Downregulation of miR-506-3p Facilitates EGFR-TKI Resistance through Induction of Sonic Hedgehog Signaling in Non-Small-Cell Lung Cancer Cell Lines


 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
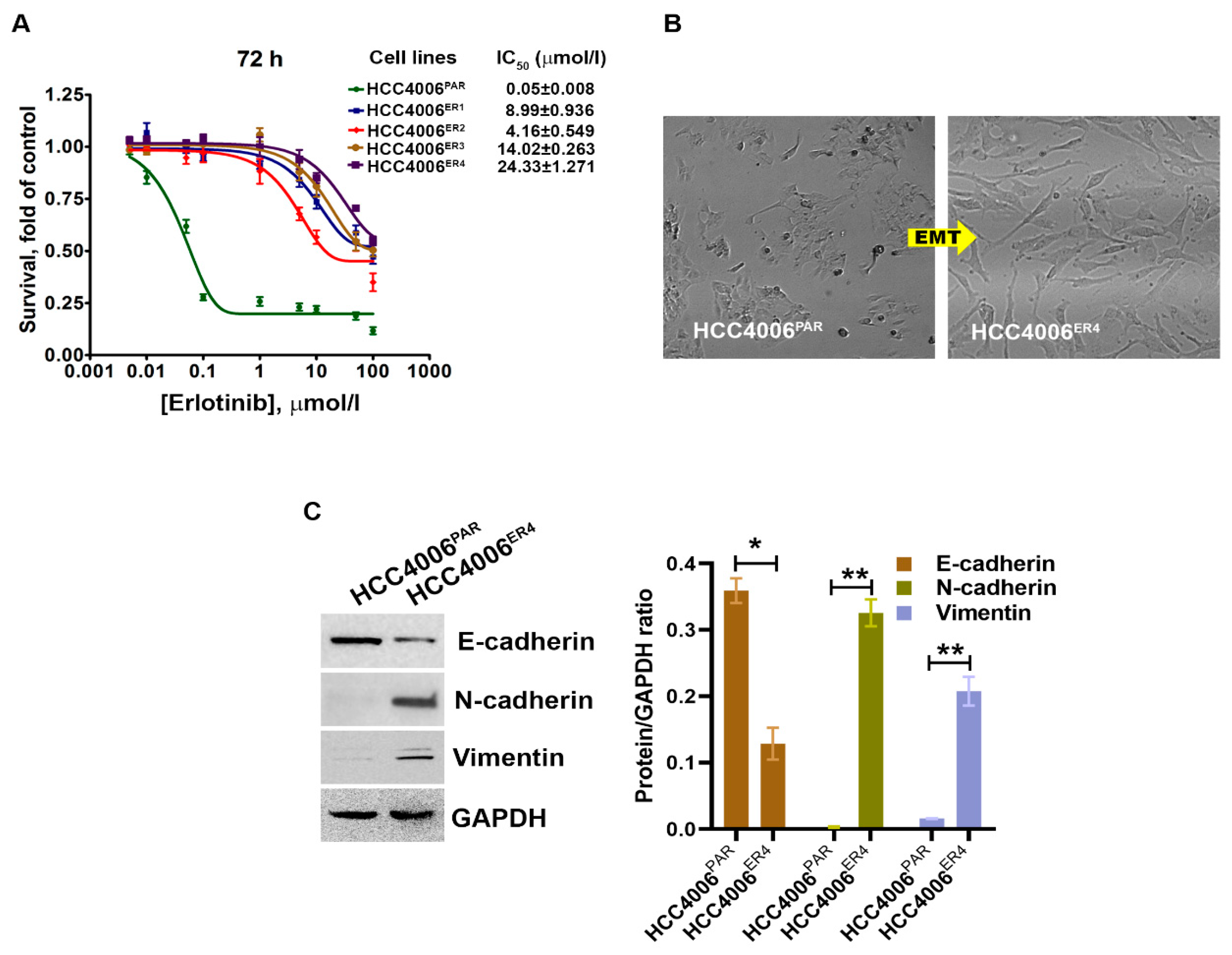
2.1. EGFR-TKI-Resistant Cells Demonstrate Cancer Stem Cell Behavior and Resist Death in Response to Erlotinib Treatment
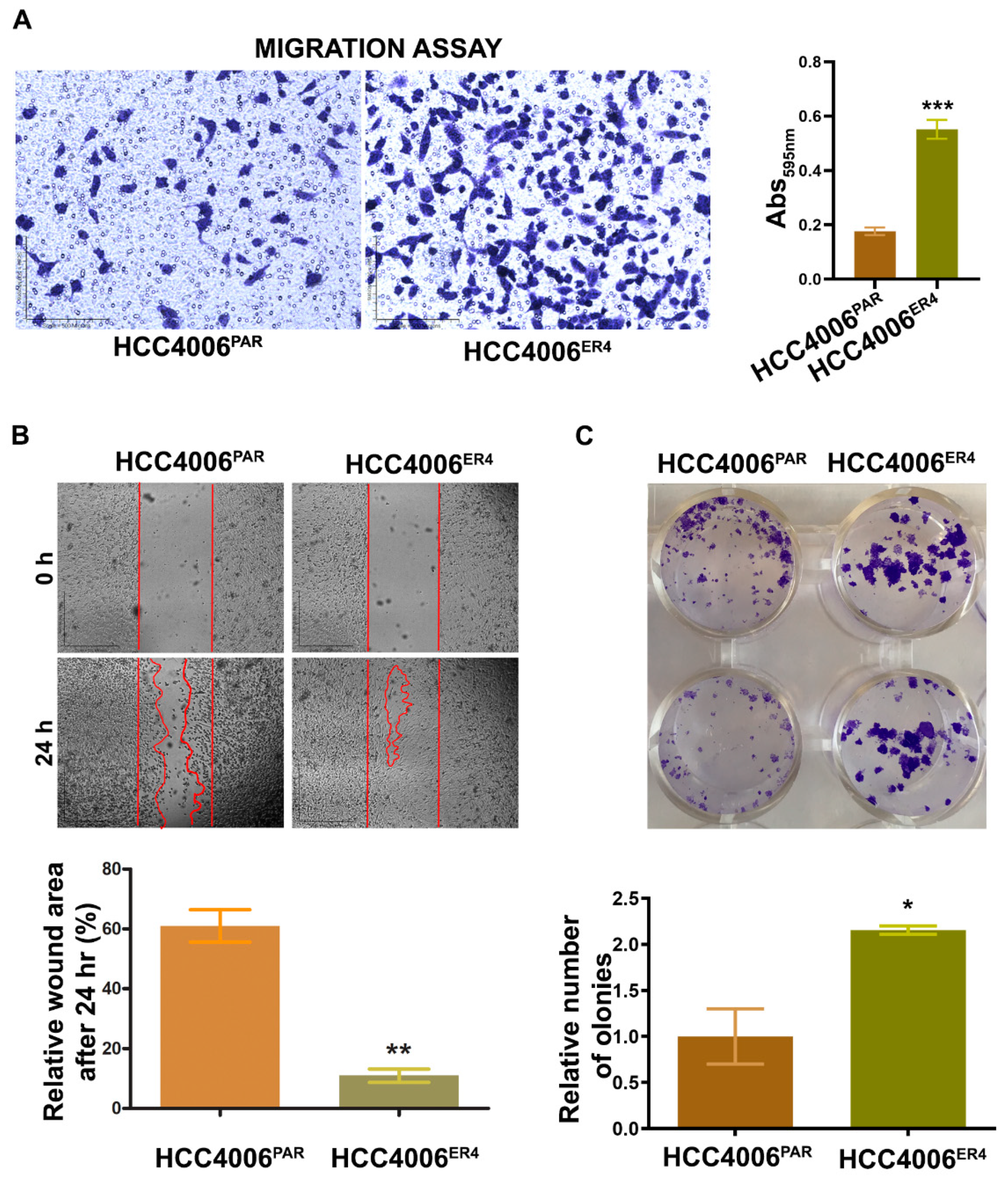
2.2. EGFR-TKI-Resistant Cells Show a Significant Increase in Migration and Colony Formation Compared to Non-Resistant Cells
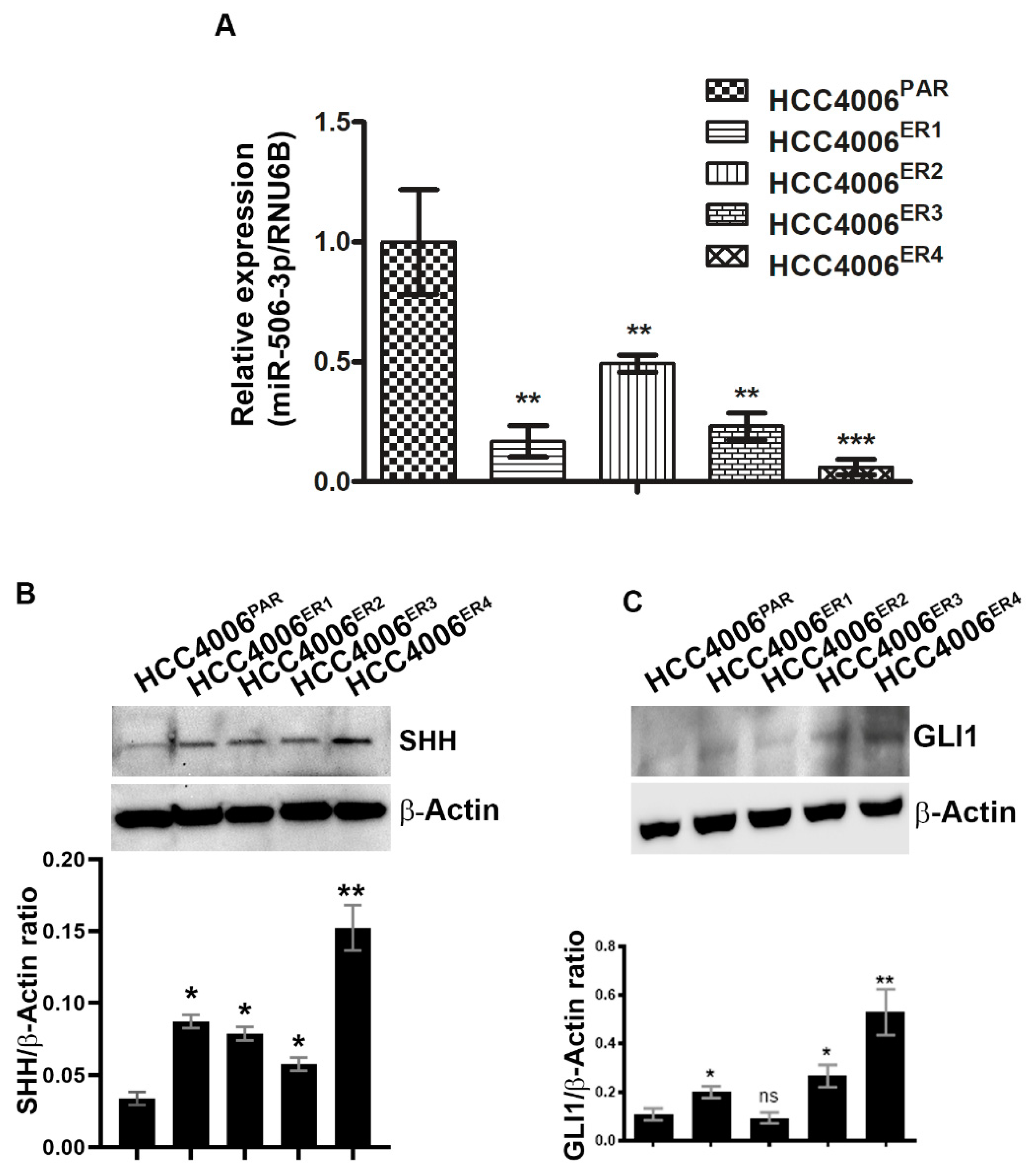
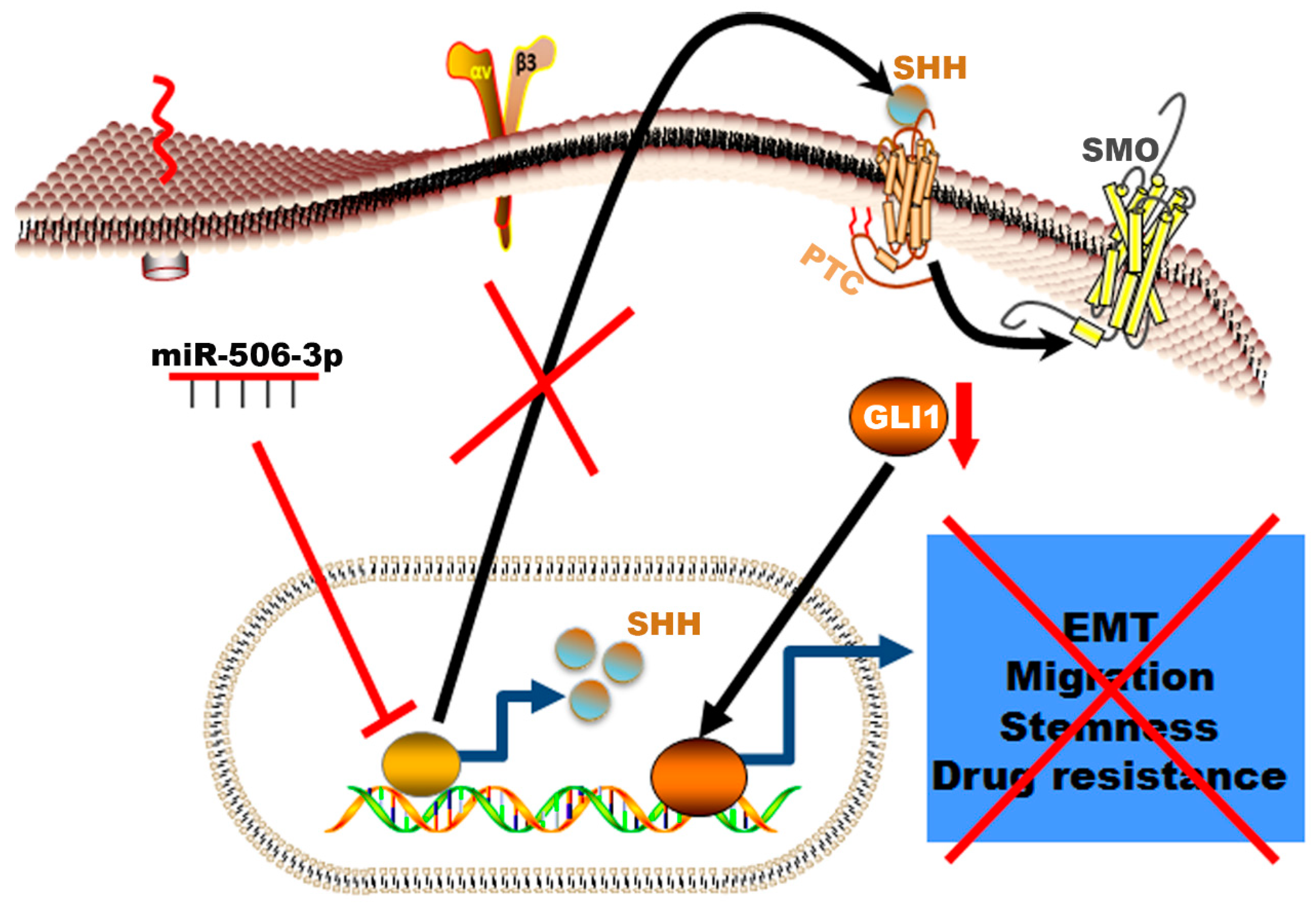
2.3. EGFR-TKI-Resistant Cells Showed Downregulation of miR-506-3p and Upregulation of Its Target SHH and Glioma-Associated Oncogene Homolog Zinc Finger Protein 1 (GLI1] Expression
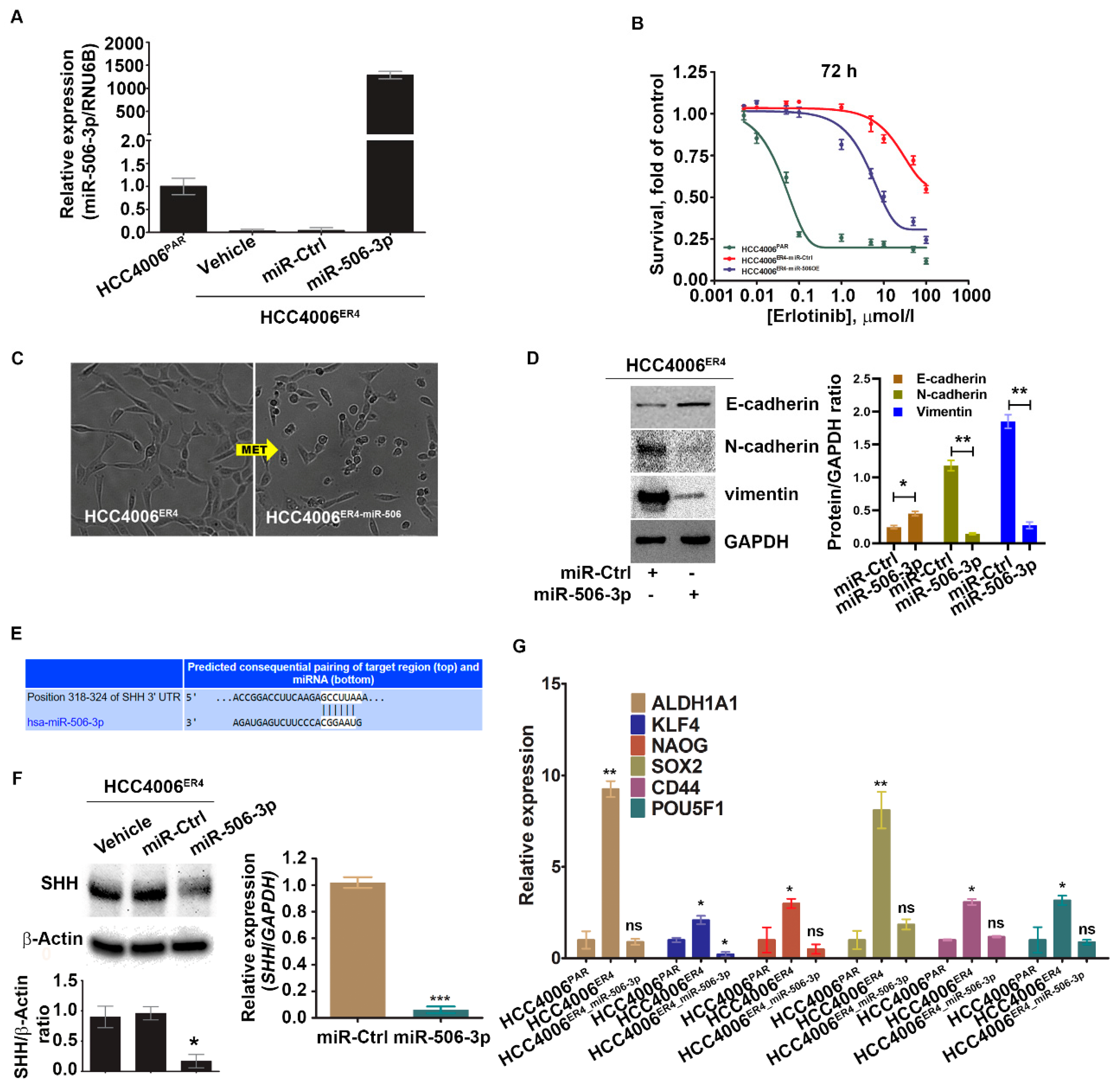
2.4. miR-506-3p Overexpression Sensitizes EGFR-TKI-Resistant Cells to Erlotinib-Induced Cell Death, Induced Mesenchymal–Epithelial Transition (MET), Downregulates SHH Expression and Stemness
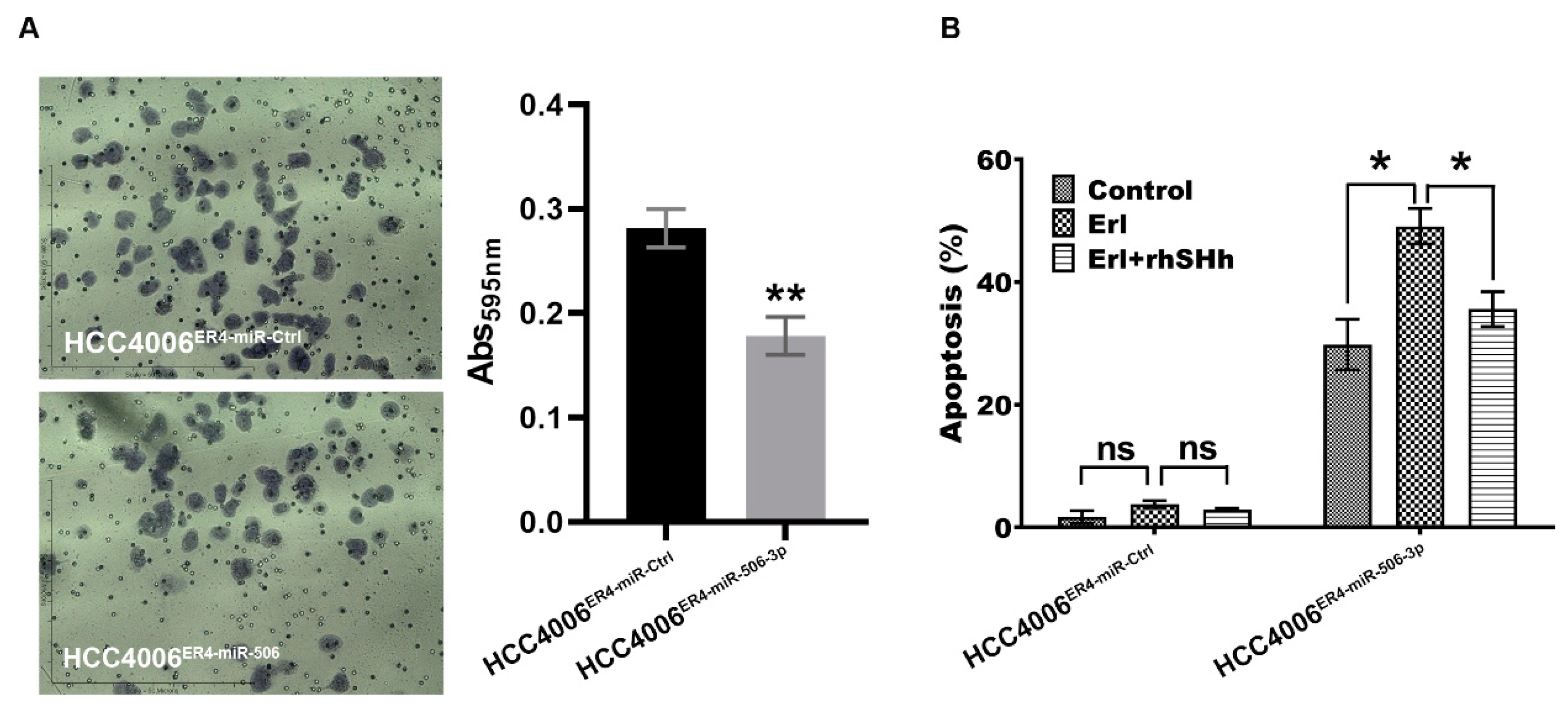
2.5. miR-506-3p Overexpression Inhibits Migration and Increases Apoptosis, Which Is Attenuated by SHH Ligand
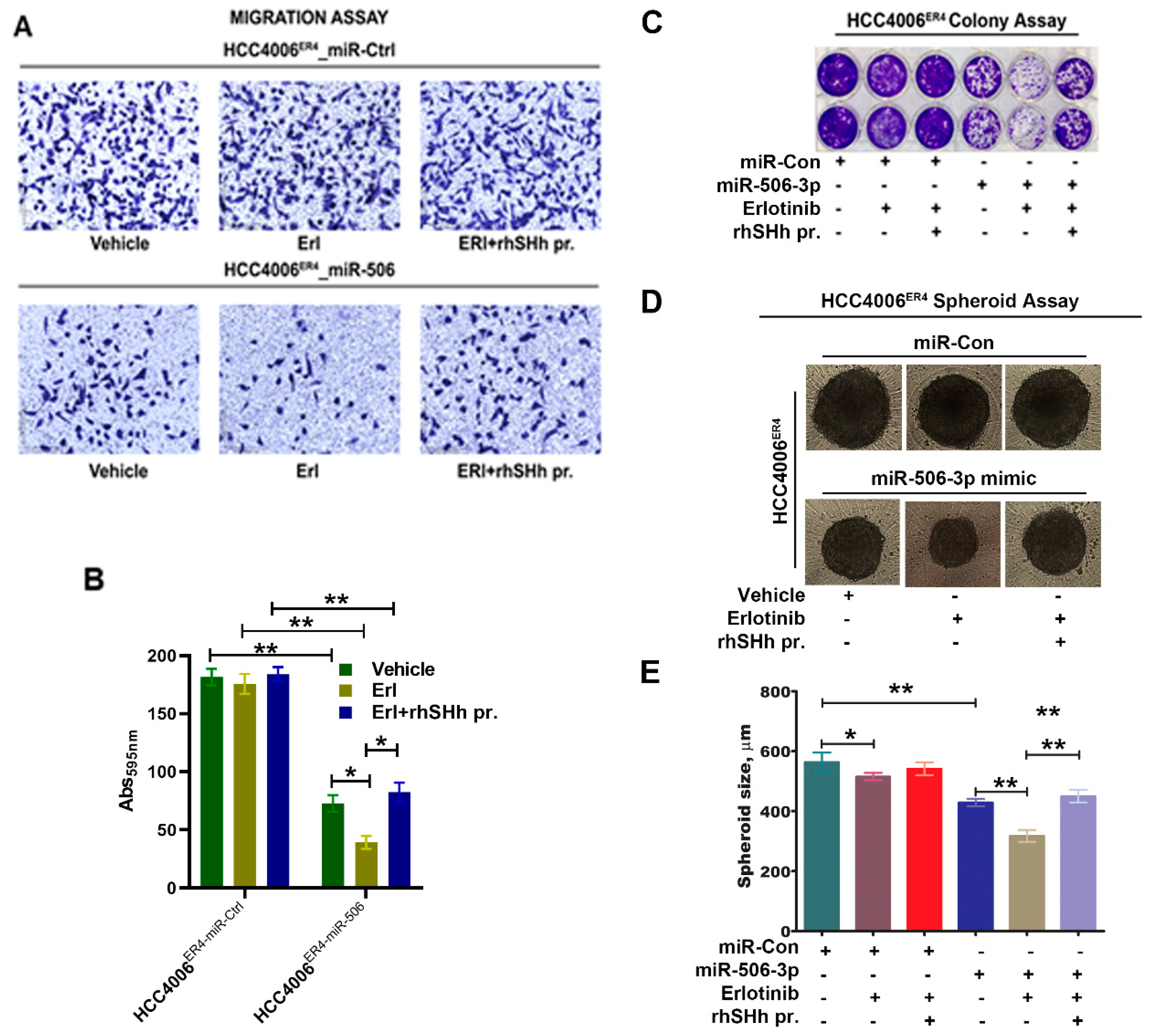
2.6. SHH Reverses miR-506-3p Overexpression-Induced Erlotinib Sensitivity
3. Discussion
4. Materials and Methods
4.1. Generation of EGFR-TKI-Resistant Cell Lines
4.2. Cell Viability Assay
4.3. Western Blot Analysis
4.4. In Vitro Boyden Chamber Migration Assay
4.5. Colony Formation Assay
4.6. Wound Healing Assay
4.7. Quantitative Real-Time PCR (qRT-PCR)
4.8. Apoptosis Assay
4.9. Spheroid Formation Assay
4.10. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NSCLC | Non-small-cell lung cancer |
| EGFR | Epidermal growth factor receptor |
| TKI | Tryrosine kinase inhibitor |
| miRNA | microRNA |
| miR-506-3p | microRNA-506-3p |
| ER | Erlotinib resistance |
| SHH | Sonic Hedgehog |
| EMT | Epithelial–mesenchymal transition |
| rhSHH Pr. | Recombinant SHH protein |
| NGS | Next-generation sequencing |
| PFS | Progression-free survival |
| OS | Overall survival |
References
- Wong, M.C.S.; Lao, X.Q.; Ho, K.F.; Goggins, W.B.; Tse, S.L.A. Incidence and mortality of lung cancer: Global trends and association with socioeconomic status. Sci. Rep. 2017, 7, 14300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Makitaro, R.; Paakko, P.; Huhti, E.; Bloigu, R.; Kinnula, V.L. Prospective population-based study on the survival of patients with lung cancer. Eur. Respir. J. 2002, 19, 1087–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. (Eds.) Seer Cancer Statistics Review, 1975–2016; National Cancer Institute: Bethesda, MD, USA, 2019.
