AGE-RAGE Axis Stimulates Oxidized LDL Uptake into Macrophages through Cyclin-Dependent Kinase 5-CD36 Pathway via Oxidative Stress Generation

 ,
,  , , , , and
, , , , and 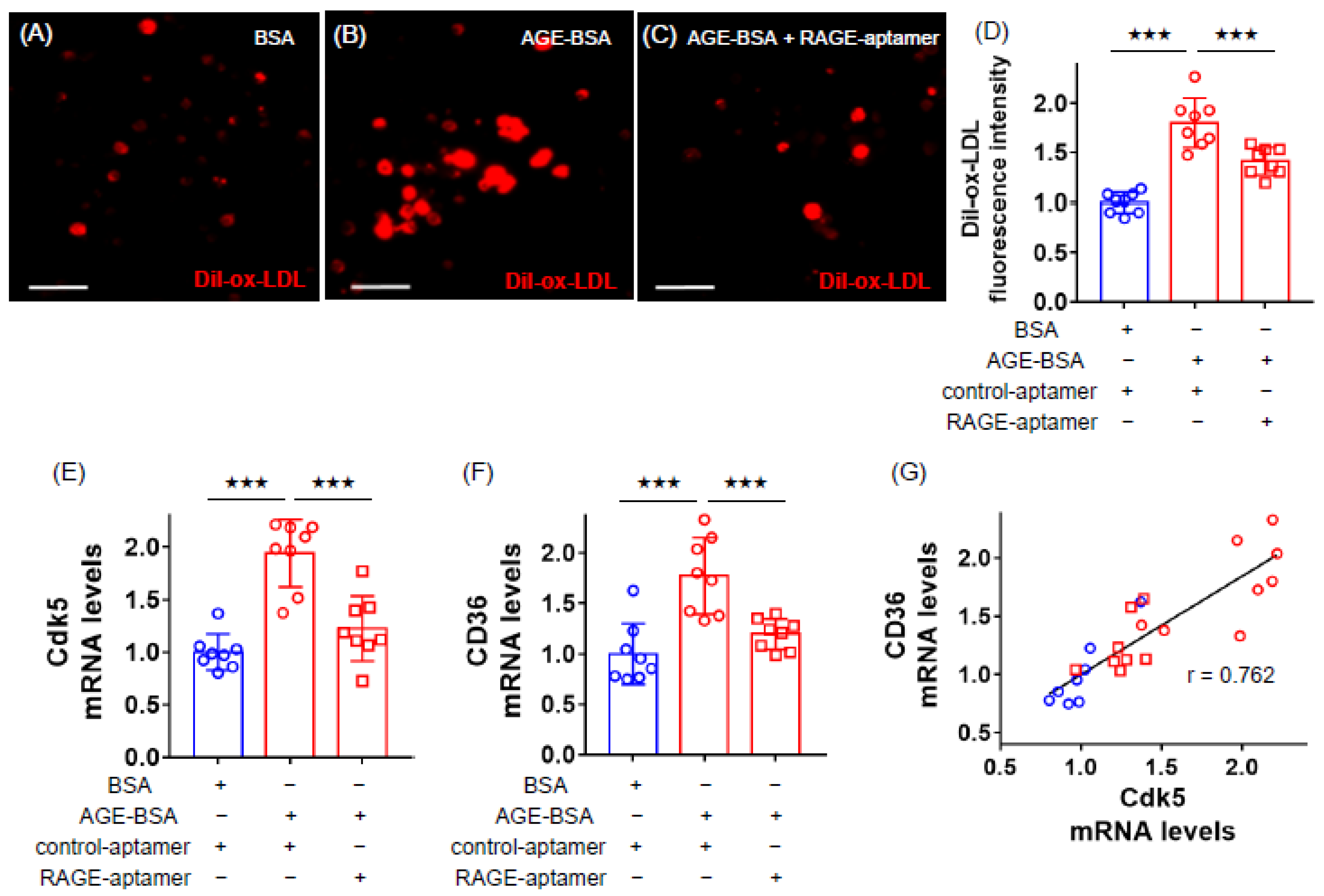{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. RAGE-Aptamer Inhibited the AGE-Induced Dil-ox-LDL Uptake, and Cdk5 and CD36 Gene Expression in U937 Cells
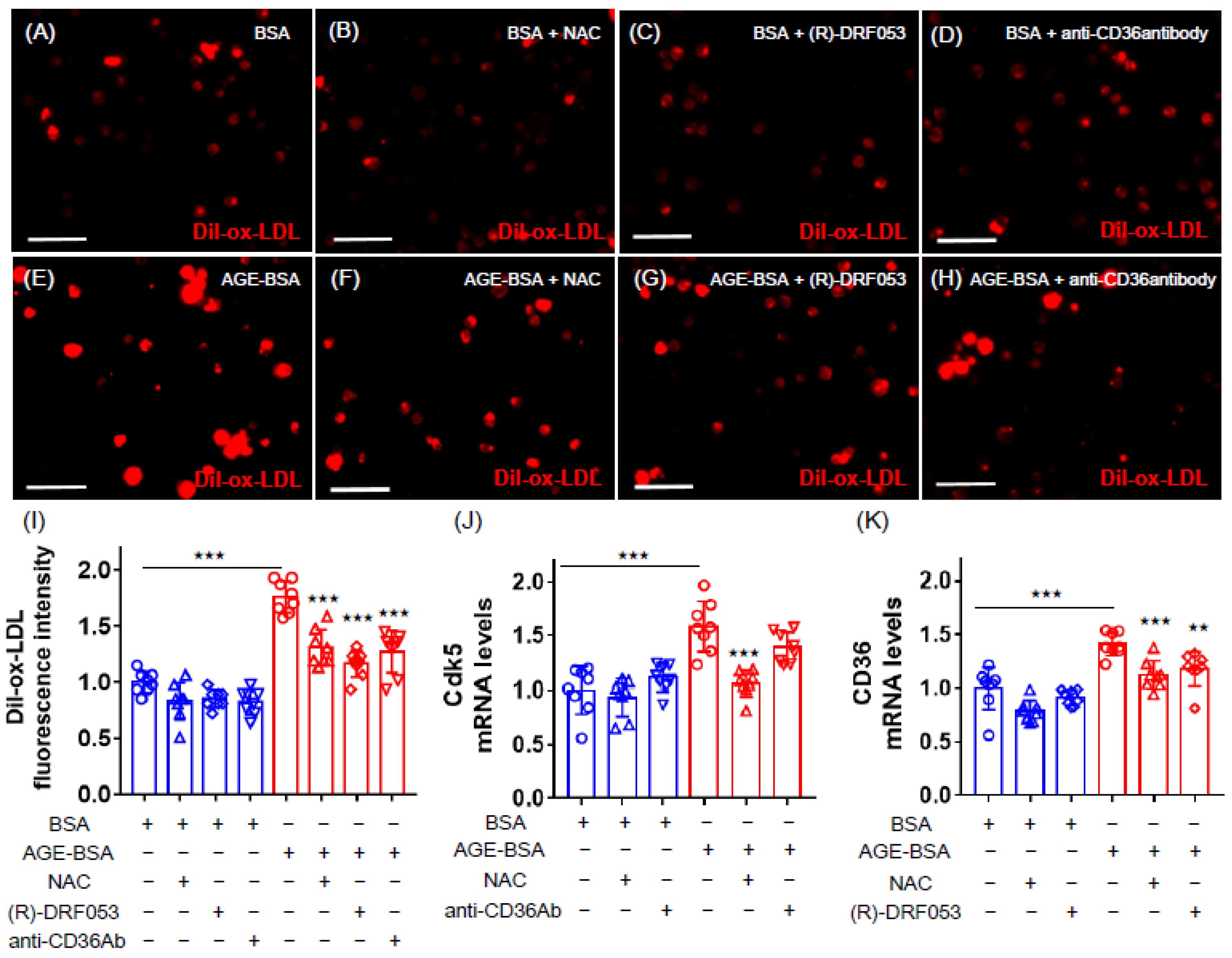
2.2. Effects of NAC, (R)-DRF053 and Anti-CD36 Antibody on AGE-Exposed U937 Cells
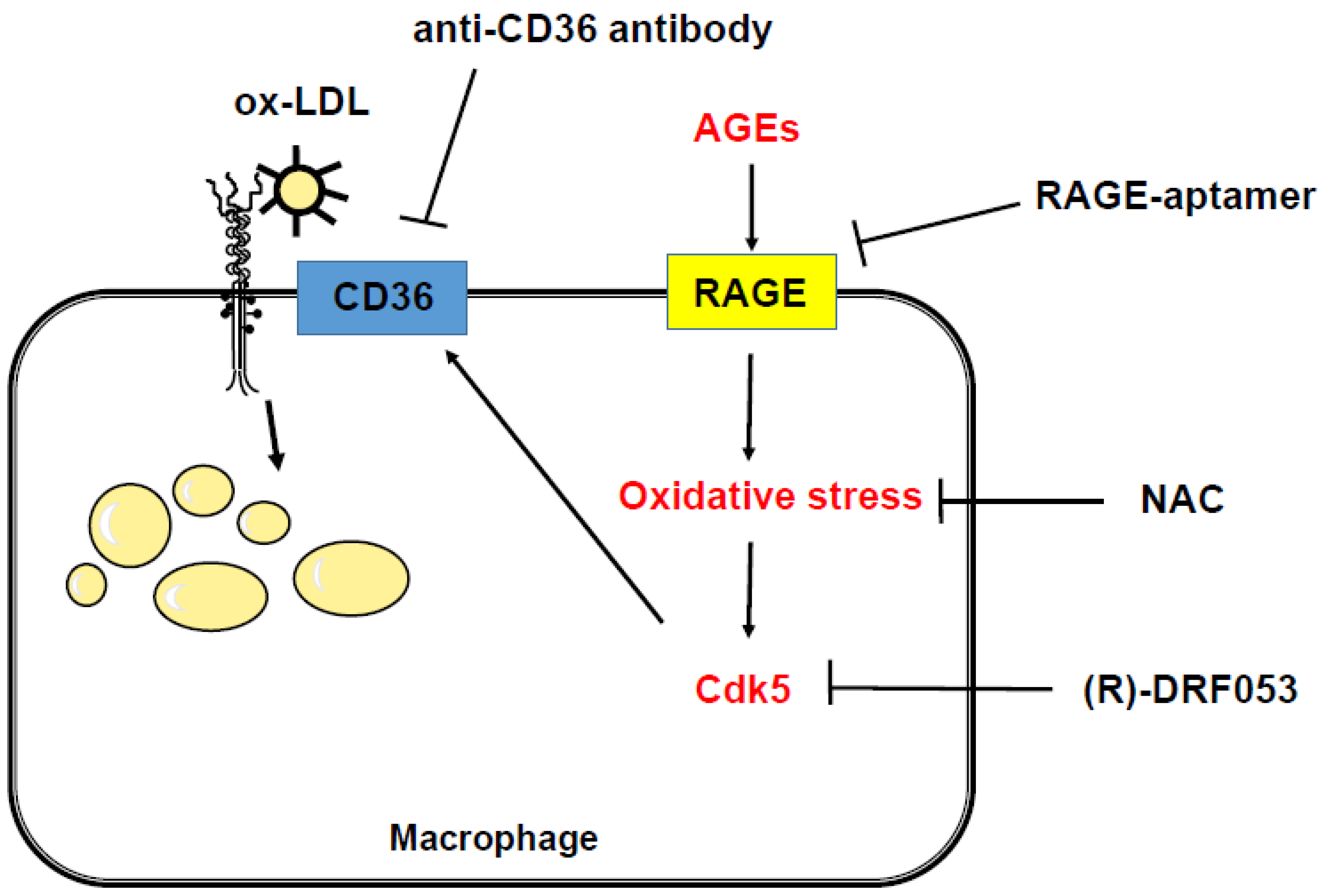
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Preparation of AGE-BSA
4.3. Preparation and Selection of RAGE-Aptamer
4.4. Experiments of U937 Macrophages
4.5. Uptake of Dil-ox-LDL into Macrophages
4.6. Gene Expression Levels
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Disclosure
References
- Rao Kondapally Seshasai, S.; Kaptoge, S.; Thompson, A.; Di Angelantonio, E.; Gao, P.; Sarwar, N.; Whincup, P.H.; Mukamal, K.J.; Gillum, R.F.; Holme, I.; et al. Diabetes mellitus, fasting glucose, and risk of cause-specific death. N. Engl. J. Med. 2011, 364, 829–841. [Google Scholar] [PubMed] [Green Version]
- Yamagishi, S.; Nakamura, N.; Suematsu, M.; Kaseda, K.; Matsui, T. Advanced glycation end products: A molecular target for vascular complications in diabetes. Mol. Med. 2015, 21, S32–S40. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Witztum, J.L. Atherosclerosis: The road ahead. Cell 2001, 104, 503–516. [Google Scholar] [CrossRef] [Green Version]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced glycation end products: Sparking the development of diabetic vascular injury. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef] [Green Version]
- Kilhovd, B.K.; Juutilainen, A.; Lehto, S.; Rönnemaa, T.; Torjesen, P.A.; Hanssen, K.F.; Laakso, M. Increased serum levels of advanced glycation endproducts predict total, cardiovascular and coronary mortality in women with type 2 diabetes: A population-based 18 year follow-up study. Diabetologia 2007, 50, 1409–1417. [Google Scholar] [CrossRef] [Green Version]
- Kilhovd, B.K.; Juutilainen, A.; Lehto, S.; Rönnemaa, T.; Torjesen, P.A.; Hanssen, K.F.; Laakso, M. Increased serum levels of methylglyoxal-derived hydroimidazolone-AGE are associated with increased cardiovascular disease mortality in nondiabetic women. Atherosclerosis 2009, 205, 590–594. [Google Scholar] [CrossRef]
