PlGF Immunological Impact during Pregnancy

 ,
,  ,
,  ,
,  , ,
, , 
Abstract
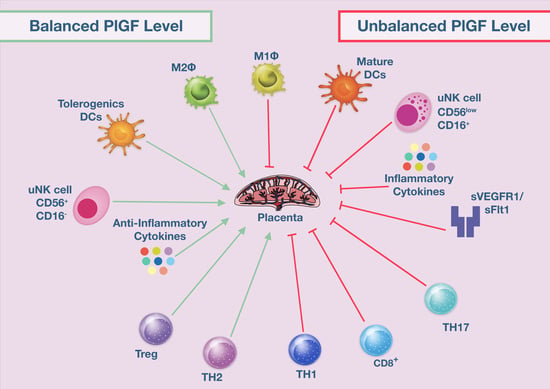
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. The Immunological Paradox of Human Pregnancy
1.2. Placental Growth Factor (PlGF)
2. Pregnancy and Inflammation
2.1. Interplay between Immune Cells and PlGF during Pregnancy
2.2. Natural Killer (NK) Cells
2.3. Macrophages
2.4. Dendritic Cells
3. Bridging PlGF and Hypertension
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Footnote
References
- Billingham, R.E.; Brent, L.; Medawar, P.B. Actively acquired tolerance of foreign cells. Nature 1953, 172, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Erlebacher, A. Immunology of the maternal-fetal interface. Annu. Rev. Immunol. 2013, 31, 387–411. [Google Scholar] [CrossRef] [PubMed]
- Mor, G.; Aldo, P.; Alvero, A.B. The unique immunological and microbial aspects of pregnancy. Nat. Rev. Immunol. 2017, 17, 469–482. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, A.; Sharkey, D.J.; Robertson, S.A.; Zenclussen, A.C. Immune Cells at the Fetomaternal Interface: How the Microenvironment Modulates Immune Cells To Foster Fetal Development. J. Immunol. 2018, 201, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Aluvihare, V.R.; Kallikourdis, M.; Betz, A.G. Regulatory T cells mediate maternal tolerance to the fetus. Nat. Immunol. 2004, 5, 266–271. [Google Scholar] [CrossRef]
- Bonney, E.A. Alternative theories: Pregnancy and immune tolerance. J. Reprod. Immunol. 2017, 123, 65–71. [Google Scholar] [CrossRef]
- Colucci, F. The immunological code of pregnancy. Science 2019, 365, 862–863. [Google Scholar] [CrossRef]
- Svensson-Arvelund, J.; Mehta, R.B.; Lindau, R.; Mirrasekhian, E.; Rodriguez-Martinez, H.; Berg, G.; Lash, G.E.; Jenmalm, M.C.; Ernerudh, J. The human fetal placenta promotes tolerance against the semiallogeneic fetus by inducing regulatory T cells and homeostatic M2 macrophages. J. Immunol. 2015, 194, 1534–1544. [Google Scholar] [CrossRef] [Green Version]
- Trowsdale, J.; Betz, A.G. Mother’s little helpers: Mechanisms of maternal-fetal tolerance. Nat. Immunol. 2006, 7, 241–246. [Google Scholar] [CrossRef]
- Valencia-Ortega, J.; Saucedo, R.; Pena-Cano, M.I.; Hernandez-Valencia, M.; Cruz-Duran, J.G. Immune tolerance at the maternal-placental interface in healthy pregnancy and pre-eclampsia. J. Obstet. Gynaecol. Res. 2020, 46, 1067–1076. [Google Scholar] [CrossRef]
- Brien, M.E.; Boufaied, I.; Bernard, N.; Forest, J.C.; Giguere, Y.; Girard, S. Specific inflammatory profile in each pregnancy complication: A comparative study. Am. J. Reprod. Immunol. 2020, e13316. [Google Scholar] [CrossRef]
- Griffith, O.W.; Chavan, A.R.; Protopapas, S.; Maziarz, J.; Romero, R.; Wagner, G.P. Embryo implantation evolved from an ancestral inflammatory attachment reaction. Proc. Natl. Acad. Sci. USA 2017, 114, E6566–E6575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadeau-Vallee, M.; Obari, D.; Palacios, J.; Brien, M.E.; Duval, C.; Chemtob, S.; Girard, S. Sterile inflammation and pregnancy complications: A review. Reproduction 2016, 152, R277–R292. [Google Scholar] [CrossRef] [PubMed]
- Nejabati, H.R.; Latifi, Z.; Ghasemnejad, T.; Fattahi, A.; Nouri, M. Placental growth factor (PlGF) as an angiogenic/inflammatory switcher: Lesson from early pregnancy losses. Gynecol. Endocrinol. 2017, 33, 668–674. [Google Scholar] [CrossRef]
- Zenclussen, A.C.; Hammerling, G.J. Cellular Regulation of the Uterine Microenvironment That Enables Embryo Implantation. Front. Immunol. 2015, 6, 321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Highet, A.R.; Khoda, S.M.; Buckberry, S.; Leemaqz, S.; Bianco-Miotto, T.; Harrington, E.; Ricciardelli, C.; Roberts, C.T. Hypoxia induced HIF-1/HIF-2 activity alters trophoblast transcriptional regulation and promotes invasion. Eur. J. Cell Biol. 2015, 94, 589–602. [Google Scholar] [CrossRef] [PubMed]
- Huppertz, B. Traditional and New Routes of Trophoblast Invasion and Their Implications for Pregnancy Diseases. Int. J. Mol. Sci. 2019, 21, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imakawa, K.; Bai, R.; Fujiwara, H.; Ideta, A.; Aoyagi, Y.; Kusama, K. Continuous model of conceptus implantation to the maternal endometrium. J. Endocrinol. 2017, 233, R53–R65. [Google Scholar] [CrossRef] [Green Version]
- Burton, G.J.; Jauniaux, E.; Murray, A.J. Oxygen and placental development; parallels and differences with tumour biology. Placenta 2017, 56, 14–18. [Google Scholar] [CrossRef]
- DaSilva-Arnold, S.; James, J.L.; Al-Khan, A.; Zamudio, S.; Illsley, N.P. Differentiation of first trimester cytotrophoblast to extravillous trophoblast involves an epithelial-mesenchymal transition. Placenta 2015, 36, 1412–1418. [Google Scholar] [CrossRef]
- Holtan, S.G.; Creedon, D.J.; Haluska, P.; Markovic, S.N. Cancer and pregnancy: Parallels in growth, invasion, and immune modulation and implications for cancer therapeutic agents. Mayo Clin. Proc. 2009, 84, 985–1000. [Google Scholar] [CrossRef] [Green Version]
- Kareva, I. Immune Suppression in Pregnancy and Cancer: Parallels and Insights. Transl. Oncol. 2020, 13, 100759. [Google Scholar] [CrossRef] [PubMed]
- Albonici, L.; Giganti, M.G.; Modesti, A.; Manzari, V.; Bei, R. Multifaceted Role of the Placental Growth Factor (PlGF) in the Antitumor Immune Response and Cancer Progression. Int. J. Mol. Sci. 2019, 20, 2970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costanzo, V.; Bardelli, A.; Siena, S.; Abrignani, S. Exploring the links between cancer and placenta development. Open Biol. 2018, 8, 180081. [Google Scholar] [CrossRef] [Green Version]
- Ferroni, P.; Palmirotta, R.; Spila, A.; Martini, F.; Formica, V.; Portarena, I.; Del Monte, G.; Buonomo, O.; Roselli, M.; Guadagni, F. Prognostic value of carcinoembryonic antigen and vascular endothelial growth factor tumor tissue content in colorectal cancer. Oncology 2006, 71, 176–184. [Google Scholar] [CrossRef] [Green Version]
- PrabhuDas, M.; Bonney, E.; Caron, K.; Dey, S.; Erlebacher, A.; Fazleabas, A.; Fisher, S.; Golos, T.; Matzuk, M.; McCune, J.M.; et al. Immune mechanisms at the maternal-fetal interface: Perspectives and challenges. Nat. Immunol. 2015, 16, 328–334. [Google Scholar] [CrossRef]
- Achen, M.G.; Gad, J.M.; Stacker, S.A.; Wilks, A.F. Placenta growth factor and vascular endothelial growth factor are co-expressed during early embryonic development. Growth Factors 1997, 15, 69–80. [Google Scholar] [CrossRef]
- Athanassiades, A.; Lala, P.K. Role of placenta growth factor (PIGF) in human extravillous trophoblast proliferation, migration and invasiveness. Placenta 1998, 19, 465–473. [Google Scholar] [CrossRef]
- Binder, N.K.; Evans, J.; Salamonsen, L.A.; Gardner, D.K.; Kaitu’u-Lino, T.J.; Hannan, N.J. Placental Growth Factor Is Secreted by the Human Endometrium and Has Potential Important Functions during Embryo Development and Implantation. PLoS ONE 2016, 11, e0163096. [Google Scholar] [CrossRef] [Green Version]
- Holme, A.M.; Roland, M.C.; Henriksen, T.; Michelsen, T.M. In vivo uteroplacental release of placental growth factor and soluble Fms-like tyrosine kinase-1 in normal and preeclamptic pregnancies. Am. J. Obstet. Gynecol. 2016, 215, 782e1–782e9. [Google Scholar] [CrossRef]
