia-Yague, A.J.; Yamamoto, M.; de Ceballos, M.L.; Cuadrado, A. The transcription factor Nrf2 is a therapeutic target against brain inflammation. J. Immunol. 2008, 181, 680–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, S.; Yang, C.; Huang, W.; Du, S.; Mai, H.; Xiao, J.; Lu, T. Sulforaphane attenuates microglia-mediated neuronal necroptosis through down-regulation of MAPK/NF-kappaB signaling pathways in LPS-activated BV-2 microglia. Pharmacol. Res. 2018, 133, 218–235. [Google Scholar] [CrossRef] [PubMed]
- Angeloni, C.; Malaguti, M.; Rizzo, B.; Barbalace, M.C.; Fabbri, D.; Hrelia, S. Neuroprotective effect of sulforaphane against methylglyoxal cytotoxicity. Chem. Res. Toxicol. 2015, 28, 1234–1245. [Google Scholar] [CrossRef]
- Pajares, M.; Jimenez-Moreno, N.; Garcia-Yague, A.J.; Escoll, M.; de Ceballos, M.L.; Van Leuven, F.; Rabano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916. [Google Scholar] [CrossRef] [Green Version]
- Jo, C.; Kim, S.; Cho, S.J.; Choi, K.J.; Yun, S.M.; Koh, Y.H.; Johnson, G.V.W.; Park, S.I. Sulforaphane induces autophagy through ERK activation in neuronal cells. FEBS Lett. 2014, 588, 3081–3088. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Jeong, J.K.; Park, S.Y. Sulforaphane-Induced Autophagy Flux Prevents Prion Protein-Mediated Neurotoxicity through Ampk Pathway. Neuroscience 2014, 278, 31–39. [Google Scholar] [CrossRef]
- Denzer, I.; Munch, G.; Friedland, K. Modulation of mitochondrial dysfunction in neurodegenerative diseases via activation of nuclear factor erythroid-2-related factor 2 by food-derived compounds. Pharmacol. Res. 2016, 103, 80–94. [Google Scholar] [CrossRef]
- Luis-Garcia, E.R.; Limon-Pacheco, J.H.; Serrano-Garcia, N.; Hernandez-Perez, A.D.; Pedraza-Chaverri, J.; Orozco-Ibarra, M. Sulforaphane prevents quinolinic acid-induced mitochondrial dysfunction in rat striatum. J. Biochem. Mol. Toxicol. 2017, 31, e21837. [Google Scholar] [CrossRef]
- Han, Z.; Xu, Q.; Li, C.; Zhao, H. Effects of sulforaphane on neural stem cell proliferation and differentiation. Genesis 2017, 55. [Google Scholar] [CrossRef]
- Tarozzi, A.; Angeloni, C.; Malaguti, M.; Morroni, F.; Hrelia, S.; Hrelia, P. Sulforaphane as a Potential Protective Phytochemical against Neurodegenerative Diseases. Oxid. Med. Cell. Longev. 2013, 2013, 415078. [Google Scholar] [CrossRef] [PubMed]
- Tannenberg, R.K.; Scott, H.L.; Tannenberg, A.E.G.; Dodd, P.R. Selective loss of synaptic proteins in Alzheimer’s disease: Evidence for an increased severity with APOE epsilon 4. Neurochem. Int. 2006, 49, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.S.; Stachowiak, A.; Al Mamun, A.; Tzvetkov, N.T.; Takeda, S.; Atanasov, A.G.; Bergantin, L.B.; Abdel-Daim, M.M.; Stankiewicz, A.M. Autophagy and Alzheimer’s Disease: From Molecular Mechanisms to Therapeutic Implications. Front. Aging Neurosci. 2018, 10, 4. [Google Scholar] [CrossRef]
- Loewen, C.A.; Feany, M.B. The unfolded protein response protects from tau neurotoxicity in vivo. PLoS ONE 2010, 5, e13084. [Google Scholar] [CrossRef]
- Hoozemans, J.J.M.; Veerhuis, R.; Van Haastert, E.S.; Rozemuller, J.M.; Baas, F.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol. 2005, 110, 165–172. [Google Scholar] [CrossRef]
- Salon, M.L.; Morelli, L.; Castano, E.M.; Soto, E.F.; Pasquini, J.M. Defective ubiquitination of cerebral proteins in Alzheimer’s disease. J. Neurosci. Res. 2000, 62, 302–310. [Google Scholar] [CrossRef]
- Hou, T.T.; Yang, H.Y.; Wang, W.; Wu, Q.Q.; Tian, Y.R.H.; Jia, J.P. Sulforaphane Inhibits the Generation of Amyloid-beta Oligomer and Promotes Spatial Learning and Memory in Alzheimer’s Disease (PS1V97L) Transgenic Mice. J. Alzheimers Dis. 2018, 62, 1803–1813. [Google Scholar] [CrossRef]
- Wang, W.; Wei, C.; Quan, M.; Li, T.; Jia, J. Sulforaphane Reverses the Amyloid-beta Oligomers Induced Depressive-Like Behavior. J. Alzheimers Dis. 2020. [Google Scholar] [CrossRef]