- Yamagishi, S.; Fukami, K.; Matsui, T. Evaluation of tissue accumulation levels of advanced glycation end products by skin autofluorescence: A novel marker of vascular complications in high-risk patients for cardiovascular disease. Int. J. Cardiol. 2015, 185, 263–268. [Google Scholar] [CrossRef]
- Yamagishi, S.; Maeda, S.; Matsui, T.; Ueda, S.; Fukami, K.; Okuda, S. Role of advanced glycation end products (AGEs) and oxidative stress in vascular complications in diabetes. Biochim. Biophys. Acta 2012, 1820, 663–671. [Google Scholar] [CrossRef]
- Yamagishi, S. Role of advanced glycation end product (AGE)-receptor for advanced glycation end product (RAGE) axis in cardiovascular disease and its therapeutic intervention. Circ. J. 2019, 83, 1822–1828. [Google Scholar] [CrossRef] [Green Version]
- Kume, S.; Takeya, M.; Mori, T.; Araki, N.; Suzuki, H.; Horiuchi, S.; Kodama, T.; Miyauchi, Y.; Takahashi, K. Immunohistochemical and ultrastructural detection of advanced glycation end products in atherosclerotic lesions of human aorta with a novel specific monoclonal antibody. Am. J. Pathol. 1995, 147, 654–667. [Google Scholar] [PubMed]
- Wang, Z.Q.; Jing, L.L.; Yan, J.C.; Sun, Z.; Bao, Z.Y.; Shao, C.; Pang, Q.W.; Geng, Y.; Zhang, L.L.; Li, L.H. Role of AGEs in the progression and regression of atherosclerotic plaques. Glycoconj. J. 2018, 35, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Hassen, N.M.; Wouters, K.; Hujiberts, M.S.; Gijbels, M.J.; Sluimer, J.C.; Scheijen, J.L.; Heeneman, S.; Biessen, E.A.; Daemen, M.J.; Brownlee, M.; et al. Higher levels of advanced glycation endproducts in human carotid atherosclerotic plaques are associated with a repture-prone phenotype. Eur. Heart J. 2014, 35, 1137–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Ma, W.; Zhu, Y.; Sun, X.; Liu, N. Advanced glycation end products enhance macrophage polarization to the Ma phenotype via the HIF-1α/PDK4 pathway. Mol. Cell. Endocrinol. 2020, 514, 110878. [Google Scholar] [CrossRef] [PubMed]
