Transthyretin Stabilization: An Emerging Strategy for the Treatment of Alzheimer’s Disease?




Abstract
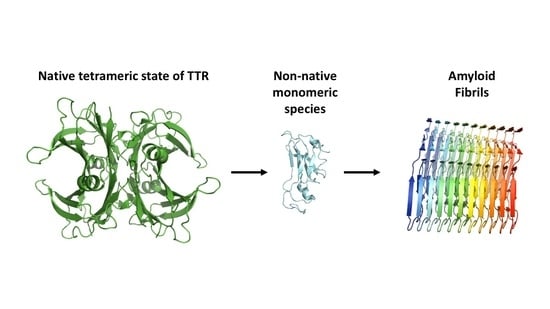
:
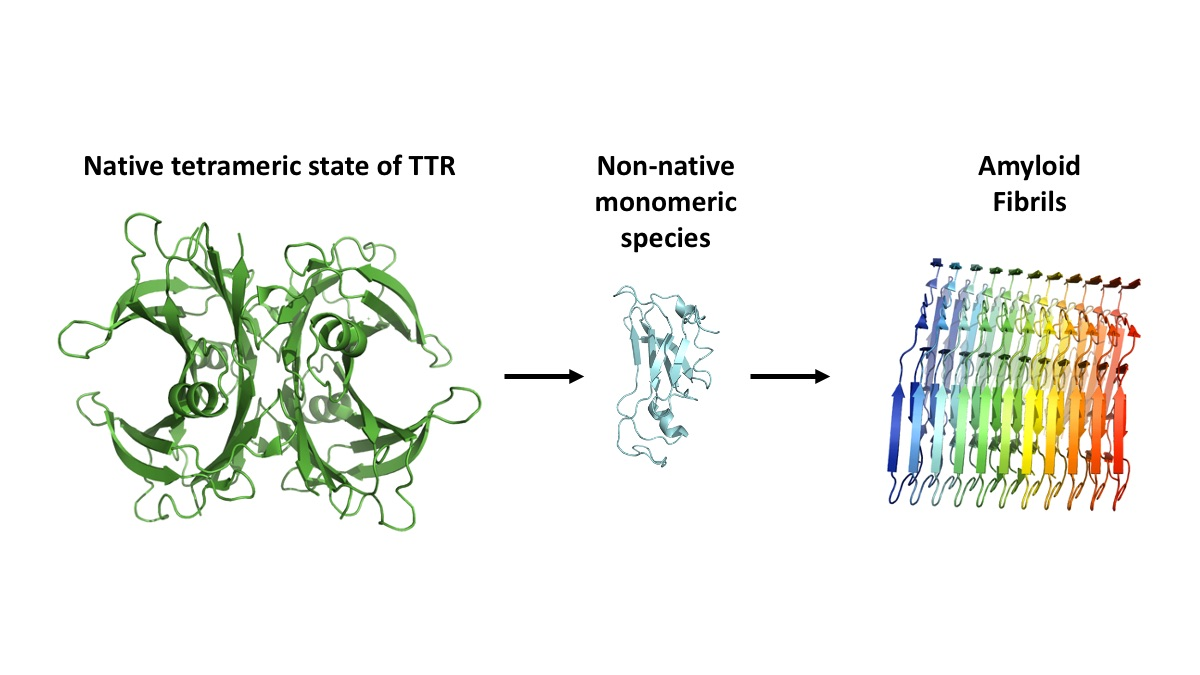{kind=link}
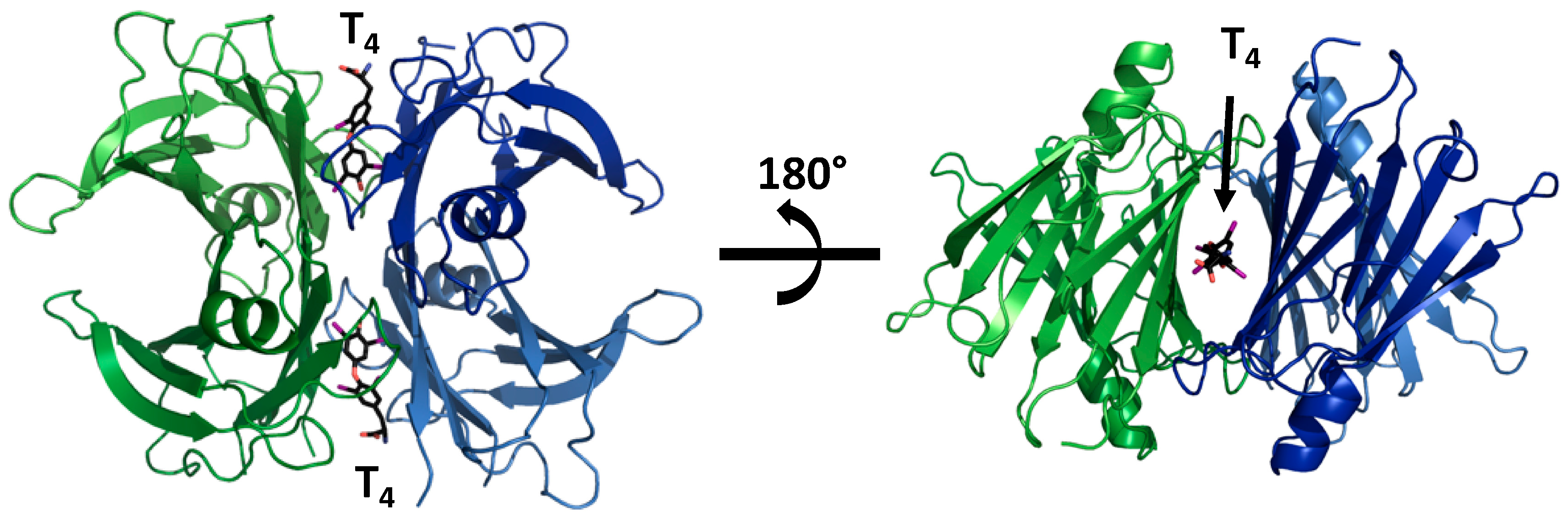{kind=link}
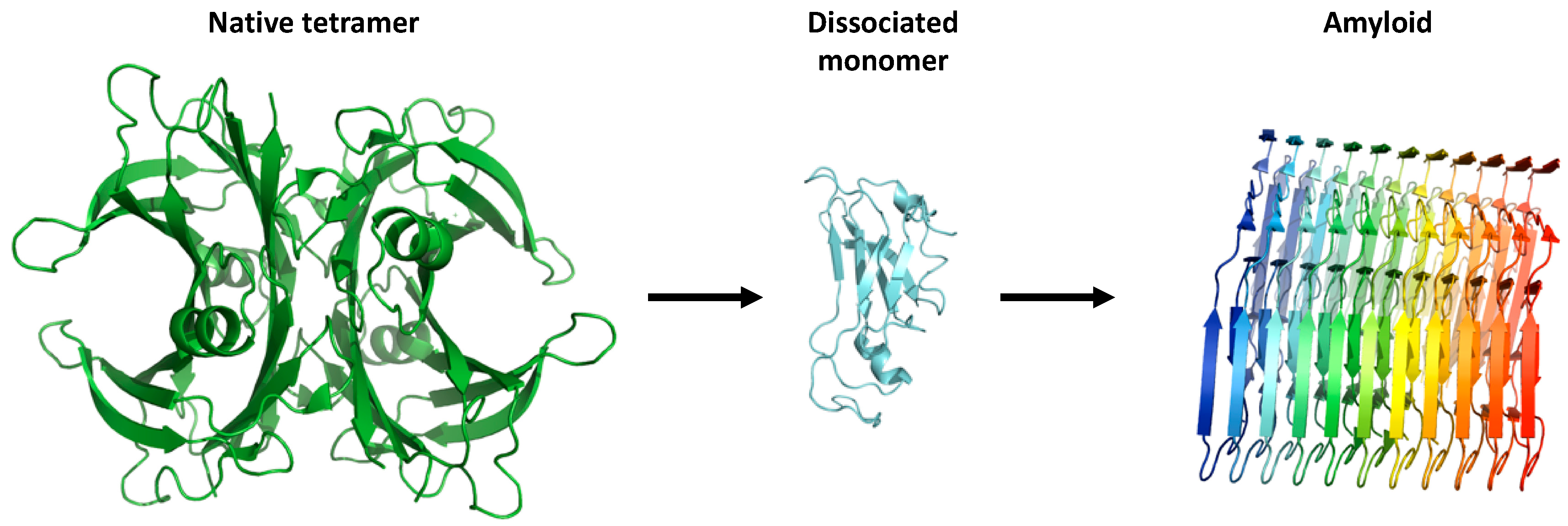{kind=link}
1. Introduction
2. TTR Physiology and Metabolism
3. The role of TTR in the Nervous System
4. The Protective Role of TTR in Alzheimer’s Disease
5. Transthyretin Stability in Alzheimer’s Disease
6. Transthyretin Tetramer Stabilizers
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Soprano, D.R.; Herbert, J.; Soprano, K.J.; Schon, E.A.; Goodman, D.S. Demonstration of transthyretin mRNA in the brain and other extrahepatic tissues in the rat. J. Biol. Chem. 1985, 260, 11793–11798. [Google Scholar] [PubMed]
- Vieira, M.; Saraiva, M.J. Transthyretin: A multifaceted protein. Biomol. Concepts 2014, 5, 45–54. [Google Scholar] [CrossRef] [PubMed]
- INGBAR, S.H. Pre-albumin: A thyroxinebinding protein of human plasma. Endocrinology 1958. [Google Scholar] [CrossRef]
- Raz, A.; Goodman, D.S. The interaction of thyroxine with human plasma prealbumin and with the prealbumin-retinol-binding protein complex. J. Biol. Chem. 1969, 244, 3230–3237. [Google Scholar] [PubMed]
- Nomenclature Committee of the International Union of Biochemistry (NC-IUB). Enzyme Nomenclature. Recommendations 1978. Supplement 1: Corrections and additions. Eur. J. Biochem. 1980. [Google Scholar] [CrossRef]
- Schreiber, G.; Richardson, S.J.; Baldwin, J. The evolution of gene expression, structure and function of transthyretin. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1997, 116, 137–160. [Google Scholar] [CrossRef]
- Power, D.M.; Elias, N.P.; Richardson, S.J.; Mendes, J.; Soares, C.M.; Santos, C.R.A. Evolution of the thyroid hormone-binding protein, transthyretin. Gen. Comp. Endocrinol. 2000, 119, 241–255. [Google Scholar] [CrossRef] [Green Version]
- Eneqvist, T.; Lundberg, E.; Nilsson, L.; Abagyan, R.; Sauer-Eriksson, A.E. The transthyretin-related protein family. Eur. J. Biochem. 2003. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, J.A.; Benson, M.D. Transthyretin: A review from a structural perspective. Cell. Mol. Life Sci. 2001, 58, 1491–1521. [Google Scholar] [CrossRef]
- Wojtczak, A. Crystal structure of rat transthyretin at 2.5 Å resolution: First report on a unique tetrameric structure. Acta Biochim. Pol. 1997. [Google Scholar] [CrossRef]
- Palaninathan, S.K. Nearly 200 X-Ray Crystal Structures of Transthyretin: What Do They Tell Us About This Protein and the Design of Drugs for TTR Amyloidoses? Curr. Med. Chem. 2012, 19, 2324–2342. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Kincaid, J.C. The molecular biology and clinical features of amyloid neuropathy. Muscle Nerve 2007, 36, 411–423. [Google Scholar] [CrossRef]
- Sekijima, Y. Transthyretin (ATTR) amyloidosis: Clinical spectrum, molecular pathogenesis and disease-modifying treatments. J. Neurol. Neurosurg. Psychiatry 2015, 86, 1036–1043. [Google Scholar] [CrossRef] [PubMed]
- Sekijima, Y. Hereditary Transthyretin Amyloidosis Summary Genetic counseling GeneReview Scope Suggestive Findings. In GeneReviews NCBI Bookshelf; Adam, M.P., Ardinger, H.H., Pagon, R.A., Eds.; University of Washington: Seattle, WA, USA, 2001; pp. 1–28. [Google Scholar]