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, S.; Yang, P.; Jiang, X.; Li, X.; Wang, Y.; Zhang, X.; Sun, B.; Zhang, Y.; Jia, Y. Genetic and epigenetic silencing of mircorna-506-3p enhances cotl1 oncogene expression to foster non-small lung cancer progression. Oncotarget 2017, 8, 644–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Yang, S.; Zhou, Q.; Wang, G.; Song, J.; Li, Z.; Zhang, Z.; Xu, J.; Xia, K.; Chang, Y.; et al. Emerging role of exosome-derived long non-coding rnas in tumor microenvironment. Mol. Cancer 2018, 17, 82. [Google Scholar] [CrossRef]
- Gong, M.; Chen, C.; Zhao, H.; Sun, M.; Song, M. Mir-506 suppresses cervical cancer cell proliferation both in vitro and in vivo. Neoplasma 2018, 65, 331–338. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.Y.; You, P.; Zhang, J.; Zhang, H.; Jiang, N. Mir-506-3p acts as a novel tumor suppressor in prostate cancer through targeting galnt4. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 5133–5138. [Google Scholar]
- Huang, M.; Xie, X.; Song, X.; Gu, S.; Chang, X.; Su, T.; Liang, B.; Huang, D. Mir-506 suppresses colorectal cancer development by inhibiting orphan nuclear receptor nr4a1 expression. J. Cancer 2019, 10, 3560–3570. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.Q.; Zhang, H.M.; Chen, S.J.; Yan, Y.; Zheng, J.H. Mir-506 is down-regulated in clear cell renal cell carcinoma and inhibits cell growth and metastasis via targeting flot1. PLoS ONE 2015, 10, e0120258. [Google Scholar] [CrossRef]
- Yao, W.J.; Wang, Y.L.; Lu, J.G.; Guo, L.; Qi, B.; Chen, Z.J. Microrna-506 inhibits esophageal cancer cell proliferation via targeting creb1. Int. J. Clin. Exp. Pathol. 2015, 8, 10868–10874. [Google Scholar]
- Yin, M.; Ren, X.; Zhang, X.; Luo, Y.; Wang, G.; Huang, K.; Feng, S.; Bao, X.; Huang, K.; He, X.; et al. Selective killing of lung cancer cells by mirna-506 molecule through inhibiting nf-kappab p65 to evoke reactive oxygen species generation and p53 activation. Oncogene 2015, 34, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, H.; Wei, G. Mir-506 regulates cell proliferation and apoptosis by affecting rhoa/rock signaling pathway in hepatocellular carcinoma cells. Int. J. Clin. Exp. Pathol. 2019, 12, 1163–1173. [Google Scholar] [PubMed]
- Zhu, X.W.; Wang, J.; Zhu, M.X.; Wang, Y.F.; Yang, S.Y.; Ke, X.Y. Microrna-506 inhibits the proliferation and invasion of mantle cell lymphoma cells by targeting b7h3. Biochem. Biophys. Res. Commun. 2019, 508, 1067–1073. [Google Scholar] [CrossRef]