- Bijnen, M.; Beelen, N.; Wetzels, S.; Gaar, J.V.; Vroomen, M.; Wijnands, E.; Scheijen, J.L.; van de Waarenburg, M.P.H.; Gijbels, M.J.; Cleutjens, J.P.; et al. RAGE deficiency dose not affect non-alcholic steatohepatitis and atherosclerosis in Western type diet-fed Ldlr(−/−) mice. Sci. Rep. 2018, 8, 15256. [Google Scholar] [CrossRef]
- Matsui, T.; Higashimoto, Y.; Nishino, Y.; Nakamura, N.; Fukami, K.; Yamagishi, S. RAGE-aptamer blocks the development and progression of experimental diabetic nephropathy. Diabetes 2017, 66, 1683–1695. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, K.; Yamagishi, S.; Yokoro, M.; Ito, S.; Kodama, G.; Kaida, Y.; Nakayama, Y.; Ando, R.; Yamada-Obara, N.; Asanuma, K.; et al. RAGE-aptamer attenuates deoxycorticosterone acetate/salt-induced renal injury in mice. Sci. Rep. 2018, 8, 2686. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, N.; Matsui, T.; Ishibashi, Y.; Sotokawauchi, A.; Fukami, K.; Higashimoto, Y.; Yamagishi, S. RAGE-aptamer attenuates the growth and liver metastasis of malignant Melanoma in nude mice. Mol. Med. 2017, 23, 295–306. [Google Scholar] [CrossRef]
- Nakamura, N.; Matsui, T.; Nishino, Y.; Sotokawauchi, A.; Higashimoto, Y.; Yamagishi, S. Long-term local injection of RAGE-aptamer suppresses the growth of malignant melanoma in nude mice. J. Oncol. 2019, 2019, 7387601. [Google Scholar] [CrossRef]
- Allahverdian, S.; Pannu, P.S.; Francis, G.A. Contribution of monocyte-derived macrophages and smooth muscle cells to arterial foam cell formation. Cardiovasc. Res. 2012, 95, 165–172. [Google Scholar] [CrossRef] [Green Version]
- Terasaki, M.; Hiromura, M.; Mori, Y.; Kohashi, K.; Kushima, H.; Koshibu, M.; Saito, T.; Yashima, H.; Watanabe, T.; Hirano, T. A dipeptidyl peptidase-4 inhibitor suppresses macrophage foam cell formation in diabetic db/db mice and type 2 diabetes patients. Int. J. Endocrinol. 2018, 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terasaki, M.; Hiromura, M.; Mori, Y.; Kohashi, K.; Nagashima, M.; Kushima, H.; Watanabe, T.; Hirano, T. Amelioration of hyperglycemia with a sodium-glucose cotransporter 2 inhibitor prevents macrophage-driven atherosclerosis through macrophage foam cell formation suppression in type 1 and type 2 diabetic mice. PLoS ONE 2015, 10, e0143396. [Google Scholar] [CrossRef] [PubMed]