- Cornwell, G.G.; Murdoch, W.L.; Kyle, R.A.; Westermark, P.; Pitkänen, P. Frequency and distribution of senile cardiovascular amyloid. A clinicopathologic correlation. Am. J. Med. 1983. [Google Scholar] [CrossRef]
- Westermark, P.; Sletten, K.; Johansson, B.; Cornwell, G.G. Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc. Natl. Acad. Sci. USA 1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmgren, G.; Steen, L.; Suhr, O.; Ericzon, B.G.; Groth, C.G.; Andersen, O.; Wallin, B.G.; Seymour, A.; Richardson, S.; Hawkins, P.N.; et al. Clinical improvement and amyloid regression after liver transplantation in hereditary transthyretin amyloidosis. Lancet 1993. [Google Scholar] [CrossRef]
- Sekijima, Y. Recent progress in the understanding and treatment of transthyretin amyloidosis. J. Clin. Pharm. Ther. 2014, 39, 225–233. [Google Scholar] [CrossRef]
- Berk, J.L.; Suhr, O.B.; Obici, L.; Sekijima, Y.; Zeldenrust, S.R.; Yamashita, T.; Heneghan, M.A.; Gorevic, P.D.; Litchy, W.J.; Wiesman, J.F.; et al. Repurposing diflunisal for familial amyloid polyneuropathy: A randomized clinical trial. JAMA-J. Am. Med. Assoc. 2013. [Google Scholar] [CrossRef] [Green Version]
- Coelho, T.; Maia, L.F.; Da Silva, A.M.; Cruz, M.W.; Planté-Bordeneuve, V.; Lozeron, P.; Suhr, O.B.; Campistol, J.M.; Conceição, I.M.; Schmidt, H.H.J.; et al. Tafamidis for transthyretin familial amyloid polyneuropathy: A randomized, controlled trial. Neurology 2012. [Google Scholar] [CrossRef]
- Silva, C.S.; Eira, J.; Ribeiro, C.A.; Oliveira, Â.; Sousa, M.M.; Cardoso, I.; Liz, M.A. Transthyretin neuroprotection in Alzheimer’s disease is dependent on proteolysis. Neurobiol. Aging 2017. [Google Scholar] [CrossRef]
- Alemi, M.; Silva, S.C.; Santana, I.; Cardoso, I. Transthyretin stability is critical in assisting beta amyloid clearance–Relevance of transthyretin stabilization in Alzheimer’s disease. CNS Neurosci. Ther. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarzman, A.L.; Gregori, L.; Vitek, M.P.; Lyubski, S.; Strittmatter, W.J.; Enghilde, J.J.; Bhasin, R.; Silverman, J.; Weisgraber, K.H.; Coyle, P.K.; et al. Transthyretin sequesters amyloid β protein and prevents amyloid formation. Proc. Natl. Acad. Sci. USA 1994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarzman, A.L.; Goldgaber, D. Interaction of transthyretin with amyloid β-protein: Binding and inhibition of amyloid formation. CIBA Found. Symp. 1996. [Google Scholar] [CrossRef]
- Buxbaum, J.N.; Ye, Z.; Reixach, N.; Friske, L.; Levy, C.; Das, P.; Golde, T.; Masliah, E.; Roberts, A.R.; Bartfai, T. Transthyretin protects Alzheimer’s mice from the behavioral and biochemical effects of Aβ toxicity. Proc. Natl. Acad. Sci. USA 2008. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, S.M.; Ribeiro, C.A.; Cardoso, I.; Saraiva, M.J. Gender-dependent transthyretin modulation of brain amyloid-β Levels: Evidence from a mouse model of alzheimer’s disease. J. Alzheimers Dis. 2011. [Google Scholar] [CrossRef] [Green Version]
- González-Marrero, I.; Giménez-Llort, L.; Johanson, C.E.; Carmona-Calero, E.M.; Castañeyra-Ruiz, L.; Brito-Armas, J.M.; Castañeyra-Perdomo, A.; Castro-Fuentes, R. Choroid plexus dysfunction impairs beta-amyloid clearance in a triple transgenic mouse model of alzheimer’s disease. Front. Cell. Neurosci. 2015. [Google Scholar] [CrossRef] [Green Version]