- Knuth, A.; Liu, L.; Nielsen, H.; Merril, D.; Torry, D.S.; Arroyo, J.A. Placenta growth factor induces invasion and activates p70 during rapamycin treatment in trophoblast cells. Am. J. Reprod. Immunol. 2015, 73, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Vrachnis, N.; Kalampokas, E.; Sifakis, S.; Vitoratos, N.; Kalampokas, T.; Botsis, D.; Iliodromiti, Z. Placental growth factor (PlGF): A key to optimizing fetal growth. J. Matern. Fetal Neonatal Med. 2013, 26, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Adini, A.; Kornaga, T.; Firoozbakht, F.; Benjamin, L.E. Placental growth factor is a survival factor for tumor endothelial cells and macrophages. Cancer Res. 2002, 62, 2749–2752. [Google Scholar] [PubMed]
- Ding, Y.; Huang, Y.; Song, N.; Gao, X.; Yuan, S.; Wang, X.; Cai, H.; Fu, Y.; Luo, Y. NFAT1 mediates placental growth factor-induced myelomonocytic cell recruitment via the induction of TNF-alpha. J. Immunol. 2010, 184, 2593–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newell, L.F.; Holtan, S.G.; Yates, J.E.; Pereira, L.; Tyner, J.W.; Burd, I.; Bagby, G.C. PlGF enhances TLR-dependent inflammatory responses in human mononuclear phagocytes. Am. J. Reprod. Immunol. 2017, 78, e12709. [Google Scholar] [CrossRef]
- Wu, M.Y.; Yang, R.S.; Lin, T.H.; Tang, C.H.; Chiu, Y.C.; Liou, H.C.; Fu, W.M. Enhancement of PLGF production by 15-(S)-HETE via PI3K-Akt, NF-kappaB and COX-2 pathways in rheumatoid arthritis synovial fibroblast. Eur. J. Pharmacol. 2013, 714, 388–396. [Google Scholar] [CrossRef]
- Yoo, S.A.; Kim, M.; Kang, M.C.; Kong, J.S.; Kim, K.M.; Lee, S.; Hong, B.K.; Jeong, G.H.; Lee, J.; Shin, M.G.; et al. Placental growth factor regulates the generation of TH17 cells to link angiogenesis with autoimmunity. Nat. Immunol. 2019, 20, 1348–1359. [Google Scholar] [CrossRef]
- Clapp, J.F., 3rd; Capeless, E. Cardiovascular function before, during, and after the first and subsequent pregnancies. Am. J. Cardiol. 1997, 80, 1469–1473. [Google Scholar] [CrossRef]
- Ridder, A.; Giorgione, V.; Khalil, A.; Thilaganathan, B. Preeclampsia: The Relationship between Uterine Artery Blood Flow and Trophoblast Function. Int. J. Mol. Sci. 2019, 20, 3263. [Google Scholar] [CrossRef] [Green Version]
- Tay, J.; Masini, G.; McEniery, C.M.; Giussani, D.A.; Shaw, C.J.; Wilkinson, I.B.; Bennett, P.R.; Lees, C.C. Uterine and fetal placental Doppler indices are associated with maternal cardiovascular function. Am. J. Obstet. Gynecol. 2019, 220, 96e1–96e8. [Google Scholar] [CrossRef] [Green Version]
- Thilaganathan, B. Pre-eclampsia and the cardiovascular-placental axis. Ultrasound Obstet. Gynecol. 2018, 51, 714–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thilaganathan, B.; Kalafat, E. Cardiovascular System in Preeclampsia and Beyond. Hypertension 2019, 73, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Yagel, S.; Verlohren, S. The Role of the Placenta in the Development of Preeclampsia: Revisited. Ultrasound Obstet. Gynecol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Borzychowski, A.M.; Croy, B.A.; Chan, W.L.; Redman, C.W.; Sargent, I.L. Changes in systemic type 1 and type 2 immunity in normal pregnancy and pre-eclampsia may be mediated by natural killer cells. Eur. J. Immunol. 2005, 35, 3054–3063. [Google Scholar] [CrossRef]
- Care, A.S.; Bourque, S.L.; Morton, J.S.; Hjartarson, E.P.; Robertson, S.A.; Davidge, S.T. Reduction in Regulatory T Cells in Early Pregnancy Causes Uterine Artery Dysfunction in Mice. Hypertension 2018, 72, 177–187. [Google Scholar] [CrossRef]
- Faas, M.M.; De Vos, P. Innate immune cells in the placental bed in healthy pregnancy and preeclampsia. Placenta 2018, 69, 125–133. [Google Scholar] [CrossRef]
- Lu, H.Q.; Hu, R. The role of immunity in the pathogenesis and development of pre-eclampsia. Scand. J. Immunol. 2019, 90, e12756. [Google Scholar] [CrossRef]
- Norlander, A.E.; Madhur, M.S.; Harrison, D.G. The immunology of hypertension. J. Exp. Med. 2018, 215, 21–33. [Google Scholar] [CrossRef]
- Rambaldi, M.P.; Weiner, E.; Mecacci, F.; Bar, J.; Petraglia, F. Immunomodulation and preeclampsia. Best Pract. Res. Clin. Obstet. Gynaecol. 2019, 60, 87–96. [Google Scholar] [CrossRef]
- Agarwal, I.; Karumanchi, S.A. Preeclampsia and the Anti-Angiogenic State. Pregnancy Hypertens. 2011, 1, 17–21. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, S.; Cerdeira, A.S.; Redman, C.; Vatish, M. Meta-Analysis and Systematic Review to Assess the Role of Soluble FMS-Like Tyrosine Kinase-1 and Placenta Growth Factor Ratio in Prediction of Preeclampsia: The SaPPPhirE Study. Hypertension 2018, 71, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Ali, L.E.; Salih, M.M.; Elhassan, E.M.; Mohmmed, A.A.; Adam, I. Placental growth factor, vascular endothelial growth factor, and hypoxia-inducible factor-1alpha in the placentas of women with pre-eclampsia. J. Matern. Fetal Neonatal Med. 2019, 32, 2628–2632. [Google Scholar] [CrossRef] [PubMed]
- Chau, K.; Hennessy, A.; Makris, A. Placental growth factor and pre-eclampsia. J. Hum. Hypertens. 2017, 31, 782–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurrell, A.; Beardmore-Gray, A.; Duhig, K.; Webster, L.; Chappell, L.C.; Shennan, A.H. Placental growth factor in suspected preterm pre-eclampsia: A review of the evidence and practicalities of implementation. BJOG 2020, 127, 1590–1597. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.J.; Maynard, S.E.; Qian, C.; Lim, K.H.; England, L.J.; Yu, K.F.; Schisterman, E.F.; Thadhani, R.; Sachs, B.P.; Epstein, F.H.; et al. Circulating angiogenic factors and the risk of preeclampsia. N. Engl. J. Med. 2004, 350, 672–683. [Google Scholar] [CrossRef] [Green Version]
- Maynard, S.E.; Min, J.Y.; Merchan, J.; Lim, K.H.; Li, J.; Mondal, S.; Libermann, T.A.; Morgan, J.P.; Sellke, F.W.; Stillman, I.E.; et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Invest. 2003, 111, 649–658. [Google Scholar] [CrossRef] [Green Version]
- Sarween, N.; Drayson, M.T.; Hodson, J.; Knox, E.M.; Plant, T.; Day, C.J.; Lipkin, G.W. Humoral immunity in late-onset Pre-eclampsia and linkage with angiogenic and inflammatory markers. Am. J. Reprod. Immunol. 2018, 80, e13041. [Google Scholar] [CrossRef]
- Sezer, S.D.; Kucuk, M.; Doger, F.K.; Yuksel, H.; Odabasi, A.R.; Turkmen, M.K.; Cakmak, B.C.; Omurlu, I.K.; Kinas, M.G. VEGF, PIGF and HIF-1alpha in placentas of early- and late-onset pre-eclamptic patients. Gynecol. Endocrinol. 2013, 29, 797–800. [Google Scholar] [CrossRef]
- Zeisler, H.; Llurba, E.; Chantraine, F.J.; Vatish, M.; Staff, A.C.; Sennstrom, M.; Olovsson, M.; Brennecke, S.P.; Stepan, H.; Allegranza, D.; et al. Soluble fms-like tyrosine kinase-1 to placental growth factor ratio: Ruling out pre-eclampsia for up to 4 weeks and value of retesting. Ultrasound Obstet. Gynecol. 2019, 53, 367–375. [Google Scholar] [CrossRef]
- Thadhani, R.; Mutter, W.P.; Wolf, M.; Levine, R.J.; Taylor, R.N.; Sukhatme, V.P.; Ecker, J.; Karumanchi, S.A. First trimester placental growth factor and soluble fms-like tyrosine kinase 1 and risk for preeclampsia. J. Clin. Endocrinol. Metab. 2004, 89, 770–775. [Google Scholar] [CrossRef]
- De Falco, S. The discovery of placenta growth factor and its biological activity. Exp. Mol. Med. 2012, 44, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maglione, D.; Guerriero, V.; Viglietto, G.; Delli-Bovi, P.; Persico, M.G. Isolation of a human placenta cDNA coding for a protein related to the vascular permeability factor. Proc. Natl. Acad. Sci. USA 1991, 88, 9267–9271. [Google Scholar] [CrossRef] [Green Version]
