The PI3K/AKT Pathway Is Activated by HGF in NT2D1 Non-Seminoma Cells and Has a Role in the Modulation of Their Malignant Behavior

 ,
,  ,
,  , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
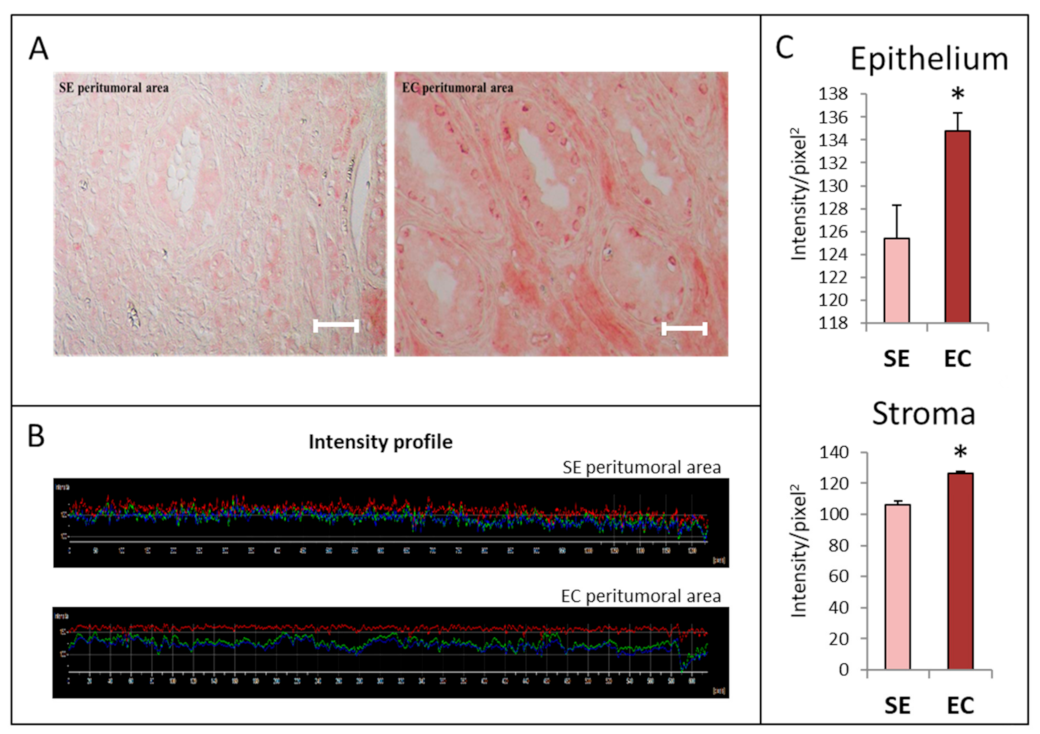
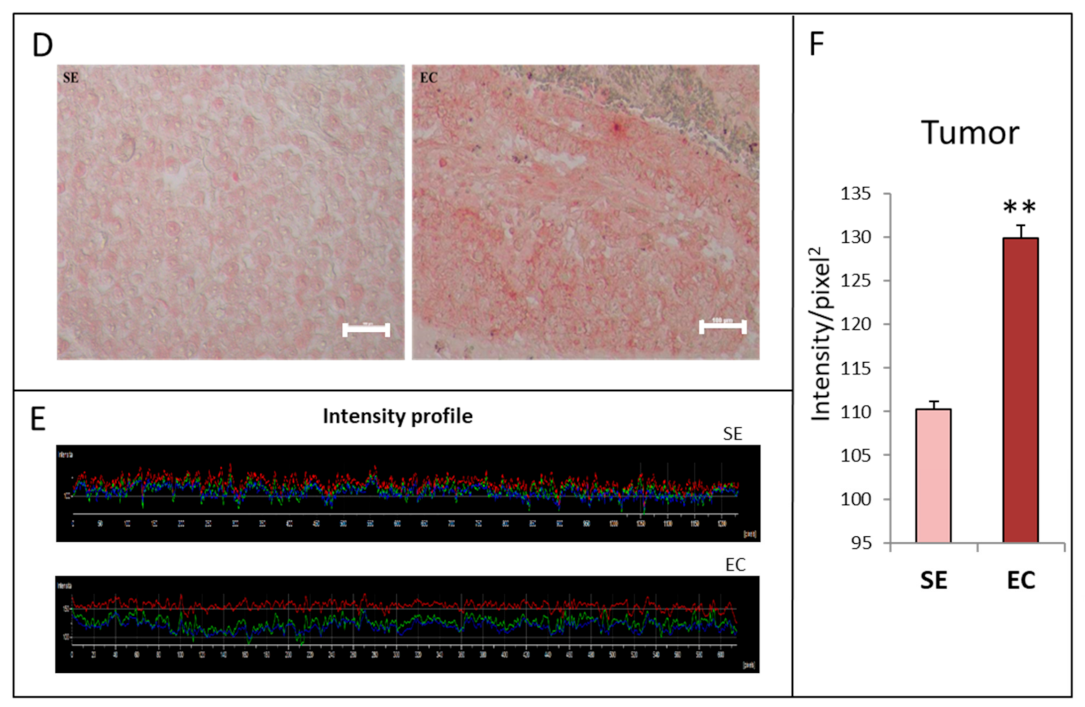
2.1. HGF Distribution Pattern in TGCT Histological Samples
2.2. The PI3K/AKT Pathway Is Activated after HGF Administration in NT2D1 Cells
2.3. Pharmacological Inhibition of PI3K/AKT in Culture Using LY294002
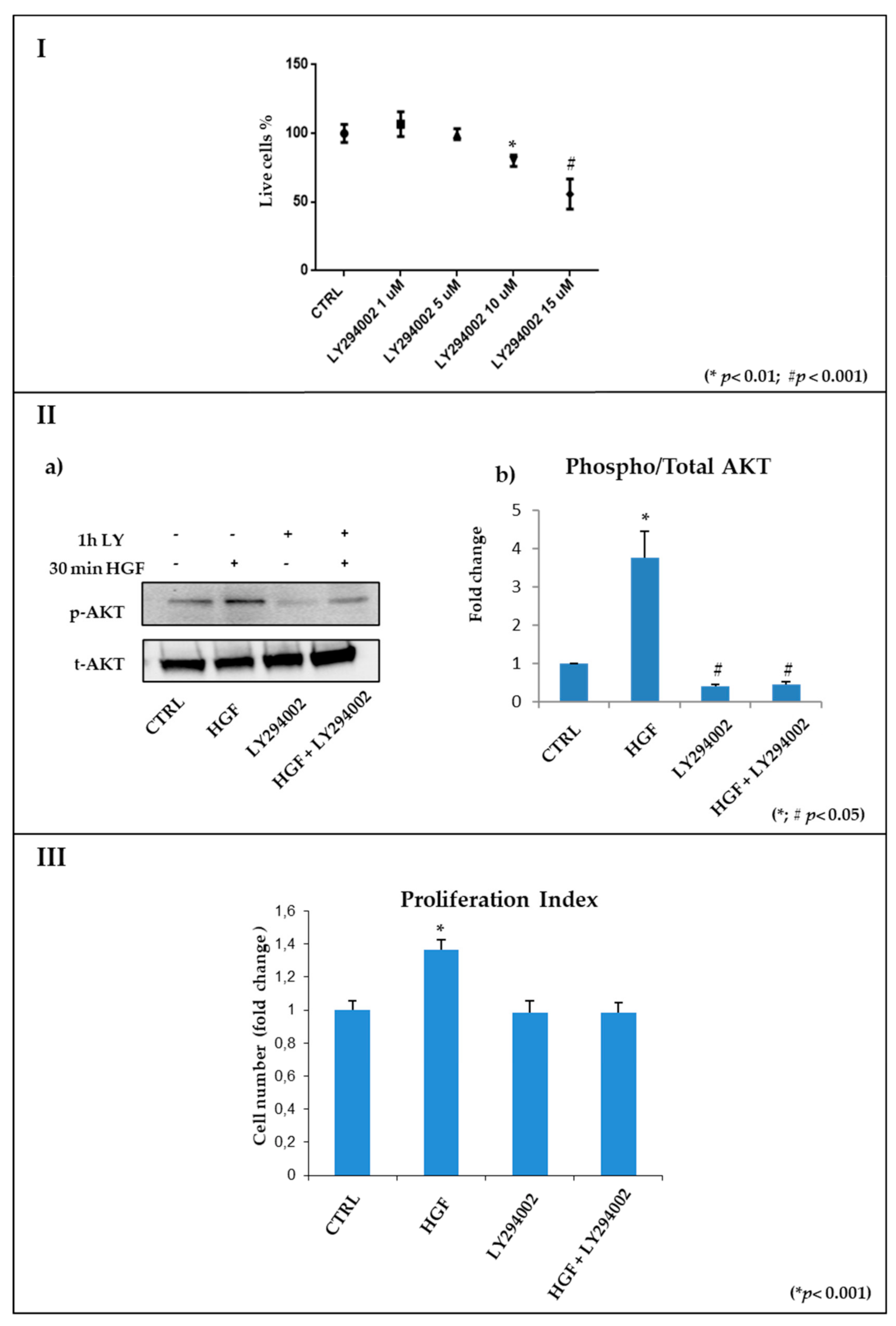
2.3.1. Identification of the Effective and Non-Toxic Concentrations of LY294002
2.3.2. The PI3K/AKT Pathway Is Inhibited by LY294002 Administration in NT2D1 Cells
2.4. HGF-Stimulated NT2D1 Cell Proliferation Depends on PI3K/AKT Activation
2.5. SEM Analysis of HGF-Induced NT2D1 Morphological Modification: Effect of PI3K Inhibition
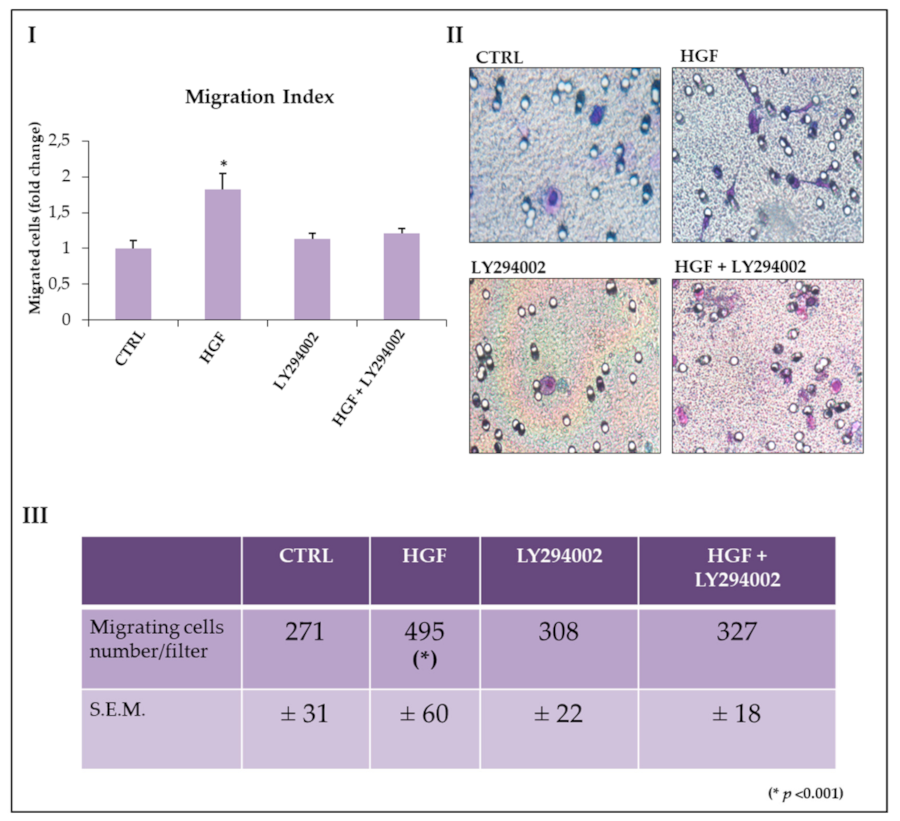
2.6. The PI3/AKT Pathway Is Involved in HGF-Dependent NT2D1 Chemotaxis
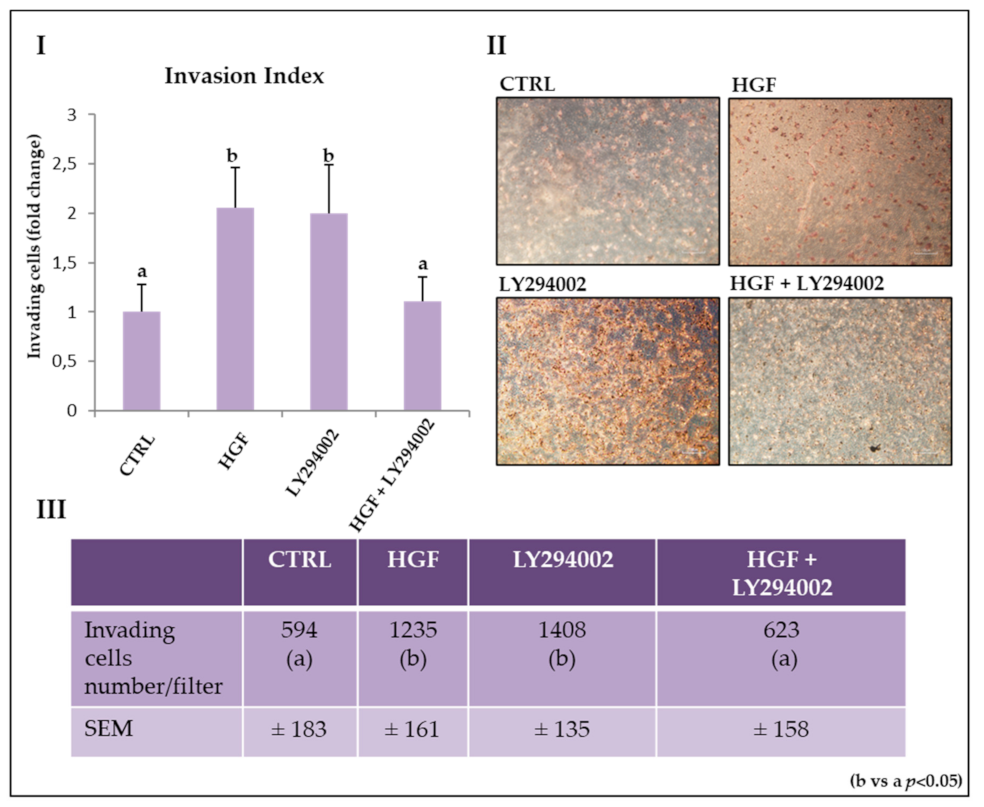
2.7. The PI3K/AKT Pathway Is Involved in HGF-Dependent and HGF-Independent NT2D1 Cell Invasion
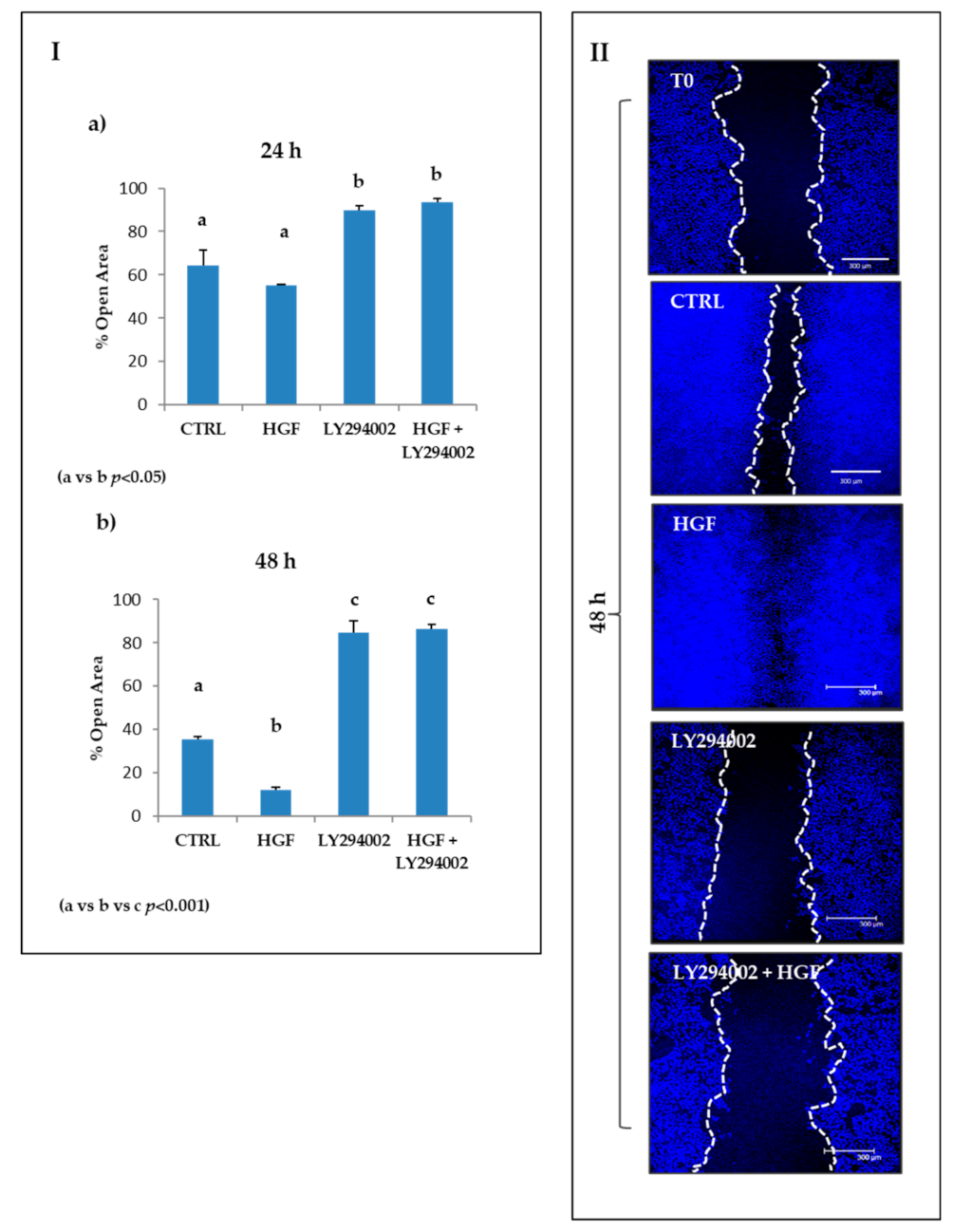
2.8. The PI3K/AKT Pathway Is Involved in the Modulation of Both Constitutive and HGF-Induced Collective Migration
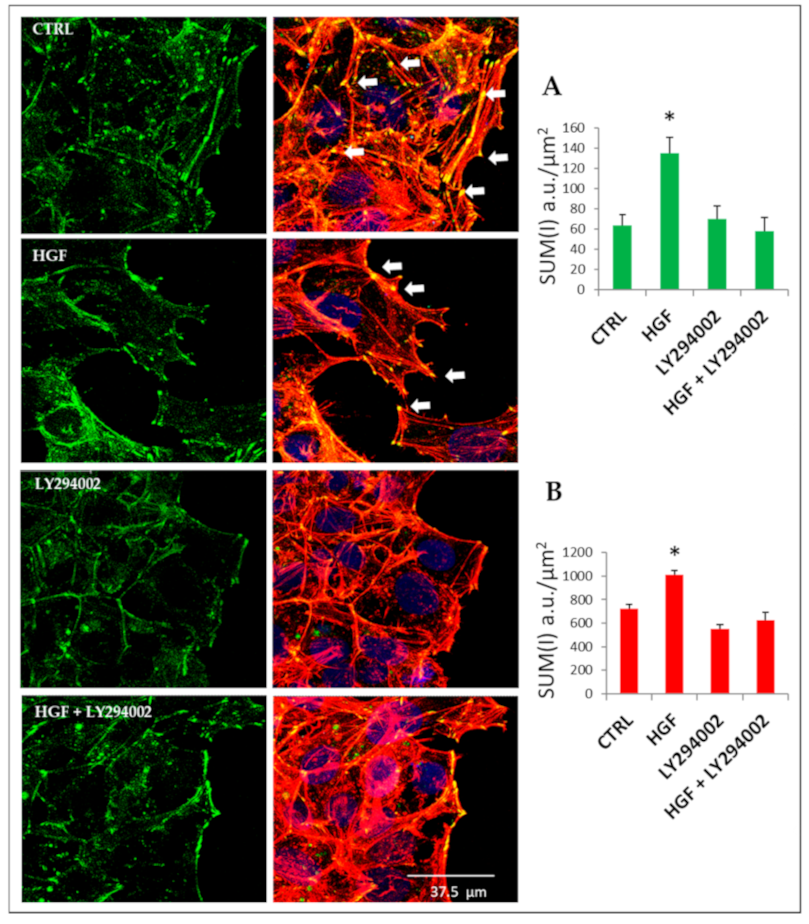
2.9. Cytoskeletal Remodeling Is Involved in HGF-Induced Collective Migration: The Role of PI3K
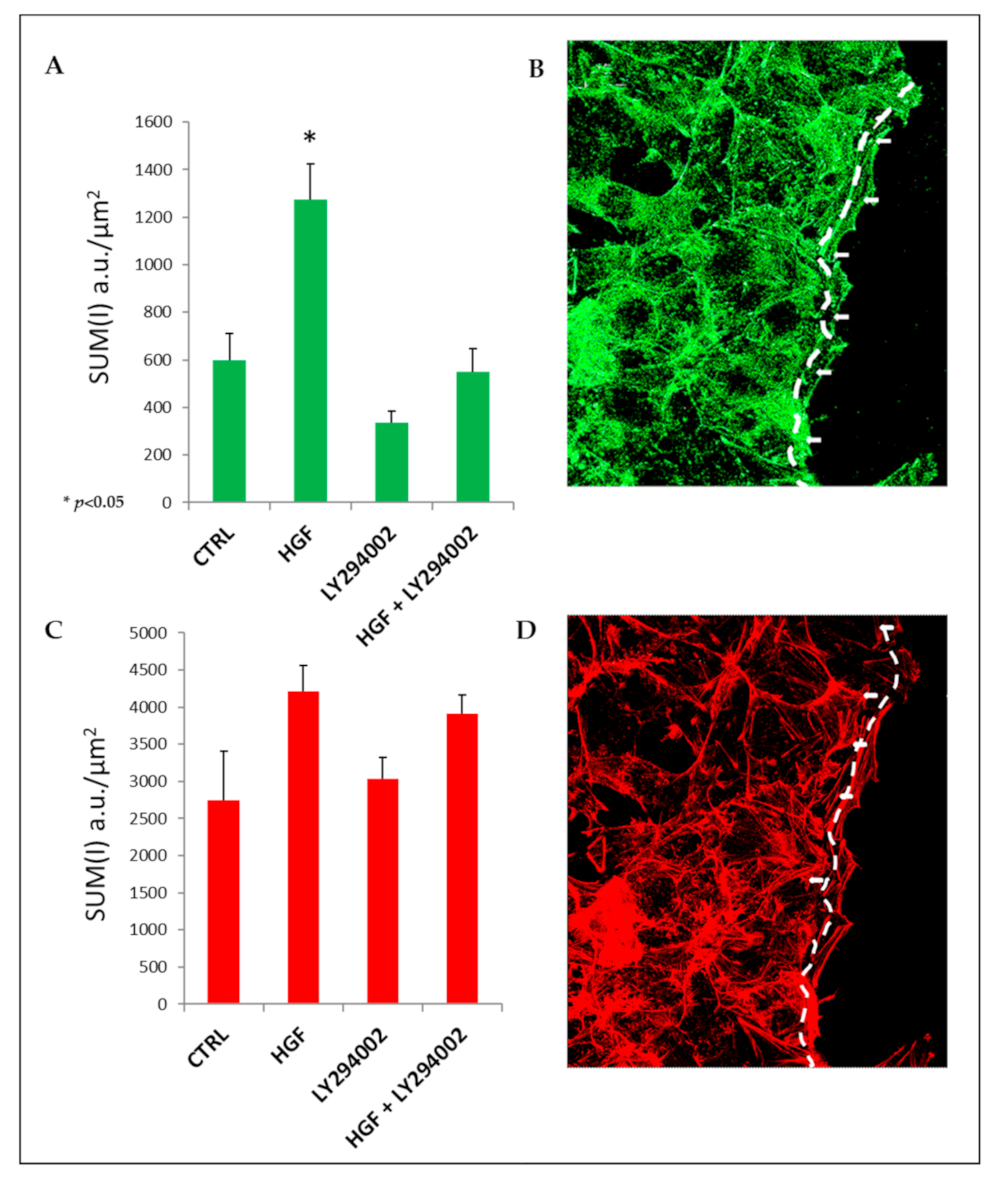
2.10. HGF Induces Focal Adhesion Formation during Collective Migration via PI3K Stimulation
3. Discussion
4. Material and Methods
4.1. Immunohistochemical Analyses
4.2. Cell Culture
4.3. Cell Death Analysis
4.4. Cell Proliferation Assay
4.5. Chemotaxis Assay
4.6. Matrigel Invasion Assay
4.7. Wound-Healing Assay (Collective Migration Assay)
4.8. Confocal Analysis of F-Actin and Vinculin Distribution Pattern
4.9. Western Blot Analyses
4.10. Scanning Electron Microscopy
4.11. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AEC | 3-amino-9-ethylcarbazole |
| AKT | Protein kinase B |
| BCA | Bicinchoninic acid |
| BSA | Bovine serum albumin |