- Li, J.; Ju, J.; Ni, B.; Wang, H. The emerging role of mir-506 in cancer. Oncotarget 2016, 7, 62778–62788. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liu, H.; Li, Y.; Wu, J.; Greenlee, A.R.; Yang, C.; Jiang, Y. The role of mir-506 in transformed 16hbe cells induced by anti-benzo[a]pyrene-trans-7,8-dihydrodiol-9,10-epoxide. Toxicol. Lett. 2011, 205, 320–326. [Google Scholar] [CrossRef]
- Yang, D.; Sun, Y.; Hu, L.; Zheng, H.; Ji, P.; Pecot, C.V.; Zhao, Y.; Reynolds, S.; Cheng, H.; Rupaimoole, R.; et al. Integrated analyses identify a master microrna regulatory network for the mesenchymal subtype in serous ovarian cancer. Cancer Cell 2013, 23, 186–199. [Google Scholar] [CrossRef] [Green Version]
- Ming, J.E.; Roessler, E.; Muenke, M. Human developmental disorders and the sonic hedgehog pathway. Mol. Med. Today 1998, 4, 343–349. [Google Scholar] [CrossRef]
- Ahmad, A.; Maitah, M.Y.; Ginnebaugh, K.R.; Li, Y.; Bao, B.; Gadgeel, S.M.; Sarkar, F.H. Inhibition of hedgehog signaling sensitizes NSCLC cells to standard therapies through modulation of EMT-regulating mirnas. J. Hematol. Oncol. 2013, 6, 77. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.Y.; Yang, J.Y. Targeting the hedgehog pathway in pediatric medulloblastoma. Cancers (Basel) 2015, 7, 2110–2123. [Google Scholar] [CrossRef] [Green Version]
- Kasper, M.; Jaks, V.; Fiaschi, M.; Toftgard, R. Hedgehog signalling in breast cancer. Carcinogenesis 2009, 30, 903–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saldanha, G. The hedgehog signalling pathway and cancer. J. Pathol. 2001, 193, 427–432. [Google Scholar] [CrossRef]
- Kim, J.E.; Kim, H.; Choe, J.Y.; Sun, P.; Jheon, S.; Chung, J.H. High expression of sonic hedgehog signaling proteins is related to the favorable outcome, egfr mutation, and lepidic predominant subtype in primary lung adenocarcinoma. Ann. Surg. Oncol. 2013, 20 Suppl. 3, S570–S576. [Google Scholar] [CrossRef]
- Della Corte, C.M.; Malapelle, U.; Vigliar, E.; Pepe, F.; Troncone, G.; Ciaramella, V.; Troiani, T.; Martinelli, E.; Belli, V.; Ciardiello, F.; et al. Efficacy of continuous egfr-inhibition and role of hedgehog in egfr acquired resistance in human lung cancer cells with activating mutation of egfr. Oncotarget 2017, 8, 23020–23032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.G.; Liu, T.; Xiong, J.X.; Wang, C.Y. Blockade of sonic hedgehog signal pathway enhances antiproliferative effect of egfr inhibitor in pancreatic cancer cells. Acta Pharmaco.l Sin. 2007, 28, 1224–1230. [Google Scholar] [CrossRef]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.G.; Lee, S.H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer stem cells (cscs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef] [Green Version]