- Autiero, M.; Luttun, A.; Tjwa, M.; Carmeliet, P. Placental growth factor and its receptor, vascular endothelial growth factor receptor-1: Novel targets for stimulation of ischemic tissue revascularization and inhibition of angiogenic and inflammatory disorders. J. Thromb. Haemost. 2003, 1, 1356–1370. [Google Scholar] [CrossRef] [PubMed]
- Dewerchin, M.; Carmeliet, P. PlGF: A multitasking cytokine with disease-restricted activity. Cold Spring Harb. Perspect. Med. 2012, 2, a011056. [Google Scholar] [CrossRef] [PubMed]
- Oura, H.; Bertoncini, J.; Velasco, P.; Brown, L.F.; Carmeliet, P.; Detmar, M. A critical role of placental growth factor in the induction of inflammation and edema formation. Blood 2003, 101, 560–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, S.; Bag, A.K.; Singh, R.K.; Talmadge, J.E.; Batra, S.K.; Datta, K. Multifaceted Role of Neuropilins in the Immune System: Potential Targets for Immunotherapy. Front. Immunol. 2017, 8, 1228. [Google Scholar] [CrossRef] [Green Version]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell. Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef]
- Torry, R.J.; Tomanek, R.J.; Zheng, W.; Miller, S.J.; Labarrere, C.A.; Torry, D.S. Hypoxia increases placenta growth factor expression in human myocardium and cultured neonatal rat cardiomyocytes. J. Heart Lung Transpl. 2009, 28, 183–190. [Google Scholar] [CrossRef] [Green Version]
- Tudisco, L.; Orlandi, A.; Tarallo, V.; De Falco, S. Hypoxia activates placental growth factor expression in lymphatic endothelial cells. Oncotarget 2017, 8, 32873–32883. [Google Scholar] [CrossRef] [Green Version]
- Gobble, R.M.; Groesch, K.A.; Chang, M.; Torry, R.J.; Torry, D.S. Differential regulation of human PlGF gene expression in trophoblast and nontrophoblast cells by oxygen tension. Placenta 2009, 30, 869–875. [Google Scholar] [CrossRef] [Green Version]
- Luttun, A.; Brusselmans, K.; Fukao, H.; Tjwa, M.; Ueshima, S.; Herbert, J.M.; Matsuo, O.; Collen, D.; Carmeliet, P.; Moons, L. Loss of placental growth factor protects mice against vascular permeability in pathological conditions. Biochem. Biophys. Res. Commun. 2002, 295, 428–434. [Google Scholar] [CrossRef]
- Tayade, C.; Hilchie, D.; He, H.; Fang, Y.; Moons, L.; Carmeliet, P.; Foster, R.A.; Croy, B.A. Genetic deletion of placenta growth factor in mice alters uterine NK cells. J. Immunol. 2007, 178, 4267–4275. [Google Scholar] [CrossRef] [PubMed]
- Ratsep, M.T.; Felker, A.M.; Kay, V.R.; Tolusso, L.; Hofmann, A.P.; Croy, B.A. Uterine natural killer cells: Supervisors of vasculature construction in early decidua basalis. Reproduction 2015, 149, R91–R102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, M.C.; Park, S.J.; Kim, H.J.; Lee, J.; Yu, D.H.; Bae, K.B.; Ji, Y.R.; Park, S.J.; Jeong, J.; Jang, W.Y.; et al. Gestational loss and growth restriction by angiogenic defects in placental growth factor transgenic mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2276–2282. [Google Scholar] [CrossRef] [Green Version]
- Kopcow, H.D.; Karumanchi, S.A. Angiogenic factors and natural killer (NK) cells in the pathogenesis of preeclampsia. J. Reprod. Immunol. 2007, 76, 23–29. [Google Scholar] [CrossRef] [Green Version]
- Ratsep, M.T.; Carmeliet, P.; Adams, M.A.; Croy, B.A. Impact of placental growth factor deficiency on early mouse implant site angiogenesis. Placenta 2014, 35, 772–775. [Google Scholar] [CrossRef]
- Yonekura Collier, A.R.; Zsengeller, Z.; Pernicone, E.; Salahuddin, S.; Khankin, E.V.; Karumanchi, S.A. Placental sFLT1 is associated with complement activation and syncytiotrophoblast damage in preeclampsia. Hypertens. Pregnancy 2019, 38, 193–199. [Google Scholar] [CrossRef]
- Mamluk, R.; Gechtman, Z.; Kutcher, M.E.; Gasiunas, N.; Gallagher, J.; Klagsbrun, M. Neuropilin-1 binds vascular endothelial growth factor 165, placenta growth factor-2, and heparin via its b1b2 domain. J. Biol. Chem. 2002, 277, 24818–24825. [Google Scholar] [CrossRef] [Green Version]
- Migdal, M.; Huppertz, B.; Tessler, S.; Comforti, A.; Shibuya, M.; Reich, R.; Baumann, H.; Neufeld, G. Neuropilin-1 is a placenta growth factor-2 receptor. J. Biol. Chem. 1998, 273, 22272–22278. [Google Scholar] [CrossRef] [Green Version]
- Neufeld, G.; Kessler, O.; Herzog, Y. The interaction of Neuropilin-1 and Neuropilin-2 with tyrosine-kinase receptors for VEGF. Adv. Exp. Med. Biol. 2002, 515, 81–90. [Google Scholar]
- Anisimov, A.; Leppanen, V.M.; Tvorogov, D.; Zarkada, G.; Jeltsch, M.; Holopainen, T.; Kaijalainen, S.; Alitalo, K. The basis for the distinct biological activities of vascular endothelial growth factor receptor-1 ligands. Sci. Signal 2013, 6, ra52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eltzschig, H.K.; Carmeliet, P. Hypoxia and inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Incio, J.; Tam, J.; Rahbari, N.N.; Suboj, P.; McManus, D.T.; Chin, S.M.; Vardam, T.D.; Batista, A.; Babykutty, S.; Jung, K.; et al. PlGF/VEGFR-1 Signaling Promotes Macrophage Polarization and Accelerated Tumor Progression in Obesity. Clin. Cancer Res. 2016, 22, 2993–3004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrotta, M.; Lori, A.; Carnevale, L.; Fardella, S.; Cifelli, G.; Iacobucci, R.; Mastroiacovo, F.; Iodice, D.; Pallante, F.; Storto, M.; et al. Deoxycorticosterone acetate-salt hypertension activates placental growth factor in the spleen to couple sympathetic drive and immune system activation. Cardiovasc. Res. 2018, 114, 456–467. [Google Scholar] [CrossRef] [PubMed]
- Sawano, A.; Iwai, S.; Sakurai, Y.; Ito, M.; Shitara, K.; Nakahata, T.; Shibuya, M. Flt-1, vascular endothelial growth factor receptor 1, is a novel cell surface marker for the lineage of monocyte-macrophages in humans. Blood 2001, 97, 785–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvaraj, S.K.; Giri, R.K.; Perelman, N.; Johnson, C.; Malik, P.; Kalra, V.K. Mechanism of monocyte activation and expression of proinflammatory cytochemokines by placenta growth factor. Blood 2003, 102, 1515–1524. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Watanabe, M.; Tanigaki, S.; Iwashita, M.; Kobayashi, Y. Tumor necrosis factor-alpha regulates angiogenesis of BeWo cells via synergy of PlGF/VEGFR1 and VEGF-A/VEGFR2 axes. Placenta 2018, 74, 20–27. [Google Scholar] [CrossRef]
- Shin, J.Y.; Yoon, I.H.; Kim, J.S.; Kim, B.; Park, C.G. Vascular endothelial growth factor-induced chemotaxis and IL-10 from T cells. Cell Immunol. 2009, 256, 72–78. [Google Scholar] [CrossRef]
- Zachary, I. Neuropilins: Role in signalling, angiogenesis and disease. Chem. Immunol. Allergy 2014, 99, 37–70. [Google Scholar]
- Lepelletier, Y.; Moura, I.C.; Hadj-Slimane, R.; Renand, A.; Fiorentino, S.; Baude, C.; Shirvan, A.; Barzilai, A.; Hermine, O. Immunosuppressive role of semaphorin-3A on T cell proliferation is mediated by inhibition of actin cytoskeleton reorganization. Eur. J. Immunol. 2006, 36, 1782–1793. [Google Scholar] [CrossRef]