- Zhang, R.; Miao, Q.W.; Zhu, C.X.; Zhao, Y.; Liu, L.; Yang, J.; An, L. Sulforaphane ameliorates neurobehavioral deficits and protects the brain from amyloid beta deposits and peroxidation in mice with Alzheimer-like lesions. Am. J. Alzheimer’s Dis. Other Dement. 2015, 30, 183–191. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, J.Z.; Fang, L.D.; Li, X.; Zhao, Y.; Shi, W.Y.; An, L. Neuroprotective Effects of Sulforaphane on Cholinergic Neurons in Mice with Alzheimer’s Disease-Like Lesions. Int. J. Mol. Sci. 2014, 15, 14396–14410. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.V.; Kim, H.Y.; Ehrlich, H.Y.; Choi, S.Y.; Kim, D.J.; Kim, Y. Amelioration of Alzheimer’s disease by neuroprotective effect of sulforaphane in animal model. Amyloid 2013, 20, 7–12. [Google Scholar] [CrossRef]
- Kanninen, K.; Malm, T.M.; Jyrkkanen, H.K.; Goldsteins, G.; Keksa-Goldsteine, V.; Tanila, H.; Yamamoto, M.; Yla-Herttuala, S.; Levonen, A.L.; Koistinaho, J. Nuclear factor erythroid 2-related factor 2 protects against beta amyloid. Mol. Cell. Neurosci. 2008, 39, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Joshi, G.; Gan, K.A.; Johnson, D.A.; Johnson, J.A. Increased Alzheimer’s disease-like pathology in the APP/PS1 Delta E9 mouse model lacking Nrf2 through modulation of autophagy. Neurobiol. Aging 2015, 36, 664–679. [Google Scholar] [CrossRef] [Green Version]
- Jo, C.; Gundemir, S.; Pritchard, S.; Jin, Y.N.; Rahman, I.; Johnson, G.V. Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat. Commun. 2014, 5, 3496. [Google Scholar] [CrossRef]
- Calkins, M.J.; Johnson, D.A.; Townsend, J.A.; Vargas, M.R.; Dowell, J.A.; Williamson, T.P.; Kraft, A.D.; Lee, J.M.; Li, J.; Johnson, J.A. The Nrf2/ARE Pathway as a Potential Therapeutic Target in Neurodegenerative Disease. Antioxid. Redox Sign. 2009, 11, 497–508. [Google Scholar] [CrossRef] [Green Version]
- Bahn, G.; Park, J.S.; Yun, U.J.; Lee, Y.J.; Choi, Y.; Park, J.S.; Baek, S.H.; Choi, B.Y.; Cho, Y.S.; Kim, H.K.; et al. NRF2/ARE pathway negatively regulates BACE1 expression and ameliorates cognitive deficits in mouse Alzheimer’s models. Proc. Natl. Acad. Sci. USA 2019, 116, 12516–12523. [Google Scholar] [CrossRef] [Green Version]
- Youn, K.; Yoon, J.H.; Lee, N.; Lim, G.; Lee, J.; Sang, S.; Ho, C.T.; Jun, M. Discovery of Sulforaphane as a Potent BACE1 Inhibitor Based on Kinetics and Computational Studies. Nutrients 2020, 12, 26. [Google Scholar] [CrossRef]
- Lee, S.; Choi, B.R.; Kim, J.; LaFerla, F.M.; Park, J.H.Y.; Han, J.S.; Lee, K.W.; Kim, J. Sulforaphane Upregulates the Heat Shock Protein Co-Chaperone CHIP and Clears Amyloid-beta and Tau in a Mouse Model of Alzheimer’s Disease. Mol. Nutr. Food Res. 2018, 62, 1800240. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K. The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol. 2011, 121, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Broca, C.; Varin, E.; Armanet, M.; Tourrel-Cuzin, C.; Bosco, D.; Dalle, S.; Wojtusciszyn, A. Proteasome Dysfunction Mediates High Glucose-Induced Apoptosis in Rodent Beta Cells and Human Islets. PLoS ONE 2014, 9, e102652. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Di Domenico, F.; Barone, E. Elevated risk of type 2 diabetes for development of Alzheimer disease: A key role for oxidative stress in brain. BBA Mol. Basis Dis. 2014, 1842, 1693–1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pu, D.; Zhao, Y.; Chen, J.; Sun, Y.; Lv, A.; Zhu, S.; Luo, C.; Zhao, K.; Xiao, Q. Protective Effects of Sulforaphane on Cognitive Impairments and AD-like Lesions in Diabetic Mice are Associated with the Upregulation of Nrf2 Transcription Activity. Neuroscience 2018, 381, 35–45. [Google Scholar] [CrossRef]
- Masci, A.; Mattioli, R.; Costantino, P.; Baima, S.; Morelli, G.; Punzi, P.; Giordano, C.; Pinto, A.; Donini, L.M.; d’Erme, M.; et al. Neuroprotective Effect of Brassica oleracea Sprouts Crude Juice in a Cellular Model of Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2015, 2015, 781938. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Park, G.H.; Lee, S.R.; Jang, J.H. Attenuation of beta-amyloid-induced oxidative cell death by sulforaphane via activation of NF-E2-related factor 2. Oxid. Med. Cell. Longev. 2013, 2013, 313510. [Google Scholar] [CrossRef] [Green Version]
