Checkpoint Kinase 1 Pharmacological Inhibition Synergizes with DNA-Damaging Agents and Overcomes Platinum Resistance in Basal-Like Breast Cancer

 , and
, and
Abstract
:
1. Introduction
2. Results
2.1. Synergistic Interactions of CHK1 Inhibitors with Standard-of-Care Therapies
2.2. CHK1 Inhibition Reduces Cell Growth in Combination with Platinum Compounds
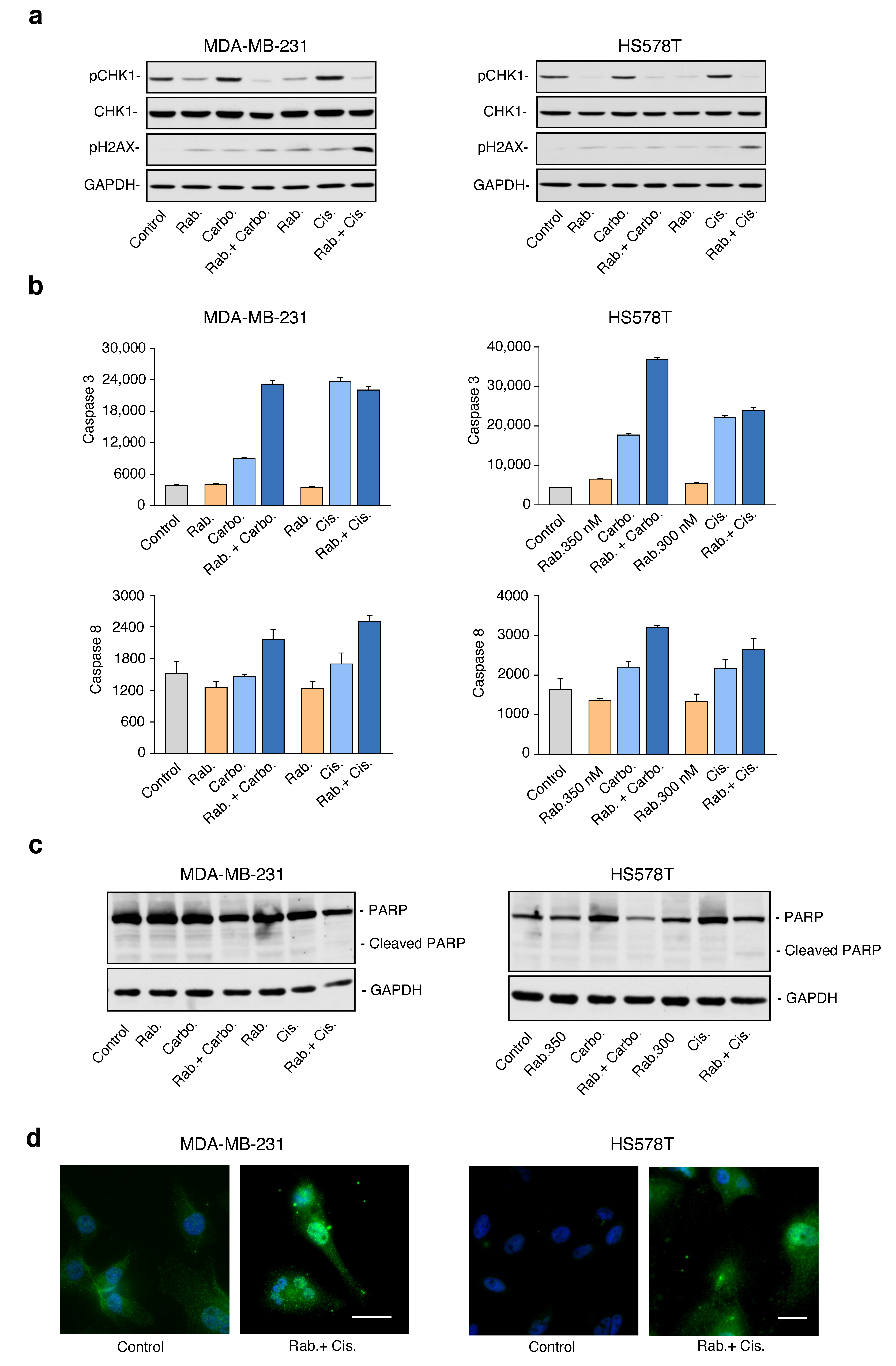
2.3. Combination of Platinum Agents and Gemcitabine with Rabusertib Induces Cell Death through a Caspase-Dependent Mechanism
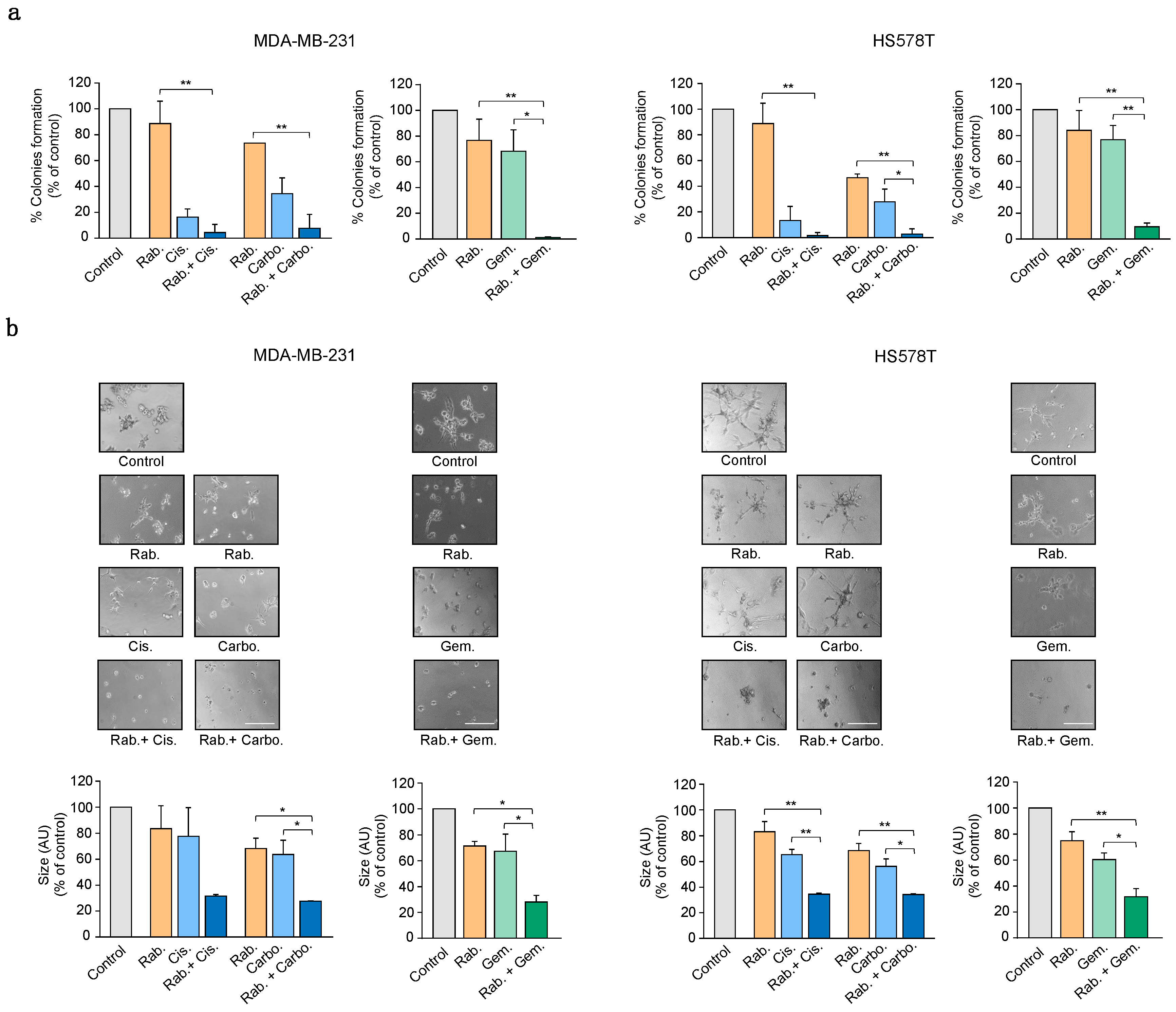
2.4. Inhibition of CHK1 Reverts Resistance to Platinum Compounds
2.5. CHK1 Inhibition Enhances the Effect of Standard-of-Care Chemotherapies
3. Discussion
4. Material and Methods
4.1. Cell Lines and Cultures
4.2. MTT and Synergistic Effect of Rabusertib and Chemotherapic Agents
4.3. Clonogenic Assays and Matrigel Embedded 3D Cultures
4.4. Cell Cycle and Apoptosis Studies
4.5. Caspase Activity Assays
4.6. Western Blotting
4.7. Immunofluorescence
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ocana, A.; Pandiella, A. Targeting oncogenic vulnerabilities in triple negative breast cancer: Biological bases and ongoing clinical studies. Oncotarget 2017, 8, 22218–22234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagliarini, R.; Shao, W.; Sellers, W.R. Oncogene addiction: Pathways of