- Kalekar, L.A.; Schmiel, S.E.; Nandiwada, S.L.; Lam, W.Y.; Barsness, L.O.; Zhang, N.; Stritesky, G.L.; Malhotra, D.; Pauken, K.E.; Linehan, J.L.; et al. CD4(+) T cell anergy prevents autoimmunity and generates regulatory T cell precursors. Nat. Immunol. 2016, 17, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.; Louvet, C.; Davini, D.; Gardner, J.M.; Martinez-Llordella, M.; Bailey-Bucktrout, S.; Anthony, B.A.; Sverdrup, F.M.; Head, R.; Kuster, D.J.; et al. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo. J. Exp. Med. 2012, 209, 1713–1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarris, M.; Andersen, K.G.; Randow, F.; Mayr, L.; Betz, A.G. Neuropilin-1 expression on regulatory T cells enhances their interactions with dendritic cells during antigen recognition. Immunity 2008, 28, 402–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arad, A.; Nammouz, S.; Nov, Y.; Ohel, G.; Bejar, J.; Vadasz, Z. The Expression of Neuropilin-1 in Human Placentas from Normal and Preeclamptic Pregnancies. Int. J. Gynecol. Pathol. 2017, 36, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Maulik, D.; De, A.; Ragolia, L.; Evans, J.; Grigoryev, D.; Lankachandra, K.; Mundy, D.; Muscat, J.; Gerkovich, M.M.; Ye, S.Q. Down-regulation of placental neuropilin-1 in fetal growth restriction. Am. J. Obstet. Gynecol. 2016, 214, 279e1–279e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moldenhauer, L.M.; Schjenken, J.E.; Hope, C.M.; Green, E.S.; Zhang, B.; Eldi, P.; Hayball, J.D.; Barry, S.C.; Robertson, S.A. Thymus-Derived Regulatory T Cells Exhibit Foxp3 Epigenetic Modification and Phenotype Attenuation after Mating in Mice. J. Immunol. 2019, 203, 647–657. [Google Scholar] [CrossRef]
- Failla, C.M.; Carbo, M.; Morea, V. Positive and Negative Regulation of Angiogenesis by Soluble Vascular Endothelial Growth Factor Receptor-1. Int. J. Mol. Sci. 2018, 19, 1306. [Google Scholar] [CrossRef] [Green Version]
- Panigrahy, D.; Adini, I.; Mamluk, R.; Levonyak, N.; Bruns, C.J.; D’Amore, P.A.; Klagsbrun, M.; Bielenberg, D.R. Regulation of soluble neuropilin 1, an endogenous angiogenesis inhibitor, in liver development and regeneration. Pathology 2014, 46, 416–423. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Jin, C.; Jiang, Y.J.; Li, J.; Di, Y.; Fu, D.L. Potential role of soluble VEGFR-1 in antiangiogenesis therapy for cancer. Expert Rev. Anticancer Ther. 2011, 11, 541–549. [Google Scholar] [CrossRef]
- Chang, Y.S.; Chen, C.N.; Jeng, S.F.; Su, Y.N.; Chen, C.Y.; Chou, H.C.; Tsao, P.N.; Hsieh, W.S. The sFlt-1/PlGF ratio as a predictor for poor pregnancy and neonatal outcomes. Pediatr Neonatol 2017, 58, 529–533. [Google Scholar] [CrossRef] [Green Version]
- Jebbink, J.; Keijser, R.; Veenboer, G.; van der Post, J.; Ris-Stalpers, C.; Afink, G. Expression of placental FLT1 transcript variants relates to both gestational hypertensive disease and fetal growth. Hypertension 2011, 58, 70–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumazaki, K.; Nakayama, M.; Suehara, N.; Wada, Y. Expression of vascular endothelial growth factor, placental growth factor, and their receptors Flt-1 and KDR in human placenta under pathologic conditions. Hum. Pathol. 2002, 33, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- McKeeman, G.C.; Ardill, J.E.; Caldwell, C.M.; Hunter, A.J.; McClure, N. Soluble vascular endothelial growth factor receptor-1 (sFlt-1) is increased throughout gestation in patients who have preeclampsia develop. Am. J. Obstet. Gynecol. 2004, 191, 1240–1246. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, A.M.; Kahane, A.; Ray, J.G. First and Second Trimester Serum sFlt-1/PlGF Ratio and Subsequent Preeclampsia: A Systematic Review. J. Obstet. Gynaecol. Can. 2018, 40, 618–626. [Google Scholar] [CrossRef]
- Palmer, K.R.; Kaitu’u-Lino, T.J.; Hastie, R.; Hannan, N.J.; Ye, L.; Binder, N.; Cannon, P.; Tuohey, L.; Johns, T.G.; Shub, A.; et al. Placental-Specific sFLT-1 e15a Protein Is Increased in Preeclampsia, Antagonizes Vascular Endothelial Growth Factor Signaling, and Has Antiangiogenic Activity. Hypertension 2015, 66, 1251–1259. [Google Scholar] [CrossRef] [PubMed]
- Palmer, K.R.; Tong, S.; Kaitu’u-Lino, T.J. Placental-specific sFLT-1: Role in pre-eclamptic pathophysiology and its translational possibilities for clinical prediction and diagnosis. Mol. Hum. Reprod. 2017, 23, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Cramer, M.; Nagy, I.; Murphy, B.J.; Gassmann, M.; Hottiger, M.O.; Georgiev, O.; Schaffner, W. NF-kappaB contributes to transcription of placenta growth factor and interacts with metal responsive transcription factor-1 in hypoxic human cells. Biol. Chem. 2005, 386, 865–872. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Armistead, B.; Kadam, L.; Drewlo, S.; Kohan-Ghadr, H.R. The Role of NFkappaB in Healthy and Preeclamptic Placenta: Trophoblasts in the Spotlight. Int. J. Mol. Sci. 2020, 21, 1775. [Google Scholar] [CrossRef] [Green Version]
- Dorrington, M.G.; Fraser, I.D.C. NF-kappaB Signaling in Macrophages: Dynamics, Crosstalk, and Signal Integration. Front. Immunol. 2019, 10, 705. [Google Scholar] [CrossRef]
- Taylor, C.T.; Cummins, E.P. The role of NF-kappaB in hypoxia-induced gene expression. Ann. N.Y. Acad. Sci. 2009, 1177, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Guney, G.; Taskin, M.I.; Tokmak, A. Increase of circulating inflammatory molecules in preeclampsia, an update. Eur. Cytokine Netw. 2020, 31, 18–31. [Google Scholar] [PubMed]
- Harmon, A.C.; Cornelius, D.C.; Amaral, L.M.; Faulkner, J.L.; Cunningham, M.W., Jr.; Wallace, K.; LaMarca, B. The role of inflammation in the pathology of preeclampsia. Clin. Sci. 2016, 130, 409–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lappas, M. Nuclear factor-kappaB mediates placental growth factor induced pro-labour mediators in human placenta. Mol. Hum. Reprod. 2012, 18, 354–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakowicz, A. The role of NFkappaB in the three stages of pregnancy—Implantation, maintenance, and labour: A review article. BJOG 2018, 125, 1379–1387. [Google Scholar] [CrossRef] [Green Version]
- D’Ignazio, L.; Rocha, S. Hypoxia Induced NF-kappaB. Cells 2016, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.S.; Ghosh, S. Regulation of NF-kappaB by TNF family cytokines. Semin. Immunol. 2014, 26, 253–266. [Google Scholar] [CrossRef] [Green Version]
- Hogan, P.G. Calcium-NFAT transcriptional signalling in T cell activation and T cell exhaustion. Cell Calcium 2017, 63, 66–69. [Google Scholar] [CrossRef] [Green Version]
- Macian, F. NFAT proteins: Key regulators of T-cell development and function. Nat. Rev. Immunol. 2005, 5, 472–484. [Google Scholar] [CrossRef]
- Fric, J.; Zelante, T.; Wong, A.Y.; Mertes, A.; Yu, H.B.; Ricciardi-Castagnoli, P. NFAT control of innate immunity. Blood 2012, 120, 1380–1389. [Google Scholar] [CrossRef]
- Jinnin, M.; Medici, D.; Park, L.; Limaye, N.; Liu, Y.; Boscolo, E.; Bischoff, J.; Vikkula, M.; Boye, E.; Olsen, B.R. Suppressed NFAT-dependent VEGFR1 expression and constitutive VEGFR2 signaling in infantile hemangioma. Nat. Med. 2008, 14, 1236–1246. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Gratton, A.; Hannan, N.J.; Cannon, P.; Deo, M.; Palmer, K.R.; Tong, S.; Kaitu’u-Lino, T.J.; Brownfoot, F.C. Nuclear factor of activated T-cells (NFAT) regulates soluble fms-like tyrosine kinase-1 secretion (sFlt-1) from human placenta. Placenta 2016, 48, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Abe, B.T.; Shin, D.S.; Mocholi, E.; Macian, F. NFAT1 supports tumor-induced anergy of CD4(+) T cells. Cancer Res. 2012, 72, 4642–4651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, M. Control of regulatory T-cell differentiation and function by T-cell receptor signalling and Foxp3 transcription factor complexes. Immunology 2020, 160, 24–37. [Google Scholar] [CrossRef] [Green Version]