- Alemi, M.; Gaiteiro, C.; Ribeiro, C.A.; Santos, L.M.; Gomes, J.R.; Oliveira, S.M.; Couraud, P.O.; Weksler, B.; Romero, I.; Saraiva, M.J.; et al. Transthyretin participates in beta-amyloid transport from the brain to the liver-involvement of the low-density lipoprotein receptor-related protein 1? Sci. Rep. 2016. [Google Scholar] [CrossRef] [Green Version]
- Santos, S.D.; Lambertsen, K.L.; Clausen, B.H.; Akinc, A.; Alvarez, R.; Finsen, B.; Saraiva, M.J. CSF transthyretin neuroprotection in a mouse model of brain ischemia. J. Neurochem. 2010. [Google Scholar] [CrossRef]
- Fleming, C.E.; Saraiva, M.J.; Sousa, M.M. Transthyretin enhances nerve regeneration. J. Neurochem. 2007. [Google Scholar] [CrossRef]
- Sousa, J.C.; Marques, F.; Dias-Ferreira, E.; Cerqueira, J.J.; Sousa, N.; Palha, J.A. Transthyretin influences spatial reference memory. Neurobiol. Learn. Mem. 2007. [Google Scholar] [CrossRef]
- Li, X.; Masliah, E.; Reixach, N.; Buxbaum, J.N. Neuronal production of transthyretin in human and murine alzheimer’s disease: Is it protective? J. Neurosci. 2011. [Google Scholar] [CrossRef] [Green Version]
- Stabilini, R.; Vergani, C.; Agostoni, A.; Agostoni, R.P.V. Influence of age and sex on prealbumin levels. Clin. Chim. Acta 1968. [Google Scholar] [CrossRef]
- Vahlquist, A.; Rask, L.; Peterson, P.A.; Berg, T. The concentrations of retinol-binding protein, prealbumin, and transferrin in the sera of newly delivered mothers and children of various ages. Scand. J. Clin. Lab. Invest. 1975. [Google Scholar] [CrossRef]
- Schreiber, G.; Aldred, A.R.; Jaworowski, A.; Nilsson, C.; Achen, M.G.; Segal, M.B. Thyroxine transport from blood to brain via transthyretin synthesis in choroid plexus. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 1990. [Google Scholar] [CrossRef]
- Pfeffer, B.A.; Becerra, S.P.; Borst, D.E.; Won, P. Expression of transthyretin and retinol binding protein mRNAs and secretion of transthyretin by cultured monkey retinal pigment epithelium. Mol. Vis. 2004, 10, 23–30. [Google Scholar] [PubMed]
- Kato, M.; Kato, K.; Blaner, W.S.; Chertow, B.S.; Goodman, D.S. Plasma and cellular retinoid-binding proteins and transthyretin (prealbumin) are all localized in the islets of Langerhans in the rat. Proc. Natl. Acad. Sci. USA 1985. [Google Scholar] [CrossRef] [Green Version]
- Jacobsson, B.; Collins, V.P.; Grimelius, L.; Pettersson, T.; Sandstedt, B.; Carlstrom, A. Transthyretin immunoreactivity in human and porcine liver, choroid plexus, and pancreatic islets. J. Histochem. Cytochem. 1989. [Google Scholar] [CrossRef]
- Hagen, G.A.; Solberg, L.A. Brain and cerebrospinal fluid permeability to intravenous thyroid hormones. Endocrinology 1974. [Google Scholar] [CrossRef]
- Palha, J.A. Transthyretin as a thyroid hormone carrier: Function revisited. Clin. Chem. Lab. Med. 2002. [Google Scholar] [CrossRef]
- Blake, C.C.F.; Geisow, M.J.; Swan, I.D.A.; Rerat, C.; Rerat, B. Structure of human plasma prealbumin at 2.5 A resolution. A preliminary report on the polypeptide chain conformation, quaternary structure and thyroxine binding. J. Mol. Biol. 1974. [Google Scholar] [CrossRef]
