Self-Assembly of pH-Labile Polymer Nanoparticles for Paclitaxel Prodrug Delivery: Formulation, Characterization, and Evaluation


Abstract
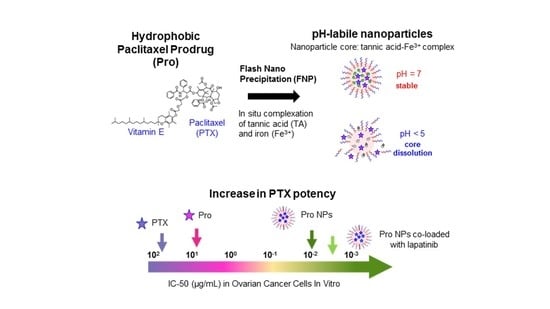
:
1. Introduction
2. Results and Discussion
2.1. Prodrug Formulation with Enhanced Potency In Vitro
2.2. Formulation of Prodrug Nanoparticles
2.3. Nanoparticle Encapsulation Efficiency and Drug Loading
2.4. Drug Release
2.5. Prodrug Nanoparticle Potency and Evaluating the Cell Cycle Distribution
2.6. Synergy of Drug Combination with the Paclitaxel Prodrug
3. Materials and Methods
3.1. Materials
3.2. Cell Culture
3.3. Prodrug Formulation
3.4. Nanoparticle Formulation
3.5. Nanoparticle Characterization
3.6. In Vitro Nanoparticle Drug Release
3.7. Cell Viability and Half Maximal Inhibitory Concentration
3.8. Cell Cycle Analysis by Flow Cytometry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Mph, K.D.M.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef]
- Ueno, N.T.; Mamounas, E.P. Neoadjuvant nab-paclitaxel in the treatment of breast cancer. Breast Cancer Res. Treat. 2016, 156, 427–440. [Google Scholar] [CrossRef] [Green Version]
- Sridhar, S.S.; Hotte, S.J.; Chin, J.L.; Hudes, G.R.; Gregg, R.; Trachtenberg, J.; Wang, L.; Tran-Thanh, D.; Pham, N.-A.; Tsao, M.-S.; et al. A Multicenter Phase II Clinical Trial of Lapatinib (GW572016) in Hormonally Untreated Advanced Prostate Cancer. Am. J. Clin. Oncol. 2010, 33, 609–613. [Google Scholar] [CrossRef]
- Varma, M.V.; Panchagnula, R. Enhanced oral paclitaxel absorption with vitamin E-TPGS: Effect on solubility and permeability in vitro, in situ and in vivo. Eur. J. Pharm. Sci. 2005, 25, 445–453. [Google Scholar] [CrossRef]
- Sohn, J.S.; Jin, J.I.; Hess, M.; Jo, B.W. Polymer prodrug approaches applied to paclitaxel. Polym. Chem. 2010, 1, 778–792. [Google Scholar] [CrossRef]
- Meng, Z.; Lv, Q.; Lu, J.; Yao, H.; Lv, X.; Jiang, F.; Lu, A.; Zhang, G. Prodrug Strategies for Paclitaxel. Int. J. Mol. Sci. 2016, 17, 796. [Google Scholar] [CrossRef] [Green Version]
- D’Addio, S.M.; Prud’Homme, R.K. Controlling drug nanoparticle formation by rapid precipitation. Adv. Drug Deliv. Rev. 2011, 63, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Pustulka, K.M.; Wohl, A.R.; Lee, H.S.; Michel, A.R.; Han, J.; Hoye, T.R.; McCormick, A.V.; Panyam, J.; Macosko, C.W. Flash Nanoprecipitation: Particle Structure and Stability. Mol. Pharm. 2013, 10, 4367–4377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Y.; Zhong, Y.; Meng, F.; Cheng, R.; Deng, C.; Zhong, Z. Acetal-Linked Paclitaxel Prodrug Micellar Nanoparticles as a Versatile and Potent Platform for Cancer Therapy. Biomacromolecules 2013, 14, 2772–2780. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.-S.; Liu, J.; Peng, T.-X.; Peng, H.; Guo, F.; Zhong, H.-J. Vitamin E-based redox-sensitive salinomycin prodrug-nanosystem with paclitaxel loaded for cancer targeted and combined chemotherapy. Colloids Surf. B Biointerfaces 2018, 172, 506–516. [Google Scholar] [CrossRef]