- Shin, D.S.; Jordan, A.; Basu, S.; Thomas, R.M.; Bandyopadhyay, S.; de Zoeten, E.F.; Wells, A.D.; Macian, F. Regulatory T cells suppress CD4+ T cells through NFAT-dependent transcriptional mechanisms. EMBO Rep. 2014, 15, 991–999. [Google Scholar] [CrossRef] [Green Version]
- Lima, P.D.; Zhang, J.; Dunk, C.; Lye, S.J.; Croy, B.A. Leukocyte driven-decidual angiogenesis in early pregnancy. Cell Mol. Immunol. 2014, 11, 522–537. [Google Scholar] [CrossRef] [Green Version]
- Racicot, K.; Kwon, J.Y.; Aldo, P.; Silasi, M.; Mor, G. Understanding the complexity of the immune system during pregnancy. Am. J. Reprod. Immunol. 2014, 72, 107–116. [Google Scholar] [CrossRef]
- Gaynor, L.M.; Colucci, F. Uterine Natural Killer Cells: Functional Distinctions and Influence on Pregnancy in Humans and Mice. Front. Immunol. 2017, 8, 467. [Google Scholar] [CrossRef] [Green Version]
- Hanna, J.; Goldman-Wohl, D.; Hamani, Y.; Avraham, I.; Greenfield, C.; Natanson-Yaron, S.; Prus, D.; Cohen-Daniel, L.; Arnon, T.I.; Manaster, I.; et al. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat. Med. 2006, 12, 1065–1074. [Google Scholar] [CrossRef]
- Jabrane-Ferrat, N.; Siewiera, J. The up side of decidual natural killer cells: New developments in immunology of pregnancy. Immunology 2014, 141, 490–497. [Google Scholar] [CrossRef] [Green Version]
- Le Bouteiller, P.; Tabiasco, J. Killers become builders during pregnancy. Nat. Med. 2006, 12, 991–992. [Google Scholar] [CrossRef] [PubMed]
- Moffett, A.; Colucci, F. Uterine NK cells: Active regulators at the maternal-fetal interface. J. Clin. Invest. 2014, 124, 1872–1879. [Google Scholar] [CrossRef] [PubMed]
- Moffett-King, A. Natural killer cells and pregnancy. Nat. Rev. Immunol. 2002, 2, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Sojka, D.K.; Yang, L.; Yokoyama, W.M. Uterine Natural Killer Cells. Front. Immunol. 2019, 10, 960. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Seillet, C.; Belz, G.T. Shaping Innate Lymphoid Cell Diversity. Front. Immunol. 2017, 8, 1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.; Motomura, K.; Garcia-Flores, V.; Romero, R.; Gomez-Lopez, N. Innate Lymphoid Cells in the Maternal and Fetal Compartments. Front. Immunol. 2018, 9, 2396. [Google Scholar] [CrossRef] [PubMed]
- Spits, H.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.; Mebius, R.E.; et al. Innate lymphoid cells--a proposal for uniform nomenclature. Nat. Rev. Immunol. 2013, 13, 145–149. [Google Scholar] [CrossRef]
- Colonna, M. Innate Lymphoid Cells: Diversity, Plasticity, and Unique Functions in Immunity. Immunity 2018, 48, 1104–1117. [Google Scholar] [CrossRef] [Green Version]
- Vacca, P.; Chiossone, L.; Mingari, M.C.; Moretta, L. Heterogeneity of NK Cells and Other Innate Lymphoid Cells in Human and Murine Decidua. Front. Immunol. 2019, 10, 170. [Google Scholar] [CrossRef] [Green Version]
- Bonanni, V.; Sciume, G.; Santoni, A.; Bernardini, G. Bone Marrow NK Cells: Origin, Distinctive Features, and Requirements for Tissue Localization. Front. Immunol. 2019, 10, 1569. [Google Scholar] [CrossRef] [Green Version]
- Klose, C.S.; Artis, D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat. Immunol. 2016, 17, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Croy, B.A.; Esadeg, S.; Chantakru, S.; van den Heuvel, M.; Paffaro, V.A.; He, H.; Black, G.P.; Ashkar, A.A.; Kiso, Y.; Zhang, J. Update on pathways regulating the activation of uterine Natural Killer cells, their interactions with decidual spiral arteries and homing of their precursors to the uterus. J. Reprod. Immunol. 2003, 59, 175–191. [Google Scholar] [CrossRef]
- Sojka, D.K.; Yang, L.; Plougastel-Douglas, B.; Higuchi, D.A.; Croy, B.A.; Yokoyama, W.M. Cutting Edge: Local Proliferation of Uterine Tissue-Resident NK Cells during Decidualization in Mice. J. Immunol. 2018, 201, 2551–2556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Heuvel, M.J.; Chantakru, S.; Xuemei, X.; Evans, S.S.; Tekpetey, F.; Mote, P.A.; Clarke, C.L.; Croy, B.A. Trafficking of circulating pro-NK cells to the decidualizing uterus: Regulatory mechanisms in the mouse and human. Immunol. Invest. 2005, 34, 273–293. [Google Scholar] [CrossRef] [Green Version]
- Bjorkstrom, N.K.; Ljunggren, H.G.; Michaelsson, J. Emerging insights into natural killer cells in human peripheral tissues. Nat. Rev. Immunol. 2016, 16, 310–320. [Google Scholar] [CrossRef]
- Tao, Y.; Li, Y.H.; Piao, H.L.; Zhou, W.J.; Zhang, D.; Fu, Q.; Wang, S.C.; Li, D.J.; Du, M.R. CD56(bright)CD25+ NK cells are preferentially recruited to the maternal/fetal interface in early human pregnancy. Cell Mol. Immunol. 2015, 12, 77–86. [Google Scholar] [CrossRef] [Green Version]
- Tayade, C.; Fang, Y.; Black, G.P.; Paffaro, V.A., Jr.; Erlebacher, A.; Croy, B.A. Differential transcription of Eomes and T-bet during maturation of mouse uterine natural killer cells. J. Leukoc. Biol. 2005, 78, 1347–1355. [Google Scholar] [CrossRef]
- Fraser, R.; Whitley, G.S.; Thilaganathan, B.; Cartwright, J.E. Decidual natural killer cells regulate vessel stability: Implications for impaired spiral artery remodelling. J. Reprod. Immunol. 2015, 110, 54–60. [Google Scholar] [CrossRef] [Green Version]
- Lash, G.E.; Schiessl, B.; Kirkley, M.; Innes, B.A.; Cooper, A.; Searle, R.F.; Robson, S.C.; Bulmer, J.N. Expression of angiogenic growth factors by uterine natural killer cells during early pregnancy. J. Leukoc. Biol. 2006, 80, 572–580. [Google Scholar] [CrossRef]
- Robson, A.; Harris, L.K.; Innes, B.A.; Lash, G.E.; Aljunaidy, M.M.; Aplin, J.D.; Baker, P.N.; Robson, S.C.; Bulmer, J.N. Uterine natural killer cells initiate spiral artery remodeling in human pregnancy. FASEB J. 2012, 26, 4876–4885. [Google Scholar] [CrossRef]
- Lockwood, C.J.; Huang, S.J.; Chen, C.P.; Huang, Y.; Xu, J.; Faramarzi, S.; Kayisli, O.; Kayisli, U.; Koopman, L.; Smedts, D.; et al. Decidual cell regulation of natural killer cell-recruiting chemokines: Implications for the pathogenesis and prediction of preeclampsia. Am. J. Pathol 2013, 183, 841–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orr, M.T.; Lanier, L.L. Natural killer cell education and tolerance. Cell 2010, 142, 847–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharkey, A.M.; Xiong, S.; Kennedy, P.R.; Gardner, L.; Farrell, L.E.; Chazara, O.; Ivarsson, M.A.; Hiby, S.E.; Colucci, F.; Moffett, A. Tissue-Specific Education of Decidual NK Cells. J. Immunol. 2015, 195, 3026–3032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukui, A.; Funamizu, A.; Fukuhara, R.; Shibahara, H. Expression of natural cytotoxicity receptors and cytokine production on endometrial natural killer cells in women with recurrent pregnancy loss or implantation failure, and the expression of natural cytotoxicity receptors on peripheral blood natural killer cells in pregnant women with a history of recurrent pregnancy loss. J. Obstet. Gynaecol. Res. 2017, 43, 1678–1686. [Google Scholar]
- Lanier, L.L. NK cell recognition. Annu. Rev. Immunol. 2005, 23, 225–274. [Google Scholar] [CrossRef]
- Ashkar, A.A.; Di Santo, J.P.; Croy, B.A. Interferon gamma contributes to initiation of uterine vascular modification, decidual integrity, and uterine natural killer cell maturation during normal murine pregnancy. J. Exp. Med. 2000, 192, 259–270. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, P.R.; Chazara, O.; Gardner, L.; Ivarsson, M.A.; Farrell, L.E.; Xiong, S.; Hiby, S.E.; Colucci, F.; Sharkey, A.M.; Moffett, A. Activating KIR2DS4 Is Expressed by Uterine NK Cells and Contributes to Successful Pregnancy. J. Immunol. 2016, 197, 4292–4300. [Google Scholar] [CrossRef] [Green Version]