- Tomar, D.; Khan, T.; Singh, R.R.; Mishra, S.; Gupta, S.; Surolia, A.; Salunke, D.M. Crystallographic Study of Novel Transthyretin Ligands Exhibiting Negative-Cooperativity between Two Thyroxine Binding Sites. PLoS ONE 2012. [Google Scholar] [CrossRef] [Green Version]
- Kanai, M.; Raz, A.; Goodman, D.S. Retinol-binding protein: The transport protein for vitamin A in human plasma. J. Clin. Invest. 1968. [Google Scholar] [CrossRef] [Green Version]
- Epstein, F.H.; Goodman, D.S. Vitamin A and Retinoids in Health and Disease. N. Engl. J. Med. 1984, 310, 1023–1031. [Google Scholar] [CrossRef]
- Noy, N.; Slosberg, E.; Scarlatal, S. Interactions of Retinol with Binding Proteins: Studies with Retinol-Binding Protein and with Transthyretin. Biochemistry 1992. [Google Scholar] [CrossRef]
- Monaco, H.L.; Rizzi, M.; Coda, A. Structure of a complex of two plasma proteins: Transthyretin and retinol-binding protein. Science 1995. [Google Scholar] [CrossRef]
- Van Bennekum, A.M.; Wei, S.; Gamble, M.V.; Vogel, S.; Piantedosi, R.; Gottesman, M.; Episkopoui, V.; Blaner, W.S. Biochemical basis for depressed serum retinol levels in transthyretin-deficient mice. J. Biol. Chem. 2001. [Google Scholar] [CrossRef] [Green Version]
- Liz, M.A.; Faro, C.J.; Saraiva, M.J.; Sousa, M.M. Transthyretin, a new cryptic protease. J. Biol. Chem. 2004. [Google Scholar] [CrossRef] [Green Version]
- Liz, M.A.; Gomes, C.M.; Saraiva, M.J.; Sousa, M.M. ApoA-I cleaved by transthyretin has reduced ability to promote cholesterol efflux and increased amyloidogenicity. J. Lipid Res. 2007. [Google Scholar] [CrossRef] [Green Version]
- Sousa, M.M.; Berglund, L.; Saraiva, M.J. Transthyretin in high density lipoproteins: Association with apolipoprotein A-I. J. Lipid Res. 2000, 41, 58–65. [Google Scholar]
- Liz, M.A.; Fleming, C.E.; Nunes, A.F.; Almeida, M.R.; Mar, F.M.; Choe, Y.; Craik, C.S.; Powers, J.C.; Bogyo, M.; Sousa, M.M. Substrate specificity of transthyretin: Identification of natural substrates in the nervous system. Biochem. J. 2009. [Google Scholar] [CrossRef] [Green Version]
- Costa, R.; Ferreira-da-Silva, F.; Saraiva, M.J.; Cardoso, I. Transthyretin protects against A-beta peptide toxicity by proteolytic cleavage of the peptide: A mechanism sensitive to the kunitz protease inhibitor. PLoS ONE 2008. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, S.M.; Cardoso, I.; Saraiva, M.J. Transthyretin: Roles in the nervous system beyond thyroxine and retinol transport. Expert Rev. Endocrinol. Metab. 2012, 7, 181–189. [Google Scholar] [CrossRef]
- Brouillette, J.; Quirion, R. Transthyretin: A key gene involved in the maintenance of memory capacities during aging. Neurobiol. Aging 2008. [Google Scholar] [CrossRef]
- Carlos Sousa, J.; Grandela, C.; Fernández-Ruiz, J.; De Miguel, R.; De Sousa, L.; Magalhães, A.I.; João Saraiva, M.; Sousa, N.; Palha, J.A. Transthyretin is involved in depression-like behaviour and exploratory activity. J. Neurochem. 2004. [Google Scholar] [CrossRef] [Green Version]
- Fleming, C.E.; Mar, F.M.; Franquinho, F.; Saraiva, M.J.; Sousa, M.M. Transthyretin internalization by sensory neurons is megalin mediated and necessary for its neuritogenic activity. J. Neurosci. 2009. [Google Scholar] [CrossRef]
- Chen, R.; Vendrell, I.; Chen, C.P.; Cash, D.; O’Toole, K.G.; Williams, S.A.; Jones, C.; Preston, J.E.; Wheeler, J.X. Proteomic analysis of rat plasma following transient focal cerebral ischemia. Biomark. Med. 2011. [Google Scholar] [CrossRef]