- Terasaki, M.; Nagashima, M.; Nohtomi, K.; Kohashi, K.; Tomoyasu, M.; Sinmura, K.; Nogi, Y.; Katayama, Y.; Sato, K.; Itoh, F.; et al. Preventive effect of dipeptidyl peptidase-4 inhibitor on atherosclerosis is mainly attributable to incretin’s actions in nondiabetic and diabetic apolipoprotein E-null mice. PLoS ONE 2013, 8, e70933. [Google Scholar] [CrossRef]
- Fukuhara-Takaki, K.; Sakai, M.; Sakamoto, Y.; Takeya, M.; Horiuchi, S. Expression of class A scavenger receptor is enhanced by high glucose in vitro and under diabetic conditions in vivo: One mechanism for an increased rate of atherosclerosis in diabetes. J. Biol. Chem. 2005, 280, 3355–3364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Sawamura, T.; Renier, G. Glucose enhances human macrophage LOX-1 expression: Role for LOX-1 in glucose-induced macrophage foam cell formation. Circ. Res. 2004, 94, 892–901. [Google Scholar] [CrossRef]
- Terasaki, M.; Yashima, H.; Mori, Y.; Saito, T.; Matsui, T.; Hiromura, M.; Kushima, H.; Osaka, N.; Ohara, M.; Fukui, T.; et al. A dipeptidyl peptidase-4 inhibitor inhibits foam cell formation of macrophages in type 1 diabetes via suppression of CD36 expression. Int. J. Mol. Sci. 2020, 21, 4811. [Google Scholar] [CrossRef]
- Ingham, M.; Schwartz, G.K. Cell-cycle therapeutics come of age. J. Clin. Oncol. 2017, 35, 2949–2959. [Google Scholar] [CrossRef]
- Cheng, W.; Yang, Z.; Wang, S.; Li, Y.; Wei, H.; Tian, X.; Kan, Q. Recent development of CDK inhibitors: An overview of CDK/inhibitor co-crystal structures. Eur. J. Med. Chem. 2019, 164, 615–639. [Google Scholar] [CrossRef]
- Bai, B.; Liang, Y.; Xu, C.; Lee, M.Y.; Xu, A.; Wu, D.; Vanhoutte, P.M.; Wang, Y. Cuclin-dependent kinase 5-mediated hyperphosphorylation of sirtuin-1 contributes to the development of endothelial senescence and atherosclerosis. Circulation 2012, 126, 729–740. [Google Scholar] [CrossRef] [Green Version]
- Na, Y.R.; Jung, D.; Gu, G.J.; Jang, A.R.; Suh, Y.H.; Seok, S.H. The early synthesis of p35 and activation of CDK5 in LPS-stimulated macrophages suppresses interleukin-10 production. Sci. Signal. 2015, 8, ra121. [Google Scholar] [CrossRef]