| c-MET | Mesenchymal–epithelial transition (HGF receptor) |
| DMEM | Dulbecco’s modified Eagle’s medium |
| EDTA | Ethylenediaminetetraacetic acid |
| FAs | Focal adhesions |
| FITC | Fluorescein Isothiocyanate |
| FBS | Fetal bovine serum |
| GCNIS | Germ cell neoplasia in situ |
| GFR | Growth factor reduced |
| HGF | Hepatocyte growth factor |
| HRP | Horseradish peroxidase |
| IGFR | Insulin-like growth factor receptor |
| PI3K | Phosphatidylinositol-3-kinase |
| PBS | Phosphate buffer saline |
| PDGFR | Platelet-derived growth factor receptors |
| PTEN | Phosphatase and tensin homolog |
| SDS | Sodium dodecyl sulfate |
| SE | Seminoma |
| S.E.M | Standard error of the mean |
| SEM | Scanning electron microscopy |
| SUM(I) | Sum of intensity |
| TGCTs | Testicular germ cell tumors |
| TRITC | Tetramethyl rhodamine iso-thiocyanate |
| VEGFR | Vascular endothelial growth factor receptor |
References
- Nason, G.J.; Chung, P.; Warde, P.; Huddart, R.; Albers, P.; Kollmannsberger, C.; Booth, C.M.; Hansen, A.R.; Bedard, P.L.; Einhorn, L.; et al. Controversies in the management of clinical stage 1 testis cancer. Can. Urol. Assoc. J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lutke Holzik, M.F.; Sijmons, R.H.; Hoekstra-Weebers, J.E.; Sleijfer, D.T.; Hoekstra, H.J. Clinical and genetic aspects of testicular germ cell tumours. Hered. Cancer Clin. Pract. 2008, 6, 3–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobo, J.; Jeronimo, C.; Henrique, R. Cisplatin Resistance in testicular germ cell tumors: Current challenges from various perspectives. Cancers 2020, 12, 1601. [Google Scholar] [CrossRef] [PubMed]
- Pichler, R.; Leonhartsberger, N.; Stohr, B.; Horninger, W.; Steiner, H. Two cycles of cisplatin-based chemotherapy for low-volume stage II seminoma: Results of a retrospective, single-center case series. Chemotherapy 2012, 58, 405–410. [Google Scholar] [CrossRef]
- Inoue, Y.; Nakamura, T.; Nakanishi, H.; Oishi, M.; Hongo, F.; Okihara, K.; Mizutani, S.; Kuroda, J.; Ukimura, O. Therapy-related acute myeloid leukemia and myelodysplastic syndrome among refractory germ cell tumor patients. Int. J. Urol. 2018, 25, 678–683. [Google Scholar] [CrossRef] [Green Version]