- Suganuma, H.; Fahey, J.W.; Bryan, K.E.; Healy, Z.R.; Talalay, P. Stimulation of phagocytosis by sulforaphane. Biochem. Biophys. Res. Commun. 2011, 405, 146–151. [Google Scholar] [CrossRef] [Green Version]
- Chilakala, R.R.; Manchikalapudi, A.L.; Kumar, A.; Sunkaria, A. Sulforaphane Attenuates A beta Oligomers Mediated Decrease in Phagocytic Activity of Microglial Cells. Neuroscience 2020, 429, 225–234. [Google Scholar] [CrossRef]
- Chen, W.W.; Zhang, X.; Huang, W.J. Role of neuroinflammation in neurodegenerative diseases. Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef] [Green Version]
- Subedi, L.; Cho, K.; Park, Y.U.; Choi, H.J.; Kim, S.Y. Sulforaphane-Enriched Broccoli Sprouts Pretreated by Pulsed Electric Fields Reduces Neuroinflammation and Ameliorates Scopolamine-Induced Amnesia in Mouse Brain through Its Antioxidant Ability via Nrf2-HO-1 Activation. Oxid. Med. Cell. Longev. 2019, 2019, 3549274. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Zhang, J.; Chang, N. Epigenetic modification of Nrf2 by sulforaphane increases the antioxidative and anti-inflammatory capacity in a cellular model of Alzheimer’s disease. Eur. J. Pharmacol. 2018, 824, 1–10. [Google Scholar] [CrossRef] [PubMed]
- An, Y.W.; Jhang, K.A.; Woo, S.Y.; Kang, J.L.; Chong, Y.H. Sulforaphane exerts its anti-inflammatory effect against amyloid-beta peptide via STAT-1 dephosphorylation and activation of Nrf2/HO-1 cascade in human THP-1 macrophages. Neurobiol. Aging 2016, 38, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Behrens, E.M.; Gadue, P.; Gong, S.Y.; Garrett, S.; Stein, P.L.; Cohen, P.L. The mer receptor tyrosine kinase: Expression and function suggest a role in innate immunity. Eur. J. Immunol. 2003, 33, 2160–2167. [Google Scholar] [CrossRef] [PubMed]
- Jhang, K.A.; Park, J.S.; Kim, H.S.; Chong, Y.H. Sulforaphane rescues amyloid-beta peptide-mediated decrease in MerTK expression through its anti-inflammatory effect in human THP-1 macrophages. J. Neuroinflamm. 2018, 15, 75. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Lee, S.; Choi, B.R.; Yang, H.; Hwang, Y.; Park, J.H.; LaFerla, F.M.; Han, J.S.; Lee, K.W.; Kim, J. Sulforaphane epigenetically enhances neuronal BDNF expression and TrkB signaling pathways. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef]
- Kordower, J.H.; Gash, D.M.; Bothwell, M.; Hersh, L.; Mufson, E.J. Nerve growth factor receptor and choline acetyltransferase remain colocalized in the nucleus basalis (Ch4) of Alzheimer’s patients. Neurobiol. Aging 1989, 10, 67–74. [Google Scholar] [CrossRef]
- Arendt, T.; Schindler, C.; Bruckner, M.K.; Eschrich, K.; Bigl, V.; Zedlick, D.; Marcova, L. Plastic neuronal remodeling is impaired in patients with Alzheimer’s disease carrying apolipoprotein epsilon 4 allele. J. Neurosci. 1997, 17, 516–529. [Google Scholar] [CrossRef] [Green Version]
- Salehi, A.; Ocampo, M.; Verhaagen, J.; Swaab, D.F. P75 neurotrophin receptor in the nucleus basalis of meynert in relation to age, sex, and Alzheimer’s disease. Exp. Neurol. 2000, 161, 245–258. [Google Scholar] [CrossRef]
- Zeng, F.; Lu, J.J.; Zhou, X.F.; Wang, Y.J. Roles of p75NTR in the pathogenesis of Alzheimer’s disease: A novel therapeutic target. Biochem. Pharmacol. 2011, 82, 1500–1509. [Google Scholar] [CrossRef]
- Yao, X.Q.; Jiao, S.S.; Saadipour, K.; Zeng, F.; Wang, Q.H.; Zhu, C.; Shen, L.L.; Zeng, G.H.; Liang, C.R.; Wang, J.; et al. p75NTR ectodomain is a physiological neuroprotective molecule against amyloid-beta toxicity in the brain of Alzheimer’s disease. Mol. Psychiatr. 2015, 20, 1301–1310. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.Z.; Zhang, R.; Zhan, Z.P.; Li, X.H.; Zhou, F.Y.; Xing, A.P.; Jiang, C.M.; Chen, Y.Q.; An, L. Beneficial Effects of Sulforaphane Treatment in Alzheimer’s Disease May Be Mediated through Reduced HDAC1/3 and Increased P75NTR Expression. Front. Aging Neurosci. 