- Fan, D.C.; Zhao, Y.R.; Qi, H.; Hou, J.X.; Zhang, T.H. Mirna-506 presents multiple tumor suppressor activities by targeting ezh2 in nasopharyngeal carcinoma. Auris Nasus Larynx 2020. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Li, P.; Zhu, M.Z.; Guo, Y.; Yang, J. Linc01308 accelerates the malignant progression of ovarian cancer by binding to mirna-506. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 3253–3260. [Google Scholar]
- Wen, S.Y.; Lin, Y.; Yu, Y.Q.; Cao, S.J.; Zhang, R.; Yang, X.M.; Li, J.; Zhang, Y.L.; Wang, Y.H.; Ma, M.Z.; et al. Mir-506 acts as a tumor suppressor by directly targeting the hedgehog pathway transcription factor gli3 in human cervical cancer. Oncogene 2015, 34, 717–725. [Google Scholar] [CrossRef]
- Akyala, A.I.; Peppelenbosch, M.P. Gastric cancer and hedgehog signaling pathway: Emerging new paradigms. Genes Cancer 2018, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Bhuria, V.; Xing, J.; Scholta, T.; Bui, K.C.; Nguyen, M.L.T.; Malek, N.P.; Bozko, P.; Plentz, R.R. Hypoxia induced sonic hedgehog signaling regulates cancer stemness, epithelial-to-mesenchymal transition and invasion in cholangiocarcinoma. Exp. Cell Res. 2019, 385, 111671. [Google Scholar] [CrossRef] [PubMed]
- Keedy, V.L.; Temin, S.; Somerfield, M.R.; Beasley, M.B.; Johnson, D.H.; McShane, L.M.; Milton, D.T.; Strawn, J.R.; Wakelee, H.A.; Giaccone, G. American society of clinical oncology provisional clinical opinion: Epidermal growth factor receptor (egfr) mutation testing for patients with advanced non-small-cell lung cancer considering first-line egfr tyrosine kinase inhibitor therapy. J. Clin. Oncol. 2011, 29, 2121–2127. [Google Scholar] [CrossRef] [PubMed]
- Kotulak-Chrzaszcz, A.; Klacz, J.; Matuszewski, M.; Kmiec, Z.; Wierzbicki, P.M. Expression of the sonic hedgehog pathway components in clear cell renal cell carcinoma. Oncol. Lett. 2019, 18, 5801–5810. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Lim, S.M.; Kim, M.J.; Park, S.Y.; Kim, J.H. Sonic hedgehog pathway as the prognostic marker in patients with extensive stage small cell lung cancer. Yonsei Med. J. 2019, 60, 898–904. [Google Scholar] [CrossRef]
- Dong, Z.; Wang, Y.; Ding, V.; Yan, X.; Lv, Y.; Zhong, M.; Zhu, F.; Zhao, P.; He, C.; Ding, F.; et al. Gli1 activation is a key mechanism of erlotinib resistance in human non-small cell lung cancer. Oncol. Lett. 2020, 20, 76. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, F.R.; Bunn, P.A., Jr. Egfr testing in lung cancer is ready for prime time. Lancet Oncol. 2009, 10, 432–433. [Google Scholar] [CrossRef]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with egfr-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef] [Green Version]