- de Vries, G.; Rosas-Plaza, X.; van Vugt, M.; Gietema, J.A.; de Jong, S. Testicular cancer: Determinants of cisplatin sensitivity and novel therapeutic opportunities. Cancer Treat. Rev. 2020, 88, 102054. [Google Scholar] [CrossRef]
- Huang, X.; Li, E.; Shen, H.; Wang, X.; Tang, T.; Zhang, X.; Xu, J.; Tang, Z.; Guo, C.; Bai, X.; et al. Targeting the HGF/MET Axis in cancer therapy: Challenges in resistance and opportunities for improvement. Front. Cell Dev. Biol. 2020, 8, 152. [Google Scholar] [CrossRef]
- Scheri, K.C.; Leonetti, E.; Laino, L.; Gigantino, V.; Gesualdi, L.; Grammatico, P.; Bizzari, M.; Franco, R.; Oosterhuis, J.W.; Stoop, H.; et al. c-MET receptor as potential biomarker and target molecule for malignant testicular germ cell tumors. Oncotarget 2018, 9, 31842–31860. [Google Scholar] [CrossRef]
- Leonetti, E.; Gesualdi, L.; Scheri, K.C.; Dinicola, S.; Fattore, L.; Masiello, M.G.; Cucina, A.; Mancini, R.; Bizzarri, M.; Ricci, G.; et al. c-Src Recruitment is involved in c-MET-Mediated malignant behaviour of NT2D1 non-seminoma cells. Int. J. Mol. Sci. 2019, 20, 320. [Google Scholar] [CrossRef] [Green Version]
- Jiang, N.; Dai, Q.; Su, X.; Fu, J.; Feng, X.; Peng, J. Role of PI3K/AKT pathway in cancer: The framework of malignant behavior. Mol. Biol. Rep. 2020, 47, 4587–4629. [Google Scholar] [CrossRef]
- Zhang, Y.; Xia, M.; Jin, K.; Wang, S.; Wei, H.; Fan, C.; Wu, Y.; Li, X.; Li, X.; Li, G.; et al. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol. Cancer 2018, 17, 45. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Solal, K.A.; Boregowda, R.K.; Lasfar, A. RUNX2 and the PI3K/AKT axis reciprocal activation as a driving force for tumor progression. Mol. Cancer 2015, 14, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djordjevic, S.; Driscoll, P.C. Structural insight into substrate specificity and regulatory mechanisms of phosphoinositide 3-kinases. Trends Biochem. Sci. 2002, 27, 426–432. [Google Scholar] [CrossRef]