2017, 9, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearce, R.K.; Owen, A.; Daniel, S.; Jenner, P.; Marsden, C.D. Alterations in the distribution of glutathione in the substantia nigra in Parkinson’s disease. J. Neural Transm. 1997, 104, 661–677. [Google Scholar] [CrossRef] [PubMed]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Parkinson’s Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Wu, J.J.; Chen, D.J.; Jin, J.; Wu, Y.; Chen, Z. Effects of sulforaphane in the central nervous system. Eur. J. Pharmacol. 2019, 853, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Tanji, K.; Maruyama, A.; Odagiri, S.; Mori, F.; Itoh, K.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Keap1 Is Localized in Neuronal and Glial Cytoplasmic Inclusions in Various Neurodegenerative Diseases. J. Neuropathol. Exp. Neurol. 2013, 72, 18–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddell, J.R. Are Astrocytes the Predominant Cell Type for Activation of Nrf2 in Aging and Neurodegeneration? Antioxidants 2017, 6, 65. [Google Scholar] [CrossRef]
- Lasser-Katz, E.; Simchovitz, A.; Chiu, W.H.; Oertel, W.H.; Sharon, R.; Soreq, H.; Roeper, J.; Goldberg, J.A. Mutant alpha-Synuclein Overexpression Induces Stressless Pacemaking in Vagal Motoneurons at Risk in Parkinson’s Disease. J. Neurosci. 2017, 37, 47–57. [Google Scholar] [CrossRef] [Green Version]
- Niso-Santano, M.; Gonzalez-Polo, R.A.; Bravo-San Pedro, J.M.; Gomez-Sanchez, R.; Lastres-Becker, I.; Ortiz-Ortiz, M.A.; Soler, G.; Moran, J.M.; Cuadrado, A.; Fuentes, J.M.; et al. Activation of apoptosis signal-regulating kinase 1 is a key factor in paraquat-induced cell death: Modulation by the Nrf2/Trx axis. Free Radic. Bio Med. 2010, 48, 1370–1381. [Google Scholar] [CrossRef]
- Bao, B.; Zhang, M.Q.; Chen, Z.Y.; Wu, X.B.; Xia, Z.B.; Chai, J.Y.; Yin, X.P. Sulforaphane prevents PC12 cells from oxidative damage via the Nrf2 pathway. Mol. Med. Rep. 2019, 19, 4890–4896. [Google Scholar] [CrossRef] [Green Version]
- Deng, C.; Tao, R.; Yu, S.Z.; Jin, H. Inhibition of 6-hydroxydopamine-induced endoplasmic reticulum stress by sulforaphane through the activation of Nrf2 nuclear translocation. Mol. Med. Rep. 2012, 6, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Tao, R.; Yu, S.Z.; Jin, H. Sulforaphane protects against 6-hydroxydopamine-induced cytotoxicity by increasing expression of heme oxygenase-1 in a PI3K/Akt-dependent manner. Mol. Med. Rep. 2012, 5, 847–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, T.T.; Ma, L.; Kandpal, G.; Warren, L.; Hess, J.F.; Seabrook, G.R. Increased nuclear factor-erythroid 2 p45-related factor 2 activity protects SH-SY5Y cells against oxidative damage. J. Neurochem. 2005, 95, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Siebert, A.; Desai, V.; Chandrasekaran, K.; Fiskum, G.; Jafri, M.S. Nrf2 Activators Provide Neuroprotection against 6-Hydroxydopamine Toxicity in Rat Organotypic Nigrostriatal Cocultures. J. Neurosci. Res. 2009, 87, 1659–1669. [Google Scholar] [CrossRef] [PubMed]
- Izumi, Y.; Kataoka, H.; Inose, Y.; Akaike, A.; Koyama, Y.; Kume, T. Neuroprotective effect of an Nrf2-ARE activator identified from a chemical library on dopaminergic neurons. Eur. J. Pharmacol. 2018, 818, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Vauzour, D.; Buonfiglio, M.; Corona, G.; Chirafisi, J.; Vafeiadou, K.; Angeloni, C.; Hrelia, S.; Hrelia, P.; Spencer, J.P.E. Sulforaphane protects cortical neurons against 5-S-cysteinyl-dopamine-induced toxicity through the activation of ERK1/2, Nrf-2 and the upregulation of detoxification enzymes. Mol. Nutr. Food Res. 2010, 54, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Wang, Y.; Yu, G.; Yuan, C.; Ma, J. Quinoprotein adducts accumulate in the substantia nigra of aged rats and correlate with dopamine-induced toxicity in SH-SY5Y cells. Neurochem. Res. 2011, 36, 2169–2175. [Google Scholar] [CrossRef]