- Arcila, M.E.; Oxnard, G.R.; Nafa, K.; Riely, G.J.; Solomon, S.B.; Zakowski, M.F.; Kris, M.G.; Pao, W.; Miller, V.A.; Ladanyi, M. Rebiopsy of lung cancer patients with acquired resistance to egfr inhibitors and enhanced detection of the t790m mutation using a locked nucleic acid-based assay. Clin. Cancer Res. 2011, 17, 1169–1180. [Google Scholar] [CrossRef] [Green Version]
- Marcoux, N.; Gettinger, S.N.; O’Kane, G.; Arbour, K.C.; Neal, J.W.; Husain, H.; Evans, T.L.; Brahmer, J.R.; Muzikansky, A.; Bonomi, P.D.; et al. EGFR-mutant adenocarcinomas that transform to small-cell lung cancer and other neuroendocrine carcinomas: Clinical outcomes. J. Clin. Oncol. 2019, 37, 278–285. [Google Scholar] [CrossRef]
- Li, J.; Wu, H.; Li, W.; Yin, L.; Guo, S.; Xu, X.; Ouyang, Y.; Zhao, Z.; Liu, S.; Tian, Y.; et al. Downregulated mir-506 expression facilitates pancreatic cancer progression and chemoresistance via sphk1/akt/nf-kappab signaling. Oncogene 2016, 35, 5501–5514. [Google Scholar] [CrossRef]
- Sakimura, S.; Sugimachi, K.; Kurashige, J.; Ueda, M.; Hirata, H.; Nambara, S.; Komatsu, H.; Saito, T.; Takano, Y.; Uchi, R.; et al. The mir-506-induced epithelial-mesenchymal transition is involved in poor prognosis for patients with gastric cancer. Ann. Surg. Oncol. 2015, 22 Suppl. 3, S1436–S1443. [Google Scholar] [CrossRef]
- Yauch, R.L.; Januario, T.; Eberhard, D.A.; Cavet, G.; Zhu, W.; Fu, L.; Pham, T.Q.; Soriano, R.; Stinson, J.; Seshagiri, S.; et al. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin. Cancer Res. 2005, 11, 8686–8698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, J.M.; Mohr, A.M.; Hollingsworth, M.A. Sonic hedgehog paracrine signaling regulates metastasis and lymphangiogenesis in pancreatic cancer. Oncogene 2009, 28, 3513–3525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karhadkar, S.S.; Bova, G.S.; Abdallah, N.; Dhara, S.; Gardner, D.; Maitra, A.; Isaacs, J.T.; Berman, D.M.; Beachy, P.A. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature 2004, 431, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Sheng, T.; Zhang, Y.; Zhang, X.; He, J.; Huang, S.; Chen, K.; Sultz, J.; Adegboyega, P.A.; Zhang, H.; et al. Hedgehog signaling is activated in subsets of esophageal cancers. Int. J. Cancer 2006, 118, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Sheng, T.; Li, C.; Zhang, X.; Chi, S.; He, N.; Chen, K.; McCormick, F.; Gatalica, Z.; Xie, J. Activation of the hedgehog pathway in advanced prostate cancer. Mol. Cancer 2004, 3, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haque, I.; De, A.; Majumder, M.; Mehta, S.; McGregor, D.; Banerjee, S.K.; Van, V.P.; Banerjee, S. The matricellular protein ccn1/cyr61 is a critical regulator of sonic hedgehog in pancreatic carcinogenesis. J. Biol. Chem. 2012, 287, 38569–38579. [Google Scholar] [CrossRef] [Green Version]
