Impact of Genetic Variations and Epigenetic Mechanisms on the Risk of Obesity



 and
and 
Abstract
:1. Introduction
2. The Role of Hormones in Appetite and Weight Regulation