- Yoon, N.S.; Cho, Y.; Lee, S.Y.; Choi, H.J.; Hwang, O. Inactivation of Aconitase by Tetrahydrobiopterin in DArgic Cells: Relevance to PD. Exp. Neurobiol. 2010, 19, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Han, J.M.; Lee, Y.J.; Lee, S.Y.; Kim, E.M.; Moon, Y.; Kim, H.W.; Hwang, O. Protective effect of sulforaphane against dopaminergic cell death. J. Pharmacol. Exp. Ther. 2007, 321, 249–256. [Google Scholar] [CrossRef]
- Tarozzi, A.; Morroni, F.; Merlicco, A.; Hrelia, S.; Angeloni, C.; Cantelli-Forti, G.; Hrelia, P. Sulforaphane as an inducer of glutathione prevents oxidative stress-induced cell death in a dopaminergic-like neuroblastoma cell line. J. Neurochem. 2009, 111, 1161–1171. [Google Scholar] [CrossRef]
- Shavali, S.; Sens, D.A. Synergistic neurotoxic effects of arsenic and dopamine in human dopaminergic neuroblastoma SH-SY5Y cells. Toxicol. Sci. Off. J. Soc. Toxicol. 2008, 102, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Morroni, F.; Sita, G.; Djemil, A.; D’Amico, M.; Pruccoli, L.; Cantelli-Forti, G.; Hrelia, P.; Tarozzi, A. Comparison of Adaptive Neuroprotective Mechanisms of Sulforaphane and its Interconversion Product Erucin in in Vitro and in Vivo Models of Parkinson’s Disease. J. Agric. Food Chem. 2018, 66, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Morroni, F.; Tarozzi, A.; Sita, G.; Bolondi, C.; Zolezzi Moraga, J.M.; Cantelli-Forti, G.; Hrelia, P. Neuroprotective effect of sulforaphane in 6-hydroxydopamine-lesioned mouse model of Parkinson’s disease. Neurotoxicology 2013, 36, 63–71. [Google Scholar] [CrossRef]
- Zhou, Q.; Chen, B.; Wang, X.D.; Wu, L.X.; Yang, Y.; Cheng, X.L.; Hu, Z.L.; Cai, X.T.; Yang, J.; Sun, X.Y.; et al. Sulforaphane protects against rotenone-induced neurotoxicity in vivo: Involvement of the mTOR, Nrf2, and autophagy pathways. Sci. Rep. 2016, 6, 32206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pu, Y.; Qu, Y.; Chang, L.; Wang, S.M.; Zhang, K.; Ushida, Y.; Suganuma, H.; Hashimoto, K. Dietary intake of glucoraphanin prevents the reduction of dopamine transporter in the mouse striatum after repeated administration of MPTP. Neuropsychopharmacol. Rep. 2019, 39, 247–251. [Google Scholar] [CrossRef] [Green Version]
- Jazwa, A.; Rojo, A.I.; Innamorato, N.G.; Hesse, M.; Fernandez-Ruiz, J.; Cuadrado, A. Pharmacological Targeting of the Transcription Factor Nrf2 at the Basal Ganglia Provides Disease Modifying Therapy for Experimental Parkinsonism. Antioxid. Redox Sign. 2011, 14, 2347–2360. [Google Scholar] [CrossRef] [Green Version]
- Frohman, E.M.; Racke, M.K.; Raine, C.S. Multiple sclerosis--the plaque and its pathogenesis. N. Engl. J. Med. 2006, 354, 942–955. [Google Scholar] [CrossRef]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Legroux, L.; Arbour, N. Multiple Sclerosis and T Lymphocytes: An Entangled Story. J. Neuroimmune Pharmacol. Off. J. Soc. Neuroimmune Pharmacol. 2015, 10, 528–546. [Google Scholar] [CrossRef] [Green Version]
- Barateiro, A.; Fernandes, A. Temporal oligodendrocyte lineage progression: In vitro models of proliferation, differentiation and myelination. Biochim. Biophys. Acta 2014, 1843, 1917–1929. [Google Scholar] [CrossRef] [Green Version]
- de Castro, F.; Bribian, A. The molecular orchestra of the migration of oligodendrocyte precursors during development. Brain Res. Brain Res. Rev. 2005, 49, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Coppi, E.; Cellai, L.; Maraula, G.; Dettori, I.; Melani, A.; Pugliese, A.M.; Pedata, F. Role of adenosine in oligodendrocyte precursor maturation. Front. Cell. Neurosci. 2015, 9, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patrikios, P.; Stadelmann, C.; Kutzelnigg, A.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Bruck, W.; Lucchinetti, C.; Lassmann, H. Remyelination is extensive in a subset of multiple sclerosis patients. Brain J. Neurol. 2006, 129, 3165–3172. [Google Scholar] [CrossRef] [Green Version]
- Franklin, R.J.; Ffrench-Constant, C. Remyelination in the CNS: From biology to therapy. Nat. Rev. Neurosci. 2008, 9, 839–855. [Google Scholar] [CrossRef] [PubMed]