- Ahmed, D.; Sharma, M. cyclin-dependent kinase 5/p35/p39: A novel and imminent therapeutic target for diabetes mellitus. Int. J. Endocrinol. 2011, 2011, 530274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roufayel, R.; Murshid, N. CDK5: Key regulation of atherosclerosis and cell survival. Biomedicines 2019, 7, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellmich, M.R.; Pant, H.C.; Wada, E.; Battey, J.F. Neuronal cdc2-like kinase: A cdc2-related protein kinase with predominantly neuronal expression. Proc. Natl. Acad. Sci. USA 1992, 89, 10867–10871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhavan, R.; Tsai, L.H. A decade of CDK5. Nat. Rev. Mol. Cell Biol. 2001, 2, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Merk, H.; Zhang, S.; Lehr, T.; Muller, C.; Ulrich, M.; Bibb, J.A.; Adams, R.H.; Bracher, F.; Zahler, S.; Vollmar, A.M.; et al. Inhibition of endothelial Cdk5 reduces tumor growth by promoting non-productive angiogenesis. Oncotarget 2016, 7, 6088–6104. [Google Scholar] [CrossRef]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef]
- Ishibashi, Y.; Matsui, T.; Isami, F.; Abe, Y.; Sakaguchi, T.; Higashimoto, Y.; Yamagishi, S. N-butanol exacts of morinda citrifolia suppress advanced glycation end products (AGE)-induced inflammatory reactions in endothelial cells through its anti-oxidative properties. BMC Complement. Altern. Med. 2017, 17, 137. [Google Scholar] [CrossRef] [Green Version]
- Cepas, V.; Collono, M.; Mayo, J.C.; Sainz, R.M. Redox signaling and advanced glycation endproducts (AGEs) in diet-related disease. Antioxidants 2020, 9, 142. [Google Scholar] [CrossRef] [Green Version]
- Wautier, M.-P.; Chappey, O.; Corda, S.; Stern, D.M.; Schmidt, A.M.; Wautier, J.L. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E685–E694. [Google Scholar] [CrossRef]
- Matheny, H.E.; Deem, T.L.; Cook-Mills, J.M. Lymphocyte migration through monolayers of endothelial cell lines involves VCAM-1 signaling via endothelial cell NADPH oxidase. J. Immunol. 2000, 164, 6550–6559. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Kim, W.; Choi, J.H.; Kim, S.H.; Lee, D.; Park, C.H.; Kim, S.; Kim, D.Y.; Kim, K.T. Stress-induced nuclear translocation of CDK5 suppresses neuronal death by downregulating ERK activation via VRK3 phosphorylation. Sci. Rep. 2016, 6, 28634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shea, T.B.; Zheng, Y.L.; Ortiz, D.; Pant, H.C. Cyclin-dependent