- Murone, M.; Rosenthal, A.; de Sauvage, F.J. Hedgehog signal transduction: From flies to vertebrates. Exp. Cell Res. 1999, 253, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The role of the hedgehog signaling pathway in cancer: A comprehensive review. Bosn. J. Basic Med. Sci. 2018, 18, 8–20. [Google Scholar] [CrossRef]
- Arora, H.; Qureshi, R.; Park, W.Y. Mir-506 regulates epithelial mesenchymal transition in breast cancer cell lines. PLoS ONE 2013, 8, e64273. [Google Scholar] [CrossRef]
- Corominas-Faja, B.; Oliveras-Ferraros, C.; Cuyas, E.; Segura-Carretero, A.; Joven, J.; Martin-Castillo, B.; Barrajon-Catalan, E.; Micol, V.; Bosch-Barrera, J.; Menendez, J.A. Stem cell-like aldh(bright) cellular states in egfr-mutant non-small cell lung cancer: A novel mechanism of acquired resistance to erlotinib targetable with the natural polyphenol silibinin. Cell Cycle 2013, 12, 3390–3404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciurea, M.E.; Georgescu, A.M.; Purcaru, S.O.; Artene, S.A.; Emami, G.H.; Boldeanu, M.V.; Tache, D.E.; Dricu, A. Cancer stem cells: Biological functions and therapeutically targeting. Int. J. Mol. Sci. 2014, 15, 8169–8185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peacock, C.D.; Wang, Q.; Gesell, G.S.; Corcoran-Schwartz, I.M.; Jones, E.; Kim, J.; Devereux, W.L.; Rhodes, J.T.; Huff, C.A.; Beachy, P.A.; et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc. Natl. Acad. Sci. USA 2007, 104, 4048–4053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bora-Singhal, N.; Perumal, D.; Nguyen, J.; Chellappan, S. Gli1-mediated regulation of Sox2 facilitates self-renewal of stem-like cells and confers resistance to EGFR inhibitors in non-small cell lung cancer. Neoplasia 2015, 17, 538–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Ma, X.; Hsiao, T.H.; Lin, G.; Kosti, A.; Yu, X.; Suresh, U.; Chen, Y.; Tomlinson, G.E.; Pertsemlidis, A.; et al. A high-content morphological screen identifies novel micrornas that regulate neuroblastoma cell differentiation. Oncotarget 2014, 5, 2499–2512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, S.; Song, R.; Xie, J. Sonidegib: Mechanism of action, pharmacology, and clinical utility for advanced basal cell carcinomas. Oncol. Targets Ther. 2017, 10, 1645–1653. [Google Scholar] [CrossRef] [Green Version]