- Setzu, A.; Lathia, J.D.; Zhao, C.; Wells, K.; Rao, M.S.; Ffrench-Constant, C.; Franklin, R.J. Inflammation stimulates myelination by transplanted oligodendrocyte precursor cells. Glia 2006, 54, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Scolding, N.; Franklin, R.; Stevens, S.; Heldin, C.H.; Compston, A.; Newcombe, J. Oligodendrocyte progenitors are present in the normal adult human CNS and in the lesions of multiple sclerosis. Brain J. Neurol. 1998, 121, 2221–2228. [Google Scholar] [CrossRef] [Green Version]
- Adamczyk, B.; Adamczyk-Sowa, M. New Insights into the Role of Oxidative Stress Mechanisms in the Pathophysiology and Treatment of Multiple Sclerosis. Oxid. Med. Cell. Longev. 2016, 2016, 1973834. [Google Scholar] [CrossRef] [Green Version]
- Fetisova, E.; Chernyak, B.; Korshunova, G.; Muntyan, M.; Skulachev, V. Mitochondria-targeted Antioxidants as a Prospective Therapeutic Strategy for Multiple Sclerosis. Curr. Med. Chem. 2017, 24, 2086–2114. [Google Scholar] [CrossRef]
- Carvalho, A.N.; Firuzi, O.; Gama, M.J.; van Horssen, J.; Saso, L. Oxidative Stress and Antioxidants in Neurological Diseases: Is There Still Hope? Curr. Drug Targets 2017, 18, 705–718. [Google Scholar] [CrossRef]
- Solleiro-Villavicencio, H.; Rivas-Arancibia, S. Effect of Chronic Oxidative Stress on Neuroinflammatory Response Mediated by CD4(+)T Cells in Neurodegenerative Diseases. Front. Cell. Neurosci. 2018, 12, 114. [Google Scholar] [CrossRef] [Green Version]
- Valacchi, G.; Virgili, F.; Cervellati, C.; Pecorelli, A. OxInflammation: From Subclinical Condition to Pathological Biomarker. Front. Physiol. 2018, 9, 858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michalickova, D.; Hrncir, T.; Canova, N.K.; Slanar, O. Targeting Keap1/Nrf2/ARE signaling pathway in multiple sclerosis. Eur. J. Pharmacol. 2020, 873, 172973. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.A.; Amirahmadi, S.; Ward, C.; Fabry, Z.; Johnson, J.A. The absence of the pro-antioxidant transcription factor Nrf2 exacerbates experimental autoimmune encephalomyelitis. Toxicol. Sci. Off. J. Soc. Toxicol. 2010, 114, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Linker, R.A.; Lee, D.H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain J. Neurol. 2011, 134, 678–692. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.M.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef]
- Li, B.; Cui, W.; Liu, J.; Li, R.; Liu, Q.; Xie, X.H.; Ge, X.L.; Zhang, J.; Song, X.J.; Wang, Y.; et al. Sulforaphane ameliorates the development of experimental autoimmune encephalomyelitis by antagonizing oxidative stress and Th17-related inflammation in mice. Exp. Neurol. 2013, 250, 239–249. [Google Scholar] [CrossRef]
- Yoo, I.H.; Kim, M.J.; Kim, J.; Sung, J.J.; Park, S.T.; Ahn, S.W. The Anti-Inflammatory Effect of Sulforaphane in Mice with Experimental Autoimmune Encephalomyelitis. J. Korean Med. Sci. 2019, 34, e197. [Google Scholar] [CrossRef]
- Lassmann, H.; van Horssen, J. The molecular basis of neurodegeneration in multiple sclerosis. FEBS Lett. 2011, 585, 3715–3723. [Google Scholar] [CrossRef] [Green Version]
- Lassmann, H.; van Horssen, J.; Mahad, D. Progressive multiple sclerosis: Pathology and pathogenesis. Nat. Rev. Neurol. 2012, 8, 647–656. [Google Scholar] [CrossRef]
- Lim, J.L.; van der Pol, S.M.; Baron, W.; McCord, J.M.; de Vries, H.E.; van Horssen, J. Protandim Protects Oligodendrocytes against an Oxidative Insult. Antioxidants 2016, 5, 30. [Google Scholar] [CrossRef] [Green Version]
- Wierinckx, A.; Breve, J.; Mercier, D.; Schultzberg, M.; Drukarch, B.; Van Dam, A.M. Detoxication enzyme inducers modify cytokine production in rat mixed glial cells. J. Neuroimmunol. 2005, 166, 132–143. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Experimental Models | Dose/Concentration Range | Route of Administration | Results | References |
|---|---|---|---|---|
| PS1V97L transgenic mice and primary cortical cells of rats | SFN (5 mg/kg) SFN (0.1 μM) | Intraperitoneal | In vivo, SFN improved cognitive deficits, inhibited Aβ aggregation and tau hyperphosphorylation, as well as reduced the oxidative stress and neuroinflammation. Instead, in vitro SFN improved cell viability and preserved dendritic length | [39] |