- Foster, F.M.; Traer, C.J.; Abraham, S.M.; Fry, M.J. The phosphoinositide (PI) 3-kinase family. J. Cell Sci. 2003, 116, 3037–3040. [Google Scholar] [CrossRef] [Green Version]
- Guillermet-Guibert, J.; Smith, L.B.; Halet, G.; Whitehead, M.A.; Pearce, W.; Rebourcet, D.; Leon, K.; Crepieux, P.; Nock, G.; Stromstedt, M.; et al. Novel role for p110beta PI 3-Kinase in male fertility through regulation of androgen receptor activity in sertoli cells. PLoS Genet. 2015, 11, e1005304. [Google Scholar] [CrossRef] [Green Version]
- di Vizio, D.; Cito, L.; Boccia, A.; Chieffi, P.; Insabato, L.; Pettinato, G.; Motti, M.L.; Schepis, F.; D’Amico, W.; Fabiani, F.; et al. Loss of the tumor suppressor gene PTEN marks the transition from intratubular germ cell neoplasias (ITGCN) to invasive germ cell tumors. Oncogene 2005, 24, 1882–1894. [Google Scholar] [CrossRef] [Green Version]
- Eder, J.P.; Vande Woude, G.F.; Boerner, S.A.; LoRusso, P.M. Novel therapeutic inhibitors of the c-Met signaling pathway in cancer. Clin. Cancer Res. 2009, 15, 2207–2214. [Google Scholar] [CrossRef] [Green Version]
- Demkova, L.; Kucerova, L. Role of the HGF/c-MET tyrosine kinase inhibitors in metastasic melanoma. Mol. Cancer 2018, 17, 26. [Google Scholar] [CrossRef]
- Qian, J.M.; Martin, A.M.; Martin, K.; Hammoudeh, L.; Catalano, P.J.; Hodi, F.S.; Cagney, D.N.; Haas-Kogan, D.A.; Schoenfeld, J.D.; Aizer, A.A. Response rate and local recurrence after concurrent immune checkpoint therapy and radiotherapy for non-small cell lung cancer and melanoma brain metastases. Cancer 2020. [Google Scholar] [CrossRef]
- Wang, C.; Xi, W.; Ji, J.; Cai, Q.; Zhao, Q.; Jiang, J.; Zhou, C.; Shi, M.; Zhang, H.; Zhu, Z.; et al. The prognostic value of HGF-c-MET signaling pathway in Gastric Cancer: A study based on TCGA and GEO databases. Int. J. Med. Sci. 2020, 17, 1946–1955. [Google Scholar] [CrossRef]