kinase 5 increases perikaryal neurofilament phosphorylation and inhibits neurofilaent axonal transport in response to oxidative stress. J. Neurosci. Res. 2004, 76, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Banks, A.S.; Estall, J.L.; Kajimura, S.; Bostrom, P.; Laznik, D.; Ruas, J.L.; Chalmers, M.J.; Kamenecka, T.M.; Bluher, M.; et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature 2010, 466, 451–456. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Fan, W.; Xu, J.; Lu, M.; Yamamoto, H.; Auwerx, J.; Sears, D.D.; Talukdar, S.; Oh, D.; Chen, A.; et al. Adiopocyte NCoR knockout decreases PPARγ phosphorylation and enhances PPARγ activity and insulin sensitivity. Cell 2011, 147, 815–826. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.; Han, J.; Pearce, S.F.; Silverstein, R.L.; Gotto, A.M.; Hajjar, D.P.; Nicholson, A.C. Induction of CD36 expression by oxidized LDL and IL-4 by a common signaling pathway dependent on protein kinase C and PPAR-gamma. J. Lipid Res. 2000, 41, 688–696. [Google Scholar] [PubMed]
- Nagy, L.; Tontonoz, P.; Alvarez, J.G.; Chen, H.; Evans, R.M. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell 1998, 93, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Kotla, S.; Rao, G.N. Reactive oxygen species (ROS) mediate p300-dependent STAT1 protein interaction with peroxisome proliferator-activated receptor (PPAR)-γ in CD36 protein expression and foam cell formation. J. Biol. Chem. 2015, 290, 30306–30320. [Google Scholar] [CrossRef] [Green Version]
- Leonarduzzi, G.; Gamba, P.; Gargiulo, S.; Sottero, B.; Kadl, A.; Biasi, F.; Chiarpotto, E.; Leitinger, N.; Vendemiale, G.; Serviddio, G.; et al. Oxidation as a crucial reaction for cholesterol to induce tissue degeneration: CD36 overexpression in human promonocytic cells treated with a biologically relevant oxysterol mixture. Aging Cell 2008, 7, 375–382. [Google Scholar] [CrossRef]