| Sprague-Dawley male rats | SFN (5 mg/kg) | Intraperitoneal | SFN improved depressive behaviors, reduced oxidative stress and neuroinflammation | [40] |
| C57BL/6 mice | SFN (25 mg/kg) | Oral | SFN treatment improved cognitive and motor deficits, reduced oxidative stress and formation of Aβ plaques both in the cortex and hippocampus | [41] |
| Kunming mice | SFN (25 mg/kg) | Gavage | SFN improved cognitive deficits as well as attenuated the loss of cholinergic neurons in the hippocampus and medial septum | [42] |
| ICR mice | SFN (30 mg/kg) | Intraperitoneal | SFN improved cognitive and memory deficits, as well as reduced the oxidative stress and prevented Aβ aggregation | [43] |
| 3 × Tg-AD mice | SFN (10 or 50 mg/kg) | Gavage | SFN improved memory and learning deficits. Moreover, SFN reduced Aβ levels in the cortex, as well as Aβ and tau levels in the hippocampus | [50] |
| type 2 diabetes mellitus transgenic mice | SFN (1 mg/kg) | Intraperitoneal | SFN improved the cognitive deficits, it also reduced the oxidative stress and Aβ aggregation as well as phosphorylated tau levels in the hippocampus | [54] |
| Experimental Models | Dose/Concentration Range | Route of Administration | Results | References |
|---|---|---|---|---|
| SH-SY5Y cells and 5xFAD and 3 × Tg-AD mice | SFN (1 μM) SFN (5 or 10 mg/kg) | . Intraperitoneal | In vitro, SFN treatment led to a reduction in the amyloidogenesis and oxidative stress. While, in vivo SFN administration, improved cognitive deficits and reduced Aβ aggregation | [48] |
| SH-SY5Y cells | Crude juices of broccoli sprouts (10 μM) | . | The treatment with broccoli juices reduced cell death and oxidative stress Aβ-induced | [55] |
| SH-SY5Y cells | SFN (1 μM, 2 μM and 5 μM) | . | SFN protected cells from cytotoxicity and apoptosis induced by Aβ25–35. Moreover, the SFN treatment reduced oxidative stress | [56] |
| Microglial cells | SFN (5 µM) | . | SFN treatment improved microglia phagocytosis, reduced due to Aβ aggregates | [58] |
| Murine microglia cell BV2 and neuroblastoma cell N2a and male ICR mice | SFN-enriched broccoli sprouts 10 Μl Broccoli sprout (200 mg/kg) | . Oral | In vitro, SFN treatment reduced the inflammation, oxidative stress and apoptosis. While in vivo, the administration of SFN improved the memory deficits | [60] |
| Neuroblastoma N2a cells | SFN (1.25 and 2.5 μM) | . | SFN treatment decreased Aβ1–40 and Aβ1–42 levels in a dose-dependent manner. Moreover, SFN reduced the oxidative stress and the neuroinflammation | [61] |
| Human THP-1 macrophages | SFN (5 μM) | . | SFN treatment reduced neuroinflammation in Aβ1–42 induced macrophages | [62] |
| Human THP-1 macrophages | SFN (5 μM) | . | SFN through Mer tyrosine kinase could exert an anti-inflammatory effect induced by Aβ1–42 | [64] |
| Cortical neurons of ICR mice and 3 × Tg-AD mice | SFN (10 or 20 Μm) SFN (10 or 50 mg/kg) | . Gavage | Both in vitro and in vivo, SFN treatment led to a reduction of neurodegeneration in the cortex and hippocampus | [65] |
| APP/presenilin1 double transgenic mouse and SH-SY5Y cells | SFN (25 mg/kg) SFN (2 μM) | Gavage . | In vivo SFN improved cognitive deficits and preserved the cortex from the increase of Aβ aggregates. While, in vitro, SFN prevented the reduction in cell viability | [71] |
| Experimental Models | Dose/Concentration Range | Route of Administration | Results | References |
|---|---|---|---|---|
| SH-SY5Y cells and Mouse embryonic fibroblasts | SFN (5 µM) | . | SFN reduced the oxidative stress and cell death | [79] |
| PC12 cells | SFN (0.5, 1.0, 2.5, 5.0 and 10 µmol/L) | . | SFN pre-treatment reduced cell damage induced by oxidative stress | [80] |
| PC12 cells | SFN (0.1, 1 and 5 µM) | . | SFN led to an increase in cell viability and inhibited cell death. In addition, SFN also reduced stress in the endoplasmic reticulum | [81] |