- Zhou, L.; Zhao, Y. B7-H3 induces ovarian cancer drugs resistance through an PI3K/AKT/BCL-2 signaling pathway. Cancer Manag. Res. 2019, 11, 10205–10214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.; Hu, Y.; Fu, Y.; Zhou, T.; You, J.; Du, J.; Zheng, L.; Cao, J.; Ying, M.; Dai, X.; et al. Targeting slug-mediated non-canonical activation of c-Met to overcome chemo-resistance in metastatic ovarian cancer cells. Acta Pharm. Sin. B 2019, 9, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.R.; Wu, J.; Sun, H.; Chi, L.Q.; Wang, J.H. PAK4 confers the malignance of cervical cancers and contributes to the cisplatin-resistance in cervical cancer cells via PI3K/AKT pathway. Diagn. Pathol. 2015, 10, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwak, H.S.; Kim, T.H.; Jo, G.H.; Kim, Y.J.; Kwak, H.J.; Kim, J.H.; Yin, J.; Yoo, H.; Lee, S.H.; Park, J.B. Silencing of microRNA-21 confers radio-sensitivity through inhibition of the PI3K/AKT pathway and enhancing autophagy in malignant glioma cell lines. PLoS ONE 2012, 7, e47449. [Google Scholar] [CrossRef]
- Selfe, J.; Goddard, N.C.; McIntyre, A.; Taylor, K.R.; Renshaw, J.; Popov, S.D.; Thway, K.; Summersgill, B.; Huddart, R.A.; Gilbert, D.C.; et al. IGF1R signalling in testicular germ cell tumour cells impacts on cell survival and acquired cisplatin resistance. J. Pathol. 2018, 244, 242–253. [Google Scholar] [CrossRef]
- Selfe, J.; Shipley, J.M. IGF signalling in germ cells and testicular germ cell tumours: Roles and therapeutic approaches. Andrology 2019, 7, 536–544. [Google Scholar] [CrossRef]
- Metzner, B.; Barbisch, M.; Bachmann, F.; Czech, W.; Norgauer, J. Evidence of the involvement of phosphatidylinositol 3-kinase in the migration, actin stress fiber formation, and alpha v beta 3-integrin-mediated adherence of human melanoma cells. J. Investig. Dermatol. 1996, 107, 597–602. [Google Scholar] [CrossRef] [Green Version]
- Carisey, A.; Ballestrem, C. Vinculin, an adapter protein in control of cell adhesion signalling. Eur. J. Cell Biol. 2011, 90, 157–163. [Google Scholar] [CrossRef]
- Hui, L.; Chen, Y. Tumor microenvironment: Sanctuary of the devil. Cancer Lett. 2015, 368, 7–13. [Google Scholar] [CrossRef]
- Looijenga, L.H.; van Agthoven, T.; Biermann, K. Development of malignant germ cells—The genvironmental hypothesis. Int. J. Dev. Biol. 2013, 57, 241–253. [Google Scholar] [CrossRef]