- Kanungo, J.; Zheng, Y.L.; Amin, N.D.; Kaur, S.; Ramchandran, R.; Pant, H.C. Specific inhibition of cyclin-dependent kinase 5 activity induces motor neuroc development in vivo. Biocham. Biophys. Res. Commun. 2009, 386, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, S.; Nakamura, K.; Matsui, T.; Inagaki, Y.; Takenaka, K.; Jinnouchi, Y.; Yoshida, Y.; Matsuura, T.; Narama, I.; Motomiya, Y.; et al. Pigment epithelium-derived factor inhibits advanced glycation end product-induced retinal vascular hyperpermeability by blocking reactive oxygen speciesmediated vascular endothelial growth factor expression. J. Biol. Chem. 2006, 281, 20213–20220. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, S.; Inagaki, Y.; Okamoto, T.; Amano, S.; Koga, K.; Takeuchi, M.; Makita, Z. Advanced glycation end product-induced apoptosis and overexpression of vascular endothelial growth factor and monocyte chemoattractant protein-1 in human-cultured mesangial cells. J. Biol. Chem. 2002, 277, 20309–20315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashimoto, Y.; Matsui, T.; Nishino, Y.; Taira, J.; Inoue, H.; Takeuchi, M.; Yamagishi, S. Blockade by phosphorothioate aptamers of advanced glycation end products-induced damage in cultured pericytes and endothelial cells. Microvasc. Res. 2013, 90, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Oda, E.; Higashimoto, Y.; Yamagishi, S. Glyceraldehyde-derived pyridinium (GLAP) evokes oxidative stress and inflammatory and thrombogenic reactions in endothelial cells via the interaction with RAGE. Cardiovasc. Diabetol. 2015, 14, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.G.; Ryoo, I.G.; Choi, H.Y.; Choi, B.H.; Kim, S.T.; Heo, T.H.; Lee, J.Y.; Park, P.H.; Kwak, M.K. NRF2 signaling negatively regulates phorbol-12-Myristate-13-acetate (PMA)-induced differentiation of human monocytic U937 cells into pro-inflammatory macrophages. PLoS ONE 2015, 10, e0134235. [Google Scholar] [CrossRef] [PubMed]
- Tusiimire, J.; Wallace, J.; Woods, N.; Dufton, M.J.; Parkinson, J.A.; Abbott, G.; Clements, C.J.; Young, L.; Park, J.K.; Jeon, J.W.; et al. Effect of bee venom and its fractions of pro-inflammatory cytokines in PMA-differentiated U937 cells co-stimulated with LPS. Vaccines 2016, 4, 11. [Google Scholar] [CrossRef] [Green Version]
- Hida, A.; Kawakami, A.; Nakashima, T.; Yamasaki, S.; Sakai, H.; Urayama, S.; Ida, H.; Nakamura, H.; Migita, K.; Kawabe, Y.; et al. Nuclear factor-kappaB and caspases co-operatively regulate the activation and apoptosis of human macrophages. Immunology 2000, 99, 553–560. [Google Scholar] [CrossRef]