| PC12 cells | SFN (1 and 5 µM) | . | The pre-treatment with SFN prevented cell damage induced by 6-OHDA | [82] |
| dopaminergic neurons of organotypic rat nigrostriatal cultures | SFN (5 μM) | . | SFN and tert-butylhydroquinone reduced nigrostriatal neurodegeneration | [84] |
| PC12 cells and primary mesencephalic cultures | TPNA10168 (0.1–30 µM) and SFN (0.1–10 µM) | . | Both TPNA10168 and SFN treatment, protected dopaminergic neurons from neurodegeneration | [85] |
| primary cortical neurons of mouse | SFN (0.01–1 μM) | . | SFN pre-treatment protected neurons both from cell death and oxidative stress | [86] |
| CATH.a and SK-N-BE (2) C and mesencephalic dopaminergic neurons | SFN (0.5, 1, 2.5 and 5 μM) | . | SFN preserved neurons from neurodegeneration, reducing oxidative stress and favouring the increase of NQO-1 activity | [89] |
| SH-SY5Y cells | SFN (0.63–5 μmol/L) | . | SFN pre-treatment reduced oxidative stress and prevented necrosis and apoptosis | [90] |
| SH-SY5Y cells and also in urothelial, human embryonic kidney cells | SFN (0.1–5 μM) | . | The treatment with SFN and N-acetylcysteine reduced oxidative stress and consequent neuronal damage induced by the mixture of arsenite and dopamine | [91] |
| SH-SY5Y cells and C57Bl/6 mice | SFN (5 μM) SFN (30 μmol/kg) | . Intraperitoneal | In vitro, SFN pre-treatment increased the cell survival. While, in vivo, SFN improved behavioral deficits, reduced the loss of dopaminergic neuron and apoptosis | [92] |
| Experimental Models | Dose/Concentration Range | Route of Administration | Results | References |
|---|---|---|---|---|
| C57Bl/6 mice | SFN (5 mg/kg) | Intraperitoneal | SFN administration improved motor deficits and protected the neurons from neurodegeneration and apoptosis | [93] |
| C57Bl/6 mice | SFN (50 mg/kg) | Intraperitoneal | SFN treatment prevented the motor deficits and loss of dopaminergic neurons. Moreover, SFN reduced oxidative stress | [94] |
| C57Bl/6 mice | 0.1% glucoraphanin pellet | Oral | The treatment with 0.1% glucoraphanin pellet preserved the dopaminergic neurons from the neurodegeneration | [95] |
| Wild-type mice and Nrf2-KO mice | SFN (50 mg/kg) | Intraperitoneal | In wild-type mice, SFN acting through Nrf2 attenuated both nigrostriatal neurodegeneration and neuroinflammation | [96] |
| Experimental Models | Dose/Concentration Range | Route of Administration | Results | References |
|---|---|---|---|---|
| EAE C57Bl/6 mice | SFN (50 mg/kg) | Intraperitoneal | SFN through its antioxidant action reduced oxidative stress and inhibited inflammation | [116] |
| EAE C57Bl/6 mice | SFN (50 mg/kg/day) | Intraperitoneal | SFN improved behavioral deficits, also favoring the reduction of oxidative stress and neuroinflammation | [117] |
| Experimental Models | Dose/Concentration Range | Results | References |
|---|---|---|---|
| OLN-93 cells | SFN (5 µM) | The treatment with SFN, monomethyl fumarate and Protandim prevented oxidative stress induced by hydrogen peroxide | [120] |
| primary co-cultures of astroglial and microglial cells of rats | SFN (1, 5, or 15 μM) | SFN and dimethyl fumarate alone or in combination reduced inflammation and enhancing the detoxifying action | [121] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schepici, G.; Bramanti, P.; Mazzon, E. Efficacy of Sulforaphane in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8637. https://doi.org/10.3390/ijms21228637
Schepici G, Bramanti P, Mazzon E. Efficacy of Sulforaphane in Neurodegenerative Diseases. International Journal of Molecular Sciences. 2020; 21(22):8637. https://doi.org/10.3390/ijms21228637
Chicago/Turabian StyleSchepici, Giovanni, Placido Bramanti, and Emanuela Mazzon. 2020. "Efficacy of Sulforaphane in Neurodegenerative Diseases" International Journal of Molecular Sciences 21, no. 22: 8637. https://doi.org/10.3390/ijms21228637
APA StyleSchepici, G., Bramanti, P., & Mazzon, E. (2020). Efficacy of Sulforaphane in Neurodegenerative Diseases. International Journal of Molecular Sciences, 21(22), 8637. https://doi.org/10.3390/ijms21228637