- Hersmus, R.; van Bever, Y.; Wolffenbuttel, K.P.; Biermann, K.; Cools, M.; Looijenga, L.H. The biology of germ cell tumors in disorders of sex development. Clin. Genet. 2017, 91, 292–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rijlaarsdam, M.A.; Looijenga, L.H. An oncofetal and developmental perspective on testicular germ cell cancer. Semin. Cancer Biol. 2014, 29, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Woldu, S.L.; Amatruda, J.F.; Bagrodia, A. Testicular germ cell tumor genomics. Curr. Opin. Urol. 2017, 27, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Ricci, G.; Catizone, A. Pleiotropic activities of HGF/c-Met system in testicular physiology: Paracrine and endocrine implications. Front. Endocrinol. 2014, 5, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uzumcu, M.; Zachow, R. Developmental exposure to environmental endocrine disruptors: Consequences within the ovary and on female reproductive function. Reprod. Toxicol. 2007, 23, 337–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depuydt, C.E.; de Potter, C.R.; Zalata, A.; Baekelandt, E.; Bosmans, E.; Comhaire, F.H. Levels of hepatocyte growth factor/scatter factor (HGF/SF) in seminal plasma of patients with andrological diseases. J. Androl. 1998, 19, 175–182. [Google Scholar] [PubMed]
- Chovanec, M.; Cierna, Z.; Miskovska, V.; Machalekova, K.; Svetlovska, D.; Kalavska, K.; Rejlekova, K.; Spanik, S.; Kajo, K.; Babal, P.; et al. Prognostic role of programmed-death ligand 1 (PD-L1) expressing tumor infiltrating lymphocytes in testicular germ cell tumors. Oncotarget 2017, 8, 21794–21805. [Google Scholar] [CrossRef] [Green Version]
- Madoz-Gurpide, J.; Zazo, S.; Chamizo, C.; Casado, V.; Carames, C.; Gavin, E.; Cristobal, I.; Garcia-Foncillas, J.; Rojo, F. Activation of MET pathway predicts poor outcome to cetuximab in patients with recurrent or metastatic head and neck cancer. J. Transl. Med. 2015, 13, 282. [Google Scholar] [CrossRef]