therapeutic response, resistance, and road maps toward a cure. EMBO Rep. 2015, 16, 280–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esparís-Ogando, A.; Montero, J.C.; Arribas, J.; Ocana, A.; Pandiella, A.O. Targeting the EGF/HER Ligand-Receptor System in Cancer. Available online: https://www.eurekaselect.com/144012/article (accessed on 21 September 2020).
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of Chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Smit, E.F.; Groen, H.J.M.; Mazieres, J.; Besse, B.; Helland, Å.; Giannone, V.; D’Amelio, A.M.; Zhang, P.; Mookerjee, B.; et al. Dabrafenib plus trametinib in patients with previously untreated BRAFV600E-mutant metastatic non-small-cell lung cancer: An open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1307–1316. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [Green Version]
- Pandiella, A.O.A. Novel Synthetic Lethality Approaches for Drug Combinations and Early Drug Development. Available online: https://www.eurekaselect.com/137544/article (accessed on 21 September 2020).
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Muller, F.L.; Aquilanti, E.A.; DePinho, R.A. Collateral lethality: A new therapeutic strategy in oncology. Trends Cancer 2015, 1, 161–173. [Google Scholar] [CrossRef] [Green Version]
- Saslow, D.; Solomon, D.; Lawson, H.W.; Killackey, M.; Kulasingam, S.L.; Cain, J.; Garcia, F.A.R.; Moriarty, A.T.; Waxman, A.G.; Wilbur, D.C.; et al. American cancer society, american society for colposcopy and cervical pathology, and american society for clinical pathology screening guidelines for the prevention and early detection of cervical cancer. Am. J. Clin. Pathol. 2012, 137, 516–542. [Google Scholar] [CrossRef]
- Harbeck, N.; Gnant, M. Breast cancer. Lancet 2017, 389, 1134–1150. [Google Scholar] [CrossRef]
- D’Andrea, A.D.; Grompe, M. The Fanconi anaemia/BRCA pathway. Nat. Rev. Cancer 2003, 3, 23–34. [Google Scholar] [CrossRef]
- Karnitz, L.M.; Zou, L. Molecular pathways: Targeting ATR in cancer therapy. Clin. Cancer Res. 2015, 21, 4780–4785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Tang, J.; Wang, J.; Ren, F.; Zheng, J.; Gragg, M.; Kiser, P.; Park, P.S.H.; Palczewski, K.; Yao, X.; et al. Conformational Change of Human Checkpoint Kinase 1 (Chk1) induced by DNA damage. J. Biol. Chem. 2016, 291, 12951–12959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Y.; Grant, S. New insights into Checkpoint Kinase 1 in the DNA damage response signaling network. Clin. Cancer Res. 2010, 16, 376–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastan, M.B.; Lim, D.S. The Many Substrates and Functions of ATM. Available online: https://pubmed.ncbi.nlm.nih.gov/11252893/ (accessed on 21 September 2020).