- Feng, S.-S.; Feng, S.-S. Enhanced Oral Bioavailability of Paclitaxel Formulated in Vitamin E-TPGS Emulsified Nanoparticles of Biodegradable Polymers: In Vitro and In Vivo Studies. J. Pharm. Sci. 2010, 99, 3552–3560. [Google Scholar] [CrossRef]
- Wohl, A.R.; Michel, A.R.; Kalscheuer, S.; Macosko, C.W.; Panyam, J.; Hoye, T.R. Silicate Esters of Paclitaxel and Docetaxel: Synthesis, Hydrophobicity, Hydrolytic Stability, Cytotoxicity, and Prodrug Potential. J. Med. Chem. 2014, 57, 2368–2379. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Johnstone, S.A.; Tardi, P.G.; Lo, L.; Xie, S.; Shu, Y.; Harasym, T.O.; Harasym, N.L.; Williams, L.; Bermudes, D.; et al. Modulating the Therapeutic Activity of Nanoparticle Delivered Paclitaxel by Manipulating the Hydrophobicity of Prodrug Conjugates. J. Med. Chem. 2008, 51, 3288–3296. [Google Scholar] [CrossRef] [PubMed]
- Levit, S.L.; Yang, H.; Tang, C. Rapid Self-Assembly of Polymer Nanoparticles for Synergistic Codelivery of Paclitaxel and Lapatinib via Flash NanoPrecipitation. Nanomaterials 2020, 10, 561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ndungu, J.M.; Lu, Y.J.; Zhu, S.; Yang, C.; Wang, X.; Chen, G.; Shin, D.M.; Snyder, J.P.; Shoji, M.; Sun, A. Targeted Delivery of Paclitaxel to Tumor Cells: Synthesis and in Vitro Evaluation. J. Med. Chem. 2010, 53, 3127–3132. [Google Scholar] [CrossRef] [Green Version]
- Shu, C.-H.; Yang, W.K.; Shih, Y.-L.; Kuo, M.-L.; Huang, T.-S. Cell cycle G2/M arrest and activation of cyclin-dependent kinases associated with low-dose paclitaxel-induced sub-G1 apoptosis. Apoptosis 1997, 2, 463–470. [Google Scholar] [CrossRef]
- Wang, T.H.; Wang, H.S.; Soong, Y.K. Paclitaxel-Induced Cell Death: Where the Cell Cycle and Apoptosis Come Together. Cancer 2000, 88, 2619–2628. [Google Scholar] [CrossRef]
- Hsiao, J.-R.; Leu, S.-F.; Huang, B.-M. Apoptotic mechanism of paclitaxel-induced cell death in human head and neck tumor cell lines. J. Oral Pathol. Med. 2009, 38, 188–197. [Google Scholar] [CrossRef]
- Li, F.; Danquah, M.; Singh, S.; Wu, H.; Mahato, R.I. Paclitaxel- and lapatinib-loaded lipopolymer micelles overcome multidrug resistance in prostate cancer. Drug Deliv. Transl. Res. 2011, 1, 420–428. [Google Scholar] [CrossRef]
- Barichello, J.M.; Morishita, M.; Takayama, K.; Nagai, T. Encapsulation of Hydrophilic and Lipophilic Drugs in PLGA Nanoparticles by the Nanoprecipitation Method. Drug Dev. Ind. Pharm. 1999, 25, 471–476. [Google Scholar] [CrossRef]
- Tang, C.; Prud’Homme, R.K.; Vauthier, C.; Ponchel, G. Targeted Theragnostic Nanoparticles Via Flash Nanoprecipitation: Principles of Material Selection. In Polymer Nanoparticles for Nanomedicines; Springer International Publishing: Cham, Switzerland, 2016; pp. 55–85. [Google Scholar]
- Tang, C.; Amin, D.; Messersmith, P.B.; Anthony, J.E.; Prud’Homme, R.K. Polymer Directed Self-Assembly of pH-Responsive Antioxidant Nanoparticles. Langmuir 2015, 31, 3612–3620. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Levit, S.; Zeevi, M.; Vasey, C.; Fromen, C. Chapter 12. Polymer Colloids Enable Medical Applications. In Polymer Colloids: Formation, Characterization, and Applications; Royal Society of Chemistry (RSC): Cambridge, UK, 2019; pp. 358–398. [Google Scholar]