- Kerdiles, Y.; Ugolini, S.; Vivier, E. T cell regulation of natural killer cells. J. Exp. Med. 2013, 210, 1065–1068. [Google Scholar] [CrossRef]
- Knorr, M.; Munzel, T.; Wenzel, P. Interplay of NK cells and monocytes in vascular inflammation and myocardial infarction. Front. Physiol 2014, 5, 295. [Google Scholar] [CrossRef]
- Lucas, M.; Schachterle, W.; Oberle, K.; Aichele, P.; Diefenbach, A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity 2007, 26, 503–517. [Google Scholar] [CrossRef] [Green Version]
- Zingoni, A.; Sornasse, T.; Cocks, B.G.; Tanaka, Y.; Santoni, A.; Lanier, L.L. NK cell regulation of T cell-mediated responses. Mol. Immunol. 2005, 42, 451–454. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Sharkey, A.M.; Kennedy, P.R.; Gardner, L.; Farrell, L.E.; Chazara, O.; Bauer, J.; Hiby, S.E.; Colucci, F.; Moffett, A. Maternal uterine NK cell-activating receptor KIR2DS1 enhances placentation. J. Clin. Invest. 2013, 123, 4264–4272. [Google Scholar] [CrossRef] [PubMed]
- Hiby, S.E.; Apps, R.; Sharkey, A.M.; Farrell, L.E.; Gardner, L.; Mulder, A.; Claas, F.H.; Walker, J.J.; Redman, C.W.; Morgan, L.; et al. Maternal activating KIRs protect against human reproductive failure mediated by fetal HLA-C2. J. Clin. Invest. 2010, 120, 4102–4110. [Google Scholar] [CrossRef] [PubMed]
- Drake, P.M.; Gunn, M.D.; Charo, I.F.; Tsou, C.L.; Zhou, Y.; Huang, L.; Fisher, S.J. Human placental cytotrophoblasts attract monocytes and CD56(bright) natural killer cells via the actions of monocyte inflammatory protein 1alpha. J. Exp. Med. 2001, 193, 1199–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Gars, M.; Seiler, C.; Kay, A.W.; Bayless, N.L.; Starosvetsky, E.; Moore, L.; Shen-Orr, S.S.; Aziz, N.; Khatri, P.; Dekker, C.L.; et al. Pregnancy-Induced Alterations in NK Cell Phenotype and Function. Front. Immunol. 2019, 10, 2469. [Google Scholar] [CrossRef] [Green Version]
- Blois, S.M.; Klapp, B.F.; Barrientos, G. Decidualization and angiogenesis in early pregnancy: Unravelling the functions of DC and NK cells. J. Reprod. Immunol. 2011, 88, 86–92. [Google Scholar] [CrossRef]
- Cartwright, J.E.; James-Allan, L.; Buckley, R.J.; Wallace, A.E. The role of decidual NK cells in pregnancies with impaired vascular remodelling. J. Reprod. Immunol. 2017, 119, 81–84. [Google Scholar] [CrossRef]
- Solocinski, K.; Padget, M.R.; Fabian, K.P.; Wolfson, B.; Cecchi, F.; Hembrough, T.; Benz, S.C.; Rabizadeh, S.; Soon-Shiong, P.; Schlom, J.; et al. Overcoming hypoxia-induced functional suppression of NK cells. J. Immunother Cancer 2020, 8. [Google Scholar] [CrossRef]
- Vacca, P.; Cantoni, C.; Vitale, M.; Prato, C.; Canegallo, F.; Fenoglio, D.; Ragni, N.; Moretta, L.; Mingari, M.C. Crosstalk between decidual NK and CD14+ myelomonocytic cells results in induction of Tregs and immunosuppression. Proc. Natl. Acad. Sci. USA 2010, 107, 11918–11923. [Google Scholar] [CrossRef] [Green Version]
- Fukui, A.; Yokota, M.; Funamizu, A.; Nakamua, R.; Fukuhara, R.; Yamada, K.; Kimura, H.; Fukuyama, A.; Kamoi, M.; Tanaka, K.; et al. Changes of NK cells in preeclampsia. Am. J. Reprod. Immunol. 2012, 67, 278–286. [Google Scholar] [CrossRef]
- Golic, M.; Haase, N.; Herse, F.; Wehner, A.; Vercruysse, L.; Pijnenborg, R.; Balogh, A.; Saether, P.C.; Dissen, E.; Luft, F.C.; et al. Natural Killer Cell Reduction and Uteroplacental Vasculopathy. Hypertension 2016, 68, 964–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuon, R.J.; Vomstein, K.; Weber, M.; Muller, F.; Seitz, C.; Wallwiener, S.; Strowitzki, T.; Schleussner, E.; Markert, U.R.; Daniel, V.; et al. The “killer cell story” in recurrent miscarriage: Association between activated peripheral lymphocytes and uterine natural killer cells. J. Reprod. Immunol. 2017, 119, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Fu, B.; Zhou, Y.; Ni, X.; Tong, X.; Xu, X.; Dong, Z.; Sun, R.; Tian, Z.; Wei, H. Natural Killer Cells Promote Fetal Development through the Secretion of Growth-Promoting Factors. Immunity 2017, 47, 1100–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretta, A.; Marcenaro, E.; Parolini, S.; Ferlazzo, G.; Moretta, L. NK cells at the interface between innate and adaptive immunity. Cell Death Differ. 2008, 15, 226–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kofod, L.; Lindhard, A.; Hviid, T.V.F. Implications of uterine NK cells and regulatory T cells in the endometrium of infertile women. Hum. Immunol. 2018, 79, 693–701. [Google Scholar] [CrossRef]
- Santner-Nanan, B.; Peek, M.J.; Khanam, R.; Richarts, L.; Zhu, E.; Fazekas de St Groth, B.; Nanan, R. Systemic increase in the ratio between Foxp3+ and IL-17-producing CD4+ T cells in healthy pregnancy but not in preeclampsia. J. Immunol. 2009, 183, 7023–7030. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, Y.; Darmochwal-Kolarz, D.; Suzuki, D.; Sakai, M.; Ito, M.; Shima, T.; Shiozaki, A.; Rolinski, J.; Saito, S. Proportion of peripheral blood and decidual CD4(+) CD25(bright) regulatory T cells in pre-eclampsia. Clin. Exp. Immunol. 2007, 149, 139–145. [Google Scholar] [CrossRef]
- Molvarec, A.; Czegle, I.; Szijarto, J.; Rigo, J., Jr. Increased circulating interleukin-17 levels in preeclampsia. J. Reprod. Immunol. 2015, 112, 53–57. [Google Scholar] [CrossRef] [Green Version]
- Vishnyakova, P.; Elchaninov, A.; Fatkhudinov, T.; Sukhikh, G. Role of the Monocyte-Macrophage System in Normal Pregnancy and Preeclampsia. Int. J. Mol. Sci. 2019, 20, 3695. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Porta, C.; Riboldi, E.; Ippolito, A.; Sica, A. Molecular and epigenetic basis of macrophage polarized activation. Semin. Immunol. 2015, 27, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Faas, M.M.; Spaans, F.; De Vos, P. Monocytes and macrophages in pregnancy and pre-eclampsia. Front. Immunol. 2014, 5, 298. [Google Scholar] [CrossRef] [Green Version]
- Lash, G.E.; Pitman, H.; Morgan, H.L.; Innes, B.A.; Agwu, C.N.; Bulmer, J.N. Decidual macrophages: Key regulators of vascular remodeling in human pregnancy. J. Leukoc. Biol. 2016, 100, 315–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, F.; Liu, H.; Lash, G.E. The Role of Decidual Macrophages during Normal and Pathological Pregnancy. Am. J. Reprod. Immunol. 2016, 75, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Xu, X.H.; Jin, L. Macrophage Polarization in Physiological and Pathological Pregnancy. Front. Immunol. 2019, 10, 792. [Google Scholar] [CrossRef]
- Zhang, Y.H.; He, M.; Wang, Y.; Liao, A.H. Modulators of the Balance between M1 and M2 Macrophages during Pregnancy. Front. Immunol. 2017, 8, 120. [Google Scholar] [CrossRef] [Green Version]
- Nagamatsu, T.; Schust, D.J. The contribution of macrophages to normal and pathological pregnancies. Am. J. Reprod. Immunol. 2010, 63, 460–471. [Google Scholar] [CrossRef]
- Muramatsu, M.; Yamamoto, S.; Osawa, T.; Shibuya, M. Vascular endothelial growth factor receptor-1 signaling promotes mobilization of macrophage lineage cells from bone marrow and stimulates solid tumor growth. Cancer Res. 2010, 70, 8211–8221. [Google Scholar] [CrossRef] [Green Version]
- Riboldi, E.; Porta, C.; Morlacchi, S.; Viola, A.; Mantovani, A.; Sica, A. Hypoxia-mediated regulation of macrophage functions in pathophysiology. Int. Immunol. 2013, 25, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Kalish, F.S.; Wong, R.J.; Stevenson, D.K. Hypoxia regulates placental angiogenesis via alternatively activated macrophages. Am. J. Reprod. Immunol. 2018, 80, e12989. [Google Scholar] [CrossRef] [PubMed]