- Babiker, H.M.; McBride, A.; Cooke, L.S.; Mahadevan, D. Therapeutic potential of investigational CHK-1 inhibitors for the treatment of solid tumors. Expert Opin. Investig. Drugs 2017, 26, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.; Eastman, A. The cancer therapeutic potential of Chk1 inhibitors: How mechanistic studies impact on clinical trial design. Br. J. Clin. Pharmacol. 2013, 76, 358–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Chou, T.-C. Drug combination studies and their synergy quantification using the chou-talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.-C. Preclinical versus clinical drug combination studies. Leuk. Lymphoma 2008, 49, 2059–2080. [Google Scholar] [CrossRef]
- Ocana, A.; Pandiella, A.; Siu, L.L.; Tannock, I.F. Preclinical development of molecular-targeted agents for cancer. Nat. Rev. Clin. Oncol. 2011, 8, 200–209. [Google Scholar] [CrossRef]
- Ben-David, U.; Siranosian, B.; Ha, G.; Tang, H.; Oren, Y.; Hinohara, K.; Strathdee, C.A.; Dempster, J.; Lyons, N.J.; Burns, R.; et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature 2018, 560, 325–330. [Google Scholar] [CrossRef]
- Massey, A.J.; Stokes, S.; Browne, H.; Foloppe, N.; Fiumana, A.; Scrace, S.; Fallowfield, M.; Bedford, S.; Webb, P.; Baker, L.; et al. Identification of novel, in vivo active Chk1 inhibitors utilizing structure guided drug design. Oncotarget 2015, 6, 35797–35812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herůdková, J.; Paruch, K.; Khirsariya, P.; Souček, K.; Krkoška, M.; Vondálová Blanářová, O.; Sova, P.; Kozubík, A.; Vaculová, A.H. Chk1 inhibitor SCH900776 effectively potentiates the cytotoxic effects of platinum-based chemotherapeutic drugs in human colon cancer cells. Neoplasia 2017, 19, 830–841. [Google Scholar] [CrossRef] [PubMed]
- Hinohara, K.; Polyak, K. Intratumoral heterogeneity: More than just mutations. Trends Cell Biol. 2019, 29, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.-H.; Zhao, X.; Zhu, J.; Kim, I.-K.; Rao, G.; McCutcheon, J.; Hsu, S.-T.; Teicher, B.; Kallakury, B.; Dowlati, A.; et al. Chk1 inhibition enhances cisplatin cytotoxicity and overcomes cisplatin resistance in small cell lung cancer by promoting mitotic cell death. J. Thorac. Oncol. 2019, 14, 1032. [Google Scholar] [CrossRef] [PubMed]
- Montero, J.C.; Esparís-Ogando, A.; Re-Louhau, M.F.; Seoane, S.; Abad, M.; Calero, R.; Ocaña, A.; Pandiella, A. Active kinase profiling, genetic and pharmacological data define mTOR as an important common target in triple-negative breast cancer. Oncogene 2014, 33, 148–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bareche, Y.; Buisseret, L.; Gruosso, T.; Girard, E.; Venet, D.; Dupont, F.; Desmedt, C.; Larsimont, D.; Park, M.; Rothé, F.; et al. Unraveling triple-negative breast cancer tumor microenvironment heterogeneity: Towards an optimized treatment approach. J. Natl. Cancer Inst. 2020, 112, 708–719. [Google Scholar] [CrossRef] [Green Version]