- Couillaud, B.M.; Espeau, P.; Mignet, N.; Corvis, Y. State of the Art of Pharmaceutical Solid Forms: From Crystal Property Issues to Nanocrystals Formulation. ChemMedChem 2019, 14, 8–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z. Flash Nanoprecipitation: Prediction and Enhancement of Particle Stability via Drug Structure. Mol. Pharm. 2014, 11, 776–786. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.K.; Prud’Homme, R.K. Chemical processing and micromixing in confined impinging jets. AIChE J. 2003, 49, 2264–2282. [Google Scholar] [CrossRef]
- Saad, W.S.; Prud’Homme, R.K. Principles of nanoparticle formation by flash nanoprecipitation. Nano Today 2016, 11, 212–227. [Google Scholar] [CrossRef]
- Zhang, C.; Pansare, V.J.; Prud’Homme, R.K.; Priestley, R.D. Flash nanoprecipitation of polystyrenenanoparticles. Soft Matter 2012, 8, 86–93. [Google Scholar] [CrossRef]
- Kim, S.; Philippot, S.; Fontanay, S.; Duval, R.E.; Lamouroux, E.; Canilho, N.; Pasc, A. pH- and glutathione-responsive release of curcumin from mesoporous silica nanoparticles coated using tannic acid–Fe(iii) complex. RSC Adv. 2015, 5, 90550–90558. [Google Scholar] [CrossRef]
- Fu, Y.; Zhang, J.; Wang, H.; Chen, J.-L.; Zhao, P.; Chen, G.-R.; He, X.-P. Intracellular pH sensing and targeted imaging of lysosome by a galactosyl naphthalimide-piperazine probe. Dye Pigment 2016, 133, 372–379. [Google Scholar] [CrossRef]
- Davoust, J.; Gruenberg, J.; Howell, K.E. Two threshold values of low pH block endocytosis at different stages. EMBO J. 1987, 6, 3601–3609. [Google Scholar] [CrossRef]
- Swietach, P.; Vaughan-Jones, R.D.; Harris, A.L.; Hulikova, A. The chemistry, physiology and pathology of pH in cancer. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130099. [Google Scholar] [CrossRef] [Green Version]
- Morton, S.W.; Lee, M.J.; Deng, Z.J.; Dreaden, E.C.; Siouve, E.; Shopsowitz, K.E.; Shah, N.J.; Yaffe, M.B.; Hammond, P.T. A Nanoparticle-Based Combination Chemotherapy Delivery System for Enhanced Tumor Killing by Dynamic Rewiring of Signaling Pathways. Sci. Signal. 2014, 7, ra44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Michel, A.R.; Lee, H.S.; Kalscheuer, S.; Wohl, A.; Hoye, T.R.; McCormick, A.V.; Panyam, J.; Macosko, C.W. Nanoparticles Containing High Loads of Paclitaxel-Silicate Prodrugs: Formulation, Drug Release, and Anticancer Efficacy. Mol. Pharm. 2015, 12, 4329–4335. [Google Scholar] [CrossRef] [Green Version]
- Butt, A.M.; Pandey, M.; Katas, H.; Sarisuta, N.; Witoonsaridsilp, W.; Benjakul, R. In VitroCharacterization of Pluronic F127 and d-Tocopheryl Polyethylene Glycol 1000 Succinate Mixed Micelles as Nanocarriers for Targeted Anticancer-Drug Delivery. J. Nanomater. 2012, 2012, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Dash, S.; Murthy, P.N.; Nath, L.; Chowdhury, P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol. Pharm. Drug Res. 2010, 67, 217–223. [Google Scholar]