- Suzuyama, K.; Shiraishi, T.; Oishi, T.; Ueda, S.; Okamoto, H.; Furuta, M.; Mineta, T.; Tabuchi, K. Combined proteomic approach with SELDI-TOF-MS and peptide mass fingerprinting identified the rapid increase of monomeric transthyretin in rat cerebrospinal fluid after transient focal cerebral ischemia. Mol. Brain Res. 2004. [Google Scholar] [CrossRef]
- Liverman, C.S.; Cui, L.; Yong, C.; Choudhuri, R.; Klein, R.M.; Welch, K.M.A.; Berman, N.E.J. Response of the brain to oligemia: Gene expression, c-Fos, and Nrf2 localization. Mol. Brain Res. 2004. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G. A century of Alzheimer’s disease. Science 2006, 314, 777–781. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Funato, H.; Enya, M.; Yoshimura, M.; Morishima-Kawashima, M.; Ihara, Y. Presence of sodium dodecyl sulfate-stable amyloid β-protein dimers in the hippocampus CA1 not exhibiting neurofibrillary tangle formation. Am. J. Pathol. 1999. [Google Scholar] [CrossRef]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.S.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N. Engl. J. Med. 2012. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Forman, M.S.; Trojanowski, J.Q.; Lee, V.M.Y. Neurodegenerative diseases: A decade of discoveries paves the way for therapeutic breakthroughs. Nat. Med. 2004, 10, 1055–1063. [Google Scholar] [CrossRef]
- Elovaara, I.; Maury, C.P.J.; Palo, J. Serum amysoid A protein, albumin and prealbumin in Alzheimer’s disease and in demented patients with Down’s syndrome. Acta Neurol. Scand. 1986. [Google Scholar] [CrossRef]
- Serot, J.M.; Christmann, D.; Dubost, T.; Couturier, M. Cerebrospinal fluid transthyretin: Aging and late onset Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 1997. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, X.; Ladiwala, A.R.A.; Du, D.; Yadav, J.K.; Tessier, P.M.; Wright, P.E.; Kelly, J.W.; Buxbaum, J.N. Mechanisms of transthyretin inhibition of β-amyloid aggregation in vitro. J. Neurosci. 2013. [Google Scholar] [CrossRef]
- Liu, L.; Murphy, R.M. Kinetics of inhibition of β-amyloid aggregation by transthyretin. Biochemistry 2006. [Google Scholar] [CrossRef]
- Gloeckner, S.F.; Meyne, F.; Wagner, F.; Heinemann, U.; Krasnianski, A.; Meissner, B.; Zerr, I. Quantitative analysis of transthyretin, tau and amyloid-β in patients with dementia. J. Alzheimers Dis. 2008. [Google Scholar] [CrossRef]
- Han, S.H.; Jung, E.S.; Sohn, J.H.; Hong, H.J.; Hong, H.S.; Kim, J.W.; Na, D.L.; Kim, M.; Kim, H.; Ha, H.J.; et al. Human serum transthyretin levels correlate inversely with Alzheimer’s disease. J. Alzheimers Dis. 2011. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, C.A.; Saraiva, M.J.; Cardoso, I. Stability of the Transthyretin Molecule as a Key Factor in the Interaction with A-Beta Peptide-Relevance in Alzheimer’s Disease. PLoS ONE 2012. [Google Scholar] [CrossRef] [Green Version]
- Velayudhan, L.; Killick, R.; Hye, A.; Kinsey, A.; Güntert, A.; Lynham, S.; Ward, M.; Leung, R.; Lourdusamy, A.; To, A.W.M.; et al. Plasma transthyretin as a candidate marker for Alzheimer’s disease. J. Alzheimers Dis. 2012. [Google Scholar] [CrossRef] [Green Version]