- Rolny, C.; Mazzone, M.; Tugues, S.; Laoui, D.; Johansson, I.; Coulon, C.; Squadrito, M.L.; Segura, I.; Li, X.; Knevels, E.; et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell 2011, 19, 31–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Qi, Y. Larynx carcinoma regulates tumor-associated macrophages through PLGF signaling. Sci. Rep. 2015, 5, 10071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doedens, A.L.; Stockmann, C.; Rubinstein, M.P.; Liao, D.; Zhang, N.; DeNardo, D.G.; Coussens, L.M.; Karin, M.; Goldrath, A.W.; Johnson, R.S. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res. 2010, 70, 7465–7475. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Patel, S.P.; Roszik, J.; Qin, Y. Hypoxia-Driven Immunosuppressive Metabolites in the Tumor Microenvironment: New Approaches for Combinational Immunotherapy. Front. Immunol. 2018, 9, 1591. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.Q.; Zhang, C.M.; Gao, W.; Wang, X.F.; Zhang, H.L.; Yang, P.C. Cancer-derived matrix metalloproteinase-9 contributes to tumor tolerance. J. Cancer Res. Clin. Oncol. 2011, 137, 1525–1533. [Google Scholar] [CrossRef]
- Jetten, N.; Verbruggen, S.; Gijbels, M.J.; Post, M.J.; De Winther, M.P.; Donners, M.M. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis 2014, 17, 109–118. [Google Scholar] [CrossRef]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef]
- Wakabayashi, S. New insights into the functions of histidine-rich glycoprotein. Int. Rev. Cell. Mol. Biol. 2013, 304, 467–493. [Google Scholar]
- Blank, M.; Shoenfeld, Y. Histidine-rich glycoprotein modulation of immune/autoimmune, vascular, and coagulation systems. Clin. Rev. Allergy Immunol. 2008, 34, 307–312. [Google Scholar] [CrossRef]
- Aksornphusitaphong, A.; Phupong, V. Combination of serum histidine-rich glycoprotein and uterine artery Doppler to predict preeclampsia. Hypertens. Res. 2018, 41, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Bolin, M.; Akerud, P.; Hansson, A.; Akerud, H. Histidine-rich glycoprotein as an early biomarker of preeclampsia. Am. J. Hypertens. 2011, 24, 496–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindgren, K.E.; Karehed, K.; Karypidis, H.; Hosseini, F.; Bremme, K.; Landgren, B.M.; Skjoldebrand-Sparre, L.; Stavreus-Evers, A.; Sundstrom-Poromaa, I.; Akerud, H. Histidine-rich glycoprotein gene polymorphism in patients with recurrent miscarriage. Acta Obstet. Gynecol. Scand. 2013, 92, 974–977. [Google Scholar] [CrossRef] [PubMed]
- Lindgren, K.E.; Hreinsson, J.; Helmestam, M.; Wanggren, K.; Poromaa, I.S.; Karehed, K.; Akerud, H. Histidine-rich glycoprotein derived peptides affect endometrial angiogenesis in vitro but has no effect on embryo development. Syst. Biol. Reprod. Med. 2016, 62, 192–200. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xie, Z.; Wang, Y.; Hu, H. Macrophage M1/M2 polarization in patients with pregnancy-induced hypertension. Can. J. Physiol. Pharmacol. 2018, 96, 922–928. [Google Scholar] [CrossRef]
- Cornelius, D.C.; Cottrell, J.; Amaral, L.M.; LaMarca, B. Inflammatory mediators: A causal link to hypertension during preeclampsia. Br. J. Pharmacol. 2019, 176, 1914–1921. [Google Scholar] [CrossRef]
- Nunes, P.R.; Romao-Veiga, M.; Peracoli, J.C.; Araujo Costa, R.A.; de Oliveira, L.G.; Borges, V.T.M.; Peracoli, M.T. Downregulation of CD163 in monocytes and its soluble form in the plasma is associated with a pro-inflammatory profile in pregnant women with preeclampsia. Immunol. Res. 2019, 67, 194–201. [Google Scholar] [CrossRef]
- Co, E.C.; Gormley, M.; Kapidzic, M.; Rosen, D.B.; Scott, M.A.; Stolp, H.A.; McMaster, M.; Lanier, L.L.; Barcena, A.; Fisher, S.J. Maternal decidual macrophages inhibit NK cell killing of invasive cytotrophoblasts during human pregnancy. Biol. Reprod. 2013, 88, 155. [Google Scholar] [CrossRef]
- Ruytinx, P.; Proost, P.; Van Damme, J.; Struyf, S. Chemokine-Induced Macrophage Polarization in Inflammatory Conditions. Front. Immunol. 2018, 9, 1930. [Google Scholar] [CrossRef] [Green Version]
- Svensson, J.; Jenmalm, M.C.; Matussek, A.; Geffers, R.; Berg, G.; Ernerudh, J. Macrophages at the fetal-maternal interface express markers of alternative activation and are induced by M-CSF and IL-10. J. Immunol. 2011, 187, 3671–3682. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Liang, H.; Zen, K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tagliani, E.; Erlebacher, A. Dendritic cell function at the maternal-fetal interface. Expert Rev. Clin. Immunol. 2011, 7, 593–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iberg, C.A.; Jones, A.; Hawiger, D. Dendritic Cells as Inducers of Peripheral Tolerance. Trends Immunol. 2017, 38, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.G.; Liu, X.; Link, H. Antigen-specific T cell functions are suppressed over the estrogen-dendritic cell-indoleamine 2,3-dioxygenase axis. Steroids 2004, 69, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Rozman, P.; Svajger, U. The tolerogenic role of IFN-gamma. Cytokine Growth Factor Rev. 2018, 41, 40–53. [Google Scholar] [CrossRef]
- Mezouar, S.; Mege, J.L. Changing the paradigm of IFN-gamma at the interface between innate and adaptive immunity: Macrophage-derived IFN-gamma. J. Leukoc. Biol. 2020, 108, 419–426. [Google Scholar] [CrossRef]
- Sayama, S.; Nagamatsu, T.; Schust, D.J.; Itaoka, N.; Ichikawa, M.; Kawana, K.; Yamashita, T.; Kozuma, S.; Fujii, T. Human decidual macrophages suppress IFN-gamma production by T cells through costimulatory B7-H1:PD-1 signaling in early pregnancy. J. Reprod. Immunol. 2013, 100, 109–117. [Google Scholar] [CrossRef]
- Svajger, U.; Rozman, P. Induction of Tolerogenic Dendritic Cells by Endogenous Biomolecules: An Update. Front. Immunol. 2018, 9, 2482. [Google Scholar] [CrossRef] [Green Version]
- Krey, G.; Frank, P.; Shaikly, V.; Barrientos, G.; Cordo-Russo, R.; Ringel, F.; Moschansky, P.; Chernukhin, I.V.; Metodiev, M.; Fernandez, N.; et al. In vivo dendritic cell depletion reduces breeding efficiency, affecting implantation and early placental development in mice. J. Mol. Med. 2008, 86, 999–1011. [Google Scholar] [CrossRef]
- Plaks, V.; Birnberg, T.; Berkutzki, T.; Sela, S.; BenYashar, A.; Kalchenko, V.; Mor, G.; Keshet, E.; Dekel, N.; Neeman, M.; et al. Uterine DCs are crucial for decidua formation during embryo implantation in mice. J. Clin. Invest. 2008, 118, 3954–3965. [Google Scholar] [CrossRef] [Green Version]
- Karsten, C.M.; Behrends, J.; Wagner, A.K.; Fuchs, F.; Figge, J.; Schmudde, I.; Hellberg, L.; Kruse, A. DC within the pregnant mouse uterus influence growth and functional properties of uterine NK cells. Eur. J. Immunol. 2009, 39, 2203–2214. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.; Santner-Nanan, B.; Dahlstrom, J.E.; Fadia, M.; Chandra, A.; Peek, M.; Nanan, R. Altered decidual DC-SIGN+ antigen-presenting cells and impaired regulatory T-cell induction in preeclampsia. Am. J. Pathol. 2012, 181, 2149–2160. [Google Scholar] [CrossRef] [PubMed]