- Korsmeyer, R.W.; Gurny, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Wu, I.Y.; Bala, S.; Škalko-Basnet, N.; Di Cagno, M.P. Interpreting non-linear drug diffusion data: Utilizing Korsmeyer-Peppas model to study drug release from liposomes. Eur. J. Pharm. Sci. 2019, 138, 105026. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, D.T.; Seitaridou, E. Simple Brownian Diffusion: An Introduction to the Standard Theoreteical Models; Oxford University Press: New York, NY, USA, 2012. [Google Scholar]
- Elmas, A.; Akyüz, G.; Bergal, A.; Andac, M.; Andac, O. Mathematical Modelling of Drug Release. Res. Eng. Struct. Mater. 2020, 63–86. [Google Scholar] [CrossRef]
- Samaha, D.; Shehayeb, R.; Kyriacos, S. Modeling and Comparison of Dissolution Profiles of Diltiazem Modified-Release Formulations. Dissolution Technol. 2009, 16, 41–46. [Google Scholar] [CrossRef]
- D’Addio, S.M.; Bukari, A.A.; Dawoud, M.; Bunjes, H.; Rinaldi, C.; Prud’Homme, R.K. Determining drug release rates of hydrophobic compounds from nanocarriers. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2016, 374, 20150128. [Google Scholar] [CrossRef] [Green Version]
- Strasdat, B.; Bunjes, H. Development of a new approach to investigating the drug transfer from colloidal carrier systems applying lipid nanosuspension-containing alginate microbeads as acceptor. Int. J. Pharm. 2015, 489, 203–209. [Google Scholar] [CrossRef]
- Abouelmagd, S.A.; Sun, B.; Chang, A.C.; Ku, Y.J.; Yeo, Y. Release Kinetics Study of Poorly Water-Soluble Drugs from Nanoparticles: Are We Doing It Right? Mol. Pharm. 2015, 12, 997–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Graeser, R.; Kratz, F.; Geckeler, K.E. Paclitaxel-Loaded Polymer Nanoparticles for the Reversal of Multidrug Resistance in Breast Cancer Cells. Adv. Funct. Mater. 2011, 21, 4211–4218. [Google Scholar] [CrossRef]
- Fonseca, C.; Simões, S.; Gaspar, R. Paclitaxel-loaded PLGA nanoparticles: Preparation, physicochemical characterization and in vitro anti-tumoral activity. J. Control. Release 2002, 83, 273–286. [Google Scholar] [CrossRef] [Green Version]
- Swanson, J.; Biology, C.; Avenue, L.; Watts, C. Macropinocytosis. Trends Cell Biol. 1995, 5, 424–428. [Google Scholar] [CrossRef]
- Hirota, K.; Ter, H. Endocytosis of Particle Formulations by Macrophages and Its Application to Clinical Treatment. In Molecular Regulation of Endocytosis; IntechOpen: London, UK, 2012; pp. 1–16. [Google Scholar]
- Luo, C.; Sun, J.; Sun, B.; He, Z. Prodrug-based nanoparticulate drug delivery strategies for cancer therapy. Trends Pharmacol. Sci. 2014, 35, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Liang, P.; Pardee, A.B. Analysing differential gene expression in cancer. Nat. Rev. Cancer 2003, 3, 869–876. [Google Scholar] [CrossRef]