- Tien, Y.T.; Lee, W.J.; Liao, Y.C.; Wang, W.F.; Jhang, K.M.; Wang, S.J.; Fuh, J.L. Plasma Transthyretin as a Predictor of Amnestic Mild Cognitive Impairment Conversion to Dementia. Sci. Rep. 2019, 9, 1–7. [Google Scholar] [CrossRef]
- Kim, H.S.; Choi, Y.; Shin, K.Y.; Joo, Y.; Lee, Y.K.; Jung, S.Y.; Suh, Y.H.; Kim, J.H. Swedish amyloid precursor protein mutation increases phosphorylation of eIF2α in vitro and in vivo. J. Neurosci. Res. 2007. [Google Scholar] [CrossRef] [PubMed]
- Tagoe, C.E.; Reixach, N.; Friske, L.; Mustra, D.; French, D.; Gallo, G.; Buxbaum, J.N. In vivo stabilization of mutant human transthyretin in transgenic mice. Amyloid 2007. [Google Scholar] [CrossRef] [PubMed]
- Link, C.D. Expression of human β-amyloid peptide in transgenic Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 1995. [Google Scholar] [CrossRef] [Green Version]
- Se, H.C.; Leight, S.N.; Lee, V.M.Y.; Li, T.; Wong, P.C.; Johnson, J.A.; Saraiva, M.J.; Sisodia, S.S. Accelerated Aβ deposition in APPswe/PS1ΔE9 mice with hemizygous deletions of TTR (transthyretin). J. Neurosci. 2007. [Google Scholar] [CrossRef] [Green Version]
- Ghadami, S.A.; Chia, S.; Ruggeri, F.S.; Meisl, G.; Bemporad, F.; Habchi, J.; Cascella, R.; Dobson, C.M.; Vendruscolo, M.; Knowles, T.P.J.; et al. Transthyretin Inhibits Primary and Secondary Nucleations of Amyloid-β Peptide Aggregation and Reduces the Toxicity of Its Oligomers. Biomacromolecules 2020. [Google Scholar] [CrossRef]
- A. Ribeiro, C.; Santana, I.; Oliveira, C.; Baldeiras, I.; Moreira, J.; Joao Saraiva, M.; Cardoso, I. Transthyretin Decrease in Plasma of MCI and AD Patients: Investigation of Mechanisms for Disease Modulation. Curr. Alzheimer Res. 2012. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Murphy, R.M. Characterization of the interaction of β-Amyloid with Transthyretin monomers and tetramers. Biochemistry 2010. [Google Scholar] [CrossRef] [Green Version]
- Costa, R.; Gonçalves, A.; Saraiva, M.J.; Cardoso, I. Transthyretin binding to A-Beta peptide-Impact on A-Beta fibrillogenesis and toxicity. FEBS Lett. 2008. [Google Scholar] [CrossRef] [Green Version]
- McCutchen, S.L.; Colon, W.; Kelly, J.W. Transthyretin Mutation Leu-55-Pro Significantly Alters Tetramer Stability and Increases Amyloidogenicity. Biochemistry 1993. [Google Scholar] [CrossRef]
- Xiang, Q.; Bi, R.; Xu, M.; Zhang, D.F.; Tan, L.; Zhang, C.; Fang, Y.; Yao, Y.G. Rare Genetic Variants of the Transthyretin Gene Are Associated with Alzheimer’s Disease in Han Chinese. Mol. Neurobiol. 2017. [Google Scholar] [CrossRef]
- Sassi, C.; Ridge, P.G.; Nalls, M.A.; Gibbs, R.; Ding, J.; Lupton, M.K.; Troakes, C.; Lunnon, K.; Al-Sarraj, S.; Brown, K.S.; et al. Influence of coding variability in APP-Aβ metabolism genes in sporadic Alzheimer’s disease. PLoS ONE 2016. [Google Scholar] [CrossRef] [Green Version]
- Hurshman, A.R.; White, J.T.; Powers, E.T.; Kelly, J.W. Transthyretin aggregation under partially denaturing conditions is a downhill polymerization. Biochemistry 2004. [Google Scholar] [CrossRef] [PubMed]
- Quintela, T.; Gonçalves, I.; Baltazar, G.; Alves, C.H.; Saraiva, M.J.; Santos, C.R.A. 17β-estradiol induces transthyretin expression in murine choroid plexus via an oestrogen receptor dependent pathway. Cell. Mol. Neurobiol. 2009. [Google Scholar] [CrossRef] [PubMed]