- Kishuku, M.; Nishioka, Y.; Abe, S.; Kishi, J.; Ogino, H.; Aono, Y.; Azuma, M.; Kinoshita, K.; Batmunkh, R.; Makino, H.; et al. Expression of soluble vascular endothelial growth factor receptor-1 in human monocyte-derived mature dendritic cells contributes to their antiangiogenic property. J. Immunol. 2009, 183, 8176–8185. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Huang, L.; Wang, S.; Zhang, Z. The prevalence of regulatory T and dendritic cells is altered in peripheral blood of women with pre-eclampsia. Pregnancy Hypertens. 2019, 17, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.L.; Liang, Y.C.; Chiang, B.L. Placental growth factor down-regulates type 1 T helper immune response by modulating the function of dendritic cells. J. Leukoc. Biol. 2007, 82, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Dikov, M.M.; Ohm, J.E.; Ray, N.; Tchekneva, E.E.; Burlison, J.; Moghanaki, D.; Nadaf, S.; Carbone, D.P. Differential roles of vascular endothelial growth factor receptors 1 and 2 in dendritic cell differentiation. J. Immunol. 2005, 174, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Chen, H.L.; Girgis, K.R.; Cunningham, H.T.; Meny, G.M.; Nadaf, S.; Kavanaugh, D.; Carbone, D.P. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 1996, 2, 1096–1103. [Google Scholar] [CrossRef]
- Laurent, J.; Hull, E.F.; Touvrey, C.; Kuonen, F.; Lan, Q.; Lorusso, G.; Doucey, M.A.; Ciarloni, L.; Imaizumi, N.; Alghisi, G.C.; et al. Proangiogenic factor PlGF programs CD11b(+) myelomonocytes in breast cancer during differentiation of their hematopoietic progenitors. Cancer Res. 2011, 71, 3781–3791. [Google Scholar] [CrossRef] [Green Version]
- Drummond, G.R.; Vinh, A.; Guzik, T.J.; Sobey, C.G. Immune mechanisms of hypertension. Nat. Rev. Immunol. 2019, 19, 517–532. [Google Scholar] [CrossRef]
- Harrison, D.G. The immune system in hypertension. Trans. Am. Clin. Climatol. Assoc. 2014, 125, 130–138, discussion 130–138. [Google Scholar]
- Laresgoiti-Servitje, E. A leading role for the immune system in the pathophysiology of preeclampsia. J. Leukoc. Biol. 2013, 94, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Mikolajczyk, T.P.; Guzik, T.J. Adaptive Immunity in Hypertension. Curr. Hypertens. Rep. 2019, 21, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Iturbe, B.; Pons, H.; Johnson, R.J. Role of the Immune System in Hypertension. Physiol. Rev. 2017, 97, 1127–1164. [Google Scholar] [CrossRef] [PubMed]
- Trott, D.W.; Thabet, S.R.; Kirabo, A.; Saleh, M.A.; Itani, H.; Norlander, A.E.; Wu, J.; Goldstein, A.; Arendshorst, W.J.; Madhur, M.S.; et al. Oligoclonal CD8+ T cells play a critical role in the development of hypertension. Hypertension 2014, 64, 1108–1115. [Google Scholar] [CrossRef] [Green Version]
- Vinh, A.; Chen, W.; Blinder, Y.; Weiss, D.; Taylor, W.R.; Goronzy, J.J.; Weyand, C.M.; Harrison, D.G.; Guzik, T.J. Inhibition and genetic ablation of the B7/CD28 T-cell costimulation axis prevents experimental hypertension. Circulation 2010, 122, 2529–2537. [Google Scholar] [CrossRef]
- Crowley, S.D.; Song, Y.S.; Lin, E.E.; Griffiths, R.; Kim, H.S.; Ruiz, P. Lymphocyte responses exacerbate angiotensin II-dependent hypertension. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1089–R1097. [Google Scholar] [CrossRef]
- Guzik, T.J.; Hoch, N.E.; Brown, K.A.; McCann, L.A.; Rahman, A.; Dikalov, S.; Goronzy, J.; Weyand, C.; Harrison, D.G. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J. Exp. Med. 2007, 204, 2449–2460. [Google Scholar] [CrossRef]
- Chatterjee, P.; Chiasson, V.L.; Kopriva, S.E.; Young, K.J.; Chatterjee, V.; Jones, K.A.; Mitchell, B.M. Interleukin 10 deficiency exacerbates toll-like receptor 3-induced preeclampsia-like symptoms in mice. Hypertension 2011, 58, 489–496. [Google Scholar] [CrossRef] [Green Version]
- Tinsley, J.H.; South, S.; Chiasson, V.L.; Mitchell, B.M. Interleukin-10 reduces inflammation, endothelial dysfunction, and blood pressure in hypertensive pregnant rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R713–R719. [Google Scholar] [CrossRef]
- Loperena, R.; Van Beusecum, J.P.; Itani, H.A.; Engel, N.; Laroumanie, F.; Xiao, L.; Elijovich, F.; Laffer, C.L.; Gnecco, J.S.; Noonan, J.; et al. Hypertension and increased endothelial mechanical stretch promote monocyte differentiation and activation: Roles of STAT3, interleukin 6 and hydrogen peroxide. Cardiovasc. Res. 2018, 114, 1547–1563. [Google Scholar] [CrossRef]
- Sharma, A.; Satyam, A.; Sharma, J.B. Leptin, IL-10 and inflammatory markers (TNF-alpha, IL-6 and IL-8) in pre-eclamptic, normotensive pregnant and healthy non-pregnant women. Am. J. Reprod. Immunol. 2007, 58, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Osol, G.; Celia, G.; Gokina, N.; Barron, C.; Chien, E.; Mandala, M.; Luksha, L.; Kublickiene, K. Placental growth factor is a potent vasodilator of rat and human resistance arteries. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1381–H1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, L.; Vergouwe, Y.; van den Meiracker, A.H.; Verdonk, K.; Russcher, H.; Bremer, H.A.; Versendaal, H.J.; Steegers, E.A.P.; Danser, A.H.J.; Visser, W. Angiogenic Markers Predict Pregnancy Complications and Prolongation in Preeclampsia: Continuous Versus Cutoff Values. Hypertension 2017, 70, 1025–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinoza, J.; Betancourt, A.; Belfort, M.A.; Shamshirsaz, A.A.; Fox, K.A.; Yallampalli, C. Placental growth factor blunts uterine artery responses to angiotensin II. BJOG 2019, 126, 1058–1064. [Google Scholar] [CrossRef]
- Carnevale, D.; Pallante, F.; Fardella, V.; Fardella, S.; Iacobucci, R.; Federici, M.; Cifelli, G.; De Lucia, M.; Lembo, G. The angiogenic factor PlGF mediates a neuroimmune interaction in the spleen to allow the onset of hypertension. Immunity 2014, 41, 737–752. [Google Scholar] [CrossRef] [Green Version]
- Phipps, E.A.; Thadhani, R.; Benzing, T.; Karumanchi, S.A. Pre-eclampsia: Pathogenesis, novel diagnostics and therapies. Nat. Rev. Nephrol. 2019, 15, 275–289. [Google Scholar] [CrossRef]
- Rana, S.; Lemoine, E.; Granger, J.P.; Karumanchi, S.A. Preeclampsia: Pathophysiology, Challenges, and Perspectives. Circ. Res. 2019, 124, 1094–1112. [Google Scholar] [CrossRef]
- Steegers, E.A.; von Dadelszen, P.; Duvekot, J.J.; Pijnenborg, R. Pre-eclampsia. Lancet 2010, 376, 631–644. [Google Scholar] [CrossRef]
- Carnevale, D.; Lembo, G. PlGF, immune system and hypertension. Oncotarget 2015, 6, 18246–18247. [Google Scholar] [CrossRef]
- Magee, L.A.; Khalil, A.; Kametas, N.; von Dadelszen, P. Toward personalized management of chronic hypertension in pregnancy. Am. J. Obstet. Gynecol. 2020, in press. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albonici, L.; Benvenuto, M.; Focaccetti, C.; Cifaldi, L.; Miele, M.T.; Limana, F.; Manzari, V.; Bei, R. PlGF Immunological Impact during Pregnancy. Int. J. Mol. Sci. 2020, 21, 8714. https://doi.org/10.3390/ijms21228714
Albonici L, Benvenuto M, Focaccetti C, Cifaldi L, Miele MT, Limana F, Manzari V, Bei R. PlGF Immunological Impact during Pregnancy. International Journal of Molecular Sciences. 2020; 21(22):8714. https://doi.org/10.3390/ijms21228714
Chicago/Turabian StyleAlbonici, Loredana, Monica Benvenuto, Chiara Focaccetti, Loredana Cifaldi, Martino Tony Miele, Federica Limana, Vittorio Manzari, and Roberto Bei. 2020. "PlGF Immunological Impact during Pregnancy" International Journal of Molecular Sciences 21, no. 22: 8714. https://doi.org/10.3390/ijms21228714
APA StyleAlbonici, L., Benvenuto, M., Focaccetti, C., Cifaldi, L., Miele, M. T., Limana, F., Manzari, V., & Bei, R. (2020). PlGF Immunological Impact during Pregnancy. International Journal of Molecular Sciences, 21(22), 8714. https://doi.org/10.3390/ijms21228714