- McFarland, J.W. Parabolic relation between drug potency and hydrophobicity. J. Med. Chem. 1970, 13, 1192–1196. [Google Scholar] [CrossRef]
- Zhang, S.; Li, J.; Lykotrafitis, G.; Bao, G.; Suresh, S. Size-Dependent Endocytosis of Nanoparticles. Adv. Mater. 2009, 21, 419–424. [Google Scholar] [CrossRef] [Green Version]
- Sahay, G.; Alakhova, D.Y.; Kabanov, A.V. Endocytosis of nanomedicines. J. Control. Release 2010, 145, 182–195. [Google Scholar] [CrossRef] [Green Version]
- Levit, S.L. Formulation and Validation of Nanoparticle Controlled Delivery for Chemotherapeutic Drug Products. Ph.D. Thesis, Virginia Commonwealth University, Richmond, VA, USA, 2020. [Google Scholar]
- Kelishady, P.D.; Saadat, E.; Ravar, F.; Akbari, H.; Dorkoosh, F. Pluronic F127 polymeric micelles for co-delivery of paclitaxel and lapatinib against metastatic breast cancer: Preparation, optimization and in vitro evaluation. Pharm. Dev. Technol. 2014, 20, 1009–1017. [Google Scholar] [CrossRef]
- Kondo, N.; Tsukuda, M.; Ishiguro, Y.; Kimura, M.; Fujita, K.; Sakakibara, A.; Takahashi, H.; Toth, G.; Matsuda, H. Antitumor Effects of Lapatinib (GW572016), a Dual Inhibitor of EGFR and HER-2, in Combination with Cisplatin or Paclitaxel on Head and Neck Squamous Cell Carcinoma. Oncol. Rep. 2010, 23, 957–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, C.-L.; Tiwari, A.K.; Wu, C.-P.; Su, X.-D.; Wang, S.-R.; Liu, D.-G.; Ashby, C.R.; Huang, Y.; Robey, R.W.; Liang, Y.-J.; et al. Lapatinib (Tykerb, GW572016) Reverses Multidrug Resistance in Cancer Cells by Inhibiting the Activity of ATP-Binding Cassette Subfamily B Member 1 and G Member 2. Cancer Res. 2008, 68, 7905–7914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bullock, J. Dissolution of Nanoparticle Drug Formulations. In Poorly Soluble Drugs: Dissolution and Drug Release; Webster, G.K., Jackson, J.D., Bell, R.G., Eds.; Jenny Stanford Publishing Ptd. Ltd.: Singapore, 2017; pp. 301–351. [Google Scholar] [CrossRef]
- Liu, P.; De Wulf, O.; Laru, J.; Heikkilä, T.; Van Veen, B.; Kiesvaara, J.; Hirvonen, J.; Peltonen, L.; Laaksonen, T. Dissolution Studies of Poorly Soluble Drug Nanosuspensions in Non-sink Conditions. AAPS PharmSciTech 2013, 14, 748–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.; An, S.-J.; Zhang, X.-C.; Dong, S.; Zhang, Y.-F.; Chen, Z.-H.; Chen, H.-J.; Guo, A.-L.; Lin, Q.-X.; Wu, Y.-L. In vitro sequence-dependent synergism between paclitaxel and gefitinib in human lung cancer cell lines. Cancer Chemother. Pharmacol. 2010, 67, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.-F.; Li, S.-S.; Zhu, X.; Dou, Q.-H.; Liu, D. Lapatinib in combination with paclitaxel plays synergistic antitumor effects on esophageal squamous cancer. Cancer Chemother. Pharmacol. 2018, 82, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Wang, Y.; Strom, A.; Gustafsson, J.-Å.; Guan, X. Lapatinib induces p27Kip1-dependent G₁ arrest through both transcriptional and post-translational mechanisms. Cell Cycle 2013, 12, 2665–2674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.Q.; Guo, X.D.; Lin, W.J.; Zhang, C.Y.; Qian, Y.; Zhang, L. Amphiphilic copolymer brush with random pH-sensitive/hydrophobic structure: Synthesis and self-assembled micelles for sustained drug delivery. Soft Matter 2011, 8, 454–464. [Google Scholar] [CrossRef]