- Candore, G.; Balistreri, C.R.; Grimaldi, M.P.; Vasto, S.; Listì, F.; Chiappelli, M.; Licastro, F.; Lio, D.; Caruso, C. Age-related inflammatory diseases: Role of genetics and gender in the pathophysiology of Alzheimer’s disease. Proc. Ann. N. Y. Acad. Sci. 2006, 10889, 472–486. [Google Scholar] [CrossRef] [PubMed]
- Munro, S.L.; Lim, C.F.; Hall, J.G.; Barlow, J.W.; Craik, D.J.; Topliss, D.J.; Stockigt, J.R. Drug competition for thyroxine binding to transthyretin (prealbumin): Comparison with effects on thyroxine-binding globulin. J. Clin. Endocrinol. Metab. 1989. [Google Scholar] [CrossRef]
- Adamski-Werner, S.L.; Palaninathan, S.K.; Sacchettini, J.C.; Kelly, J.W. Diflunisal Analogues Stabilize the Native State of Transthyretin. Potent Inhibition of Amyloidogenesis. J. Med. Chem. 2004. [Google Scholar] [CrossRef]
- Miller, S.R.; Sekijima, Y.; Kelly, J.W. Native state stabilization by NSAIDs inhibits transthyretin amyloidogenesis from the most common familial disease variants. Lab. Investig. 2004. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, C.A.; Oliveira, S.M.; Guido, L.F.; Magalhães, A.; Valencia, G.; Arsequell, G.; Saraiva, M.J.; Cardoso, I. Transthyretin stabilization by iododiflunisal promotes amyloid-β peptide clearance, decreases its deposition, and ameliorates cognitive deficits in an Alzheimer’s disease mouse model. J. Alzheimers Dis. 2014. [Google Scholar] [CrossRef] [PubMed]
- Rios, X.; Gómez-Vallejo, V.; Martín, A.; Cossío, U.; Morcillo, M.Á.; Alemi, M.; Cardoso, I.; Quintana, J.; Jiménez-Barbero, J.; Cotrina, E.Y.; et al. Radiochemical examination of transthyretin (TTR) brain penetration assisted by iododiflunisal, a TTR tetramer stabilizer and a new candidate drug for AD. Sci. Rep. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer, M.S.; Schwartz, J.H.; Gundapaneni, B.; Elliott, P.M.; Merlini, G.; Waddington-Cruz, M.; Kristen, A.V.; Grogan, M.; Witteles, R.; Damy, T.; et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N. Engl. J. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Waddington-Cruz, M.; Berk, J.L.; Polydefkis, M.; Dyck, P.J.; Wang, A.K.; Planté-Bordeneuve, V.; Barroso, F.A.; Merlini, G.; Obici, L.; et al. Inotersen treatment for patients with Hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Coelho, T.; Adams, D.; Silva, A.; Lozeron, P.; Hawkins, P.N.; Mant, T.; Perez, J.; Chiesa, J.; Warrington, S.; Tranter, E.; et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N. Engl. J. Med. 2013. [Google Scholar] [CrossRef]
- Butler, J.S.; Chan, A.; Costelha, S.; Fishman, S.; Willoughby, J.L.S.; Borland, T.D.; Milstein, S.; Foster, D.J.; Gonçalves, P.; Chen, Q.; et al. Preclinical evaluation of RNAi as a treatment for transthyretin-mediated amyloidosis. Amyloid 2016. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Goel, V.; Robbie, G.J. Pharmacokinetics of Patisiran, the First Approved RNA Interference Therapy in Patients With Hereditary Transthyretin-Mediated Amyloidosis. J. Clin. Pharmacol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Jayaraman, M.; Ansell, S.M.; Mui, B.L.; Tam, Y.K.; Chen, J.; Du, X.; Butler, D.; Eltepu, L.; Matsuda, S.; Narayanannair, J.K.; et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chemie-Int. Ed. 2012. [Google Scholar] [CrossRef]