- Jankowski, K.; Kucia, M.; Wysoczynski, M.; Reca, R.; Zhao, D.; Trzyna, E.; Trent, J.; Peiper, S.; Zembala, M.; Ratajczak, J.; et al. Both hepatocyte growth factor (HGF) and stromal-derived factor-1 regulate the metastatic behavior of human rhabdomyosarcoma cells, but only HGF enhances their resistance to radiochemotherapy. Cancer Res. 2003, 63, 7926–7935. [Google Scholar]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef]
- Chen, H.T.; Ruan, N.Y.; Chen, J.C.; Lin, T.Y. Dopamine D2 receptor-mediated Akt/PKB signalling: Initiation by the D2S receptor and role in quinpirole-induced behavioural activation. ASN Neuro 2012, 4, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Arnold, L.; Enders, J.; Thomas, S.M. Activated HGF-c-Met axis in head and neck cancer. Cancers 2017, 9, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johanson, S.O.; Naccache, P.A.; Crouch, M.F. A p85 subunit-independent p110alpha PI 3-kinase colocalizes with p70 S6 kinase on actin stress fibers and regulates thrombin-stimulated stress fiber formation in swiss 3T3 cells. Exp. Cell Res. 1999, 248, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.S.; Rose, D.W.; Saltiel, A.R.; Klippel, A.; Williams, L.T.; Olefsky, J.M. Phosphatidylinositol 3-kinase is necessary and sufficient for insulin-stimulated stress fiber breakdown. Endocrinology 1996, 137, 5045–5054. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, C.L.; Cantley, L.C. Phosphoinositide kinases. Curr. Opin. Cell Biol. 1996, 8, 153–158. [Google Scholar] [CrossRef]
- Das, M.K.; Furu, K.; Evensen, H.F.; Haugen, O.P.; Haugen, T.B. Knockdown of SPRY4 and SPRY4-IT1 inhibits cell growth and phosphorylation of Akt in human testicular germ cell tumours. Sci. Rep. 2018, 8, 2462. [Google Scholar] [CrossRef] [Green Version]
- Das, M.K.; Evensen, H.S.F.; Furu, K.; Haugen, T.B. miRNA-302s may act as oncogenes in human testicular germ cell tumours. Sci. Rep. 2019, 9, 9189. [Google Scholar] [CrossRef] [Green Version]
- Feldman, D.R.; Iyer, G.; Van Alstine, L.; Patil, S.; Al-Ahmadie, H.; Reuter, V.E.; Bosl, G.J.; Chaganti, R.S.; Solit, D.B. Presence of somatic mutations within PIK3CA, AKT, RAS, and FGFR3 but not BRAF in cisplatin-resistant germ cell tumors. Clin. Cancer Res. 2014, 20, 3712–3720. [Google Scholar] [CrossRef] [Green Version]