- Jager, T.; Ocker, M.; Kiesslich, T.; Neureiter, E.; Neureiter, D. Thoughts on investigational hedgehog pathway inhibitors for the treatment of cancer. Expert Opin. Investig. Drugs 2017, 26, 133–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raven, P.A.; Lysakowski, S.; Tan, Z.; D’Costa, N.M.; Moskalev, I.; Frees, S.; Struss, W.; Matsui, Y.; Narita, S.; Buttyan, R.; et al. Inhibition of gli2 with antisense-oligonucleotides: A potential therapy for the treatment of bladder cancer. J. Cell Physiol. 2019, 234, 20634–20647. [Google Scholar] [CrossRef]
- Zhang, M.; Tan, S.; Yu, D.; Zhao, Z.; Zhang, B.; Zhang, P.; Lv, C.; Zhou, Q.; Cao, Z. Triptonide inhibits lung cancer cell tumorigenicity by selectively attenuating the shh-gli1 signaling pathway. Toxicol. Appl. Pharmacol. 2019, 365, 1–8. [Google Scholar] [CrossRef]
- Liu, G.; Yang, D.; Rupaimoole, R.; Pecot, C.V.; Sun, Y.; Mangala, L.S.; Li, X.; Ji, P.; Cogdell, D.; Hu, L.; et al. Augmentation of response to chemotherapy by microrna-506 through regulation of rad51 in serous ovarian cancers. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, G.; Yan, X.; Lee, A.G.; Kron, S.J.; Palecek, S.P. Quantifying the sensitivities of egf receptor (egfr) tyrosine kinase inhibitors in drug resistant non-small cell lung cancer (nsclc) cells using hydrogel-based peptide array. Biosens. Bioelectron. 2010, 26, 424–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haque, I.; Ghosh, A.; Acup, S.; Banerjee, S.; Dhar, K.; Ray, A.; Sarkar, S.; Kambhampati, S.; Banerjee, S.K. Leptin-induced er-alpha-positive breast cancer cell viability and migration is mediated by suppressing ccn5-signaling via activating jak/akt/stat-pathway. BMC Cancer 2018, 18, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maity, G.; Haque, I.; Ghosh, A.; Dhar, G.; Gupta, V.; Sarkar, S.; Azeem, I.; McGregor, D.; Choudhary, A.; Campbell, D.R.; et al. The MAZ transcription factor is a downstream target of the oncoprotein Cyr61/CCN1 and promotes pancreatic cancer cell invasion via CRAF-ERK signaling. J. Biol. Chem. 2018, 293, 4334–4349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. Nih image to imagej: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Haque, I.; Mehta, S.; Majumder, M.; Dhar, K.; De, A.; McGregor, D.; Van Veldhuizen, P.J.; Banerjee, S.K.; Banerjee, S. Cyr61/CCN1 signaling is critical for epithelial-mesenchymal transition and stemness and promotes pancreatic carcinogenesis. Mol. Cancer 2011, 10, 8. [Google Scholar] [CrossRef] [Green Version]
- Maity, G.; De, A.; Das, A.; Banerjee, S.; Sarkar, S.; Banerjee, S.K. Aspirin blocks growth of breast tumor cells and tumor-initiating cells and induces reprogramming factors of mesenchymal to epithelial transition. Lab. Investig. 2015, 95, 702–717. [Google Scholar] [CrossRef] [Green Version]