- Garber, K. Alnylam terminates revusiran program, stock plunges. Nat. Biotechnol. 2016. [Google Scholar] [CrossRef]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018. [Google Scholar] [CrossRef]
- Wood, H. FDA approves patisiran to treat hereditary transthyretin amyloidosis. Nat. Rev. Neurol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kristen, A.V.; Ajroud-Driss, S.; Conceição, I.; Gorevic, P.; Kyriakides, T.; Obici, L. Patisiran, an RNAi therapeutic for the treatment of hereditary transthyretin-mediated amyloidosis. Neurodegener. Dis. Manag. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciccone, L.; Tonali, N.; Nencetti, S.; Orlandini, E. Natural compounds as inhibitors of transthyretin amyloidosis and neuroprotective agents: Analysis of structural data for future drug design. J. Enzyme Inhib. Med. Chem. 2020, 335, 1145–1162. [Google Scholar] [CrossRef] [PubMed]
- Marambaud, P.; Zhao, H.; Davies, P. Resveratrol promotes clearance of Alzheimer’s disease amyloid-β peptides. J. Biol. Chem. 2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngoungoure, V.L.N.; Schluesener, J.; Moundipa, P.F.; Schluesener, H. Natural polyphenols binding to amyloid: A broad class of compounds to treat different human amyloid diseases. Mol. Nutr. Food Res. 2015, 59, 8–20. [Google Scholar] [CrossRef]
- Ortore, G.; Orlandini, E.; Braca, A.; Ciccone, L.; Rossello, A.; Martinelli, A.; Nencetti, S. Targeting Different Transthyretin Binding Sites with Unusual Natural Compounds. ChemMedChem 2016. [Google Scholar] [CrossRef] [Green Version]
- Kristen, A.V.; Lehrke, S.; Buss, S.; Mereles, D.; Steen, H.; Ehlermann, P.; Hardt, S.; Giannitsis, E.; Schreiner, R.; Haberkorn, U.; et al. Green tea halts progression of cardiac transthyretin amyloidosis: An observational report. Clin. Res. Cardiol. 2012. [Google Scholar] [CrossRef] [Green Version]
- Aus dem Siepen, F.; Bauer, R.; Aurich, M.; Buss, S.J.; Steen, H.; Altland, K.; Katus, H.A.; Kristen, A.V. Green tea extract as a treatment for patients with wild-type transthyretin amyloidosis: An observational study. Drug Des. Devel. Ther. 2015. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, N.; Gonçalves, N.P.; Saraiva, M.J.; Almeida, M.R. Curcumin: A multi-Target disease-modifying agent for late-stage transthyretin amyloidosis. Sci. Rep. 2016. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saponaro, F.; Kim, J.H.; Chiellini, G. Transthyretin Stabilization: An Emerging Strategy for the Treatment of Alzheimer’s Disease? Int. J. Mol. Sci. 2020, 21, 8672. https://doi.org/10.3390/ijms21228672
Saponaro F, Kim JH, Chiellini G. Transthyretin Stabilization: An Emerging Strategy for the Treatment of Alzheimer’s Disease? International Journal of Molecular Sciences. 2020; 21(22):8672. https://doi.org/10.3390/ijms21228672
Chicago/Turabian StyleSaponaro, Federica, Jin Hae Kim, and Grazia Chiellini. 2020. "Transthyretin Stabilization: An Emerging Strategy for the Treatment of Alzheimer’s Disease?" International Journal of Molecular Sciences 21, no. 22: 8672. https://doi.org/10.3390/ijms21228672
APA StyleSaponaro, F., Kim, J. H., & Chiellini, G. (2020). Transthyretin Stabilization: An Emerging Strategy for the Treatment of Alzheimer’s Disease? International Journal of Molecular Sciences, 21(22), 8672. https://doi.org/10.3390/ijms21228672