- Liu, G.; Lovell, J.F.; Zhang, L.; Zhang, Y. Stimulus-Responsive Nanomedicines for Disease Diagnosis and Treatment. Int. J. Mol. Sci. 2020, 21, 6380. [Google Scholar] [CrossRef]
- Levit, S.L.; Stwodah, R.M.; Tang, C. Rapid, Room Temperature Nanoparticle Drying and Low-Energy Reconstitution via Electrospinning. J. Pharm. Sci. 2018, 107, 807–813. [Google Scholar] [CrossRef]
- Fang, X.; Jin, X.; Xu, H.-J.; Liu, L.; Peng, H.-Q.; Hogg, D.; A Roth, J.; Yu, Y.; Xu, F.; Bast, R.C.; et al. Expression of p16 induces transcriptional downregulation of the RB gene. Oncogene 1998, 16, 1–8. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Encapsulation efficiency (EE%) | Drug loading (DL%) |
| PTX/Prodrug | PTX/Prodrug | |
| PTX NPs | 37.6 ± 14.4 | 3.11 ± 1.88 |
| Pro NPs | 45.3 ± 1.8 | 1.25 ± 0.22 |
| Samples | Encapsulation efficiency (EE%) | Drug loading (DL%) |
| PTX NPs | 37.6 ± 14.4 | 3.11 ± 1.88 |
| Pro NPs | 45.3 ± 1.8 | 1.25 ± 0.22 |
| Sample | Diffusion Exponent (n) | Rate Constant (a) | R2 |
|---|---|---|---|
| PTX NPs | 0.3 | 0.7 | 0.99 |
| Pro NPs | 0.9 | 1.3 | 0.98 |
| Sample | Burst Release | Sustained Release | ||
|---|---|---|---|---|
| Rate Constant (KS) | R2 | Rate Constant (KS) | R2 | |
| PTX NPs | 1.2 | 0.89 | 0.0072 | 0.82 |
| Pro NPs | 1.2 | 0.89 | 0.012 | 0.91 |
| Drug Treatment | IC50 (μM) | |
|---|---|---|
| Free PTX | 83 ± 6 | |
| Free Prodrug | 10 ± 5 | |
| PTX NPs | 0.047 ± 0.004 | |
| Pro NPs | 0.009 ± 0.002 | |
| Pro-LAP NPs: | Prodrug | 0.00442 ± 0.00001 |
| LAP | 0.00740 ± 0.00002 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levit, S.L.; Gade, N.R.; Roper, T.D.; Yang, H.; Tang, C. Self-Assembly of pH-Labile Polymer Nanoparticles for Paclitaxel Prodrug Delivery: Formulation, Characterization, and Evaluation. Int. J. Mol. Sci. 2020, 21, 9292. https://doi.org/10.3390/ijms21239292
Levit SL, Gade NR, Roper TD, Yang H, Tang C. Self-Assembly of pH-Labile Polymer Nanoparticles for Paclitaxel Prodrug Delivery: Formulation, Characterization, and Evaluation. International Journal of Molecular Sciences. 2020; 21(23):9292. https://doi.org/10.3390/ijms21239292
Chicago/Turabian StyleLevit, Shani L., Narendar Reddy Gade, Thomas D. Roper, Hu Yang, and Christina Tang. 2020. "Self-Assembly of pH-Labile Polymer Nanoparticles for Paclitaxel Prodrug Delivery: Formulation, Characterization, and Evaluation" International Journal of Molecular Sciences 21, no. 23: 9292. https://doi.org/10.3390/ijms21239292
APA StyleLevit, S. L., Gade, N. R., Roper, T. D., Yang, H., & Tang, C. (2020). Self-Assembly of pH-Labile Polymer Nanoparticles for Paclitaxel Prodrug Delivery: Formulation, Characterization, and Evaluation. International Journal of Molecular Sciences, 21(23), 9292. https://doi.org/10.3390/ijms21239292