- Juliachs, M.; Munoz, C.; Moutinho, C.A.; Vidal, A.; Condom, E.; Esteller, M.; Graupera, M.; Casanovas, O.; Germa, J.R.; Villanueva, A.; et al. The PDGFRbeta-AKT pathway contributes to CDDP-acquired resistance in testicular germ cell tumors. Clin. Cancer Res. 2014, 20, 658–667. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Jang, H.; Nussinov, R. PI3K inhibitors: Review and new strategies. Chem. Sci. 2020, 11, 5855–5865. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gesualdi, L.; Leonetti, E.; Cucina, A.; Scicchitano, B.M.; Sorrentino, S.; Tarsitano, M.G.; Isidori, A.; Bizzarri, M.; Filippini, A.; Riccioli, A.; et al. The PI3K/AKT Pathway Is Activated by HGF in NT2D1 Non-Seminoma Cells and Has a Role in the Modulation of Their Malignant Behavior. Int. J. Mol. Sci. 2020, 21, 8669. https://doi.org/10.3390/ijms21228669
Gesualdi L, Leonetti E, Cucina A, Scicchitano BM, Sorrentino S, Tarsitano MG, Isidori A, Bizzarri M, Filippini A, Riccioli A, et al. The PI3K/AKT Pathway Is Activated by HGF in NT2D1 Non-Seminoma Cells and Has a Role in the Modulation of Their Malignant Behavior. International Journal of Molecular Sciences. 2020; 21(22):8669. https://doi.org/10.3390/ijms21228669
Chicago/Turabian StyleGesualdi, Luisa, Erica Leonetti, Alessandra Cucina, Bianca Maria Scicchitano, Silvia Sorrentino, Maria Grazia Tarsitano, Andrea Isidori, Mariano Bizzarri, Antonio Filippini, Anna Riccioli, and et al. 2020. "The PI3K/AKT Pathway Is Activated by HGF in NT2D1 Non-Seminoma Cells and Has a Role in the Modulation of Their Malignant Behavior" International Journal of Molecular Sciences 21, no. 22: 8669. https://doi.org/10.3390/ijms21228669
APA StyleGesualdi, L., Leonetti, E., Cucina, A., Scicchitano, B. M., Sorrentino, S., Tarsitano, M. G., Isidori, A., Bizzarri, M., Filippini, A., Riccioli, A., Cammarota, M., Gigantino, V., Ricci, G., & Catizone, A. (2020). The PI3K/AKT Pathway Is Activated by HGF in NT2D1 Non-Seminoma Cells and Has a Role in the Modulation of Their Malignant Behavior. International Journal of Molecular Sciences, 21(22), 8669. https://doi.org/10.3390/ijms21228669