- Haque, I.; Banerjee, S.; Mehta, S.; De, A.; Majumder, M.; Mayo, M.S.; Kambhampati, S.; Campbell, D.R.; Banerjee, S.K. Cysteine-rich 61-connective tissue growth factor-nephroblastoma-overexpressed 5 (ccn5)/wnt-1-induced signaling protein-2 (wisp-2) regulates microrna-10b via hypoxia-inducible factor-1alpha-twist signaling networks in human breast cancer cells. J. Biol. Chem. 2011, 286, 43475–43485. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta c(t)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Maity, G.; Ghosh, A.; Gupta, V.; Haque, I.; Sarkar, S.; Das, A.; Dhar, K.; Bhavanasi, S.; Gunewardena, S.S.; Von Hoff, D.D.; et al. Cyr61/CCN1 regulates dCK and CTGF and causes gemcitabine-resistant phenotype in pancreatic ductal adenocarcinoma. Mol. Cancer Ther. 2019, 18, 788–800. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Dhar, K.; Maity, G.; Sarkar, S.; Ghosh, A.; Haque, I.; Dhar, G.; Banerjee, S.; Banerjee, S.K. Deficiency of CCN5/WISP-2-driven program in breast cancer promotes cancer epithelial cells to mesenchymal stem cells and breast cancer growth. Sci. Rep. 2017, 7, 1220. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haque, I.; Kawsar, H.I.; Motes, H.; Sharma, M.; Banerjee, S.; Banerjee, S.K.; Godwin, A.K.; Huang, C.H. Downregulation of miR-506-3p Facilitates EGFR-TKI Resistance through Induction of Sonic Hedgehog Signaling in Non-Small-Cell Lung Cancer Cell Lines. Int. J. Mol. Sci. 2020, 21, 9307. https://doi.org/10.3390/ijms21239307
Haque I, Kawsar HI, Motes H, Sharma M, Banerjee S, Banerjee SK, Godwin AK, Huang CH. Downregulation of miR-506-3p Facilitates EGFR-TKI Resistance through Induction of Sonic Hedgehog Signaling in Non-Small-Cell Lung Cancer Cell Lines. International Journal of Molecular Sciences. 2020; 21(23):9307. https://doi.org/10.3390/ijms21239307
Chicago/Turabian StyleHaque, Inamul, Hameem I. Kawsar, Hannah Motes, Mukut Sharma, Snigdha Banerjee, Sushanta K. Banerjee, Andrew K. Godwin, and Chao H. Huang. 2020. "Downregulation of miR-506-3p Facilitates EGFR-TKI Resistance through Induction of Sonic Hedgehog Signaling in Non-Small-Cell Lung Cancer Cell Lines" International Journal of Molecular Sciences 21, no. 23: 9307. https://doi.org/10.3390/ijms21239307
APA StyleHaque, I., Kawsar, H. I., Motes, H., Sharma, M., Banerjee, S., Banerjee, S. K., Godwin, A. K., & Huang, C. H. (2020). Downregulation of miR-506-3p Facilitates EGFR-TKI Resistance through Induction of Sonic Hedgehog Signaling in Non-Small-Cell Lung Cancer Cell Lines. International Journal of Molecular Sciences, 21(23), 9307. https://doi.org/10.3390/ijms21239307