- Ocaña, A.; García-Alonso, S.; Amir, E.; Pandiella, A. Refining early antitumoral drug development. Trends Pharmacol. Sci. 2018, 39, 922–925. [Google Scholar] [CrossRef]
- Pegram, M.D.; Konecny, G.E.; O’Callaghan, C.; Beryt, M.; Pietras, R.; Slamon, D.J. Rational combinations of trastuzumab with chemotherapeutic drugs used in the treatment of breast cancer. J. Natl. Cancer Inst. 2004, 96, 739–749. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | Origin | Receptor Expression | Mutations | ||||
|---|---|---|---|---|---|---|---|
| Tissue | Disease | EGFR | ER | PR | HER2 | ||
| MDA-MB-231 | Mammary gland/breast; derived from metastatic site: pleural effusion | Epithelial adenocarcinoma | Positive | Negative | Negative | Positive (low expression) | BRAF, CD79A, CRTC3, NF2, PCSK7, PDGFRA, TP53 |
| BT549 | Mammary gland/breast | Epithelial ductal carcinoma | Positive | Negative | Negative | Negative | PTEN, TP53, RB1 |
| HS578T | Mammary gland/breast | Epithelial carcinoma | Positive | Negative | Negative | Negative | HNF1A, NF1, PIK3R1, TP53 |
| HCC1353 | Mammary gland/breast | Ductal carcinoma | Positive | Negative | Negative | Negative | BRCA1 |
| MCF10A | Mammary gland/breast | Fibrocystic disease | Positive | Negative | Negative | Negative | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nieto-Jimenez, C.; Alcaraz-Sanabria, A.; Martinez-Canales, S.; Corrales-Sanchez, V.; Montero, J.C.; Burgos, M.; Nuncia-Cantarero, M.; Pandiella, A.; Galan-Moya, E.M.; Ocaña, A. Checkpoint Kinase 1 Pharmacological Inhibition Synergizes with DNA-Damaging Agents and Overcomes Platinum Resistance in Basal-Like Breast Cancer. Int. J. Mol. Sci. 2020, 21, 9034. https://doi.org/10.3390/ijms21239034
Nieto-Jimenez C, Alcaraz-Sanabria A, Martinez-Canales S, Corrales-Sanchez V, Montero JC, Burgos M, Nuncia-Cantarero M, Pandiella A, Galan-Moya EM, Ocaña A. Checkpoint Kinase 1 Pharmacological Inhibition Synergizes with DNA-Damaging Agents and Overcomes Platinum Resistance in Basal-Like Breast Cancer. International Journal of Molecular Sciences. 2020; 21(23):9034. https://doi.org/10.3390/ijms21239034
Chicago/Turabian StyleNieto-Jimenez, Cristina, Ana Alcaraz-Sanabria, Sandra Martinez-Canales, Veronica Corrales-Sanchez, Juan Carlos Montero, Miguel Burgos, Miriam Nuncia-Cantarero, Atanasio Pandiella, Eva M. Galan-Moya, and Alberto Ocaña. 2020. "Checkpoint Kinase 1 Pharmacological Inhibition Synergizes with DNA-Damaging Agents and Overcomes Platinum Resistance in Basal-Like Breast Cancer" International Journal of Molecular Sciences 21, no. 23: 9034. https://doi.org/10.3390/ijms21239034
APA StyleNieto-Jimenez, C., Alcaraz-Sanabria, A., Martinez-Canales, S., Corrales-Sanchez, V., Montero, J. C., Burgos, M., Nuncia-Cantarero, M., Pandiella, A., Galan-Moya, E. M., & Ocaña, A. (2020). Checkpoint Kinase 1 Pharmacological Inhibition Synergizes with DNA-Damaging Agents and Overcomes Platinum Resistance in Basal-Like Breast Cancer. International Journal of Molecular Sciences, 21(23), 9034. https://doi.org/10.3390/ijms21239034