- Whyte, J.; Roberts, A.D.; Morley, K.A.; Sharp, R.J.; Marsh, P.D. Phagocytosis of mycobacteria by U937 cells: A rapid method for monitoring uptake and separating phagocytosed and free bacteria by magnetic beads. Lett. Appl. Microbiol. 2000, 30, 90–94. [Google Scholar] [CrossRef]
- Terasaki, M.; Hiromura, M.; Mori, Y.; Kohashi, K.; Kushima, H.; Ohara, M.; Watanabe, T.; Andersson, O.; Hirano, T. Combination therapy with a sodium-glucose cotransporter 2 inhibitor and a dipeptidyl-peptidase-4 inhibitor additively suppresses macrophage foam cell formation and atherosclerosis in diabetic mice. Int. J. Endocrinol. 2017, 2017, 1365209. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yashima, H.; Terasaki, M.; Sotokawauchi, A.; Matsui, T.; Mori, Y.; Saito, T.; Osaka, N.; Kushima, H.; Hiromura, M.; Ohara, M.; et al. AGE-RAGE Axis Stimulates Oxidized LDL Uptake into Macrophages through Cyclin-Dependent Kinase 5-CD36 Pathway via Oxidative Stress Generation. Int. J. Mol. Sci. 2020, 21, 9263. https://doi.org/10.3390/ijms21239263
Yashima H, Terasaki M, Sotokawauchi A, Matsui T, Mori Y, Saito T, Osaka N, Kushima H, Hiromura M, Ohara M, et al. AGE-RAGE Axis Stimulates Oxidized LDL Uptake into Macrophages through Cyclin-Dependent Kinase 5-CD36 Pathway via Oxidative Stress Generation. International Journal of Molecular Sciences. 2020; 21(23):9263. https://doi.org/10.3390/ijms21239263
Chicago/Turabian StyleYashima, Hironori, Michishige Terasaki, Ami Sotokawauchi, Takanori Matsui, Yusaku Mori, Tomomi Saito, Naoya Osaka, Hideki Kushima, Munenori Hiromura, Makoto Ohara, and et al. 2020. "AGE-RAGE Axis Stimulates Oxidized LDL Uptake into Macrophages through Cyclin-Dependent Kinase 5-CD36 Pathway via Oxidative Stress Generation" International Journal of Molecular Sciences 21, no. 23: 9263. https://doi.org/10.3390/ijms21239263
APA StyleYashima, H., Terasaki, M., Sotokawauchi, A., Matsui, T., Mori, Y., Saito, T., Osaka, N., Kushima, H., Hiromura, M., Ohara, M., Fukui, T., & Yamagishi, S. -i. (2020). AGE-RAGE Axis Stimulates Oxidized LDL Uptake into Macrophages through Cyclin-Dependent Kinase 5-CD36 Pathway via Oxidative Stress Generation. International Journal of Molecular Sciences, 21(23), 9263. https://doi.org/10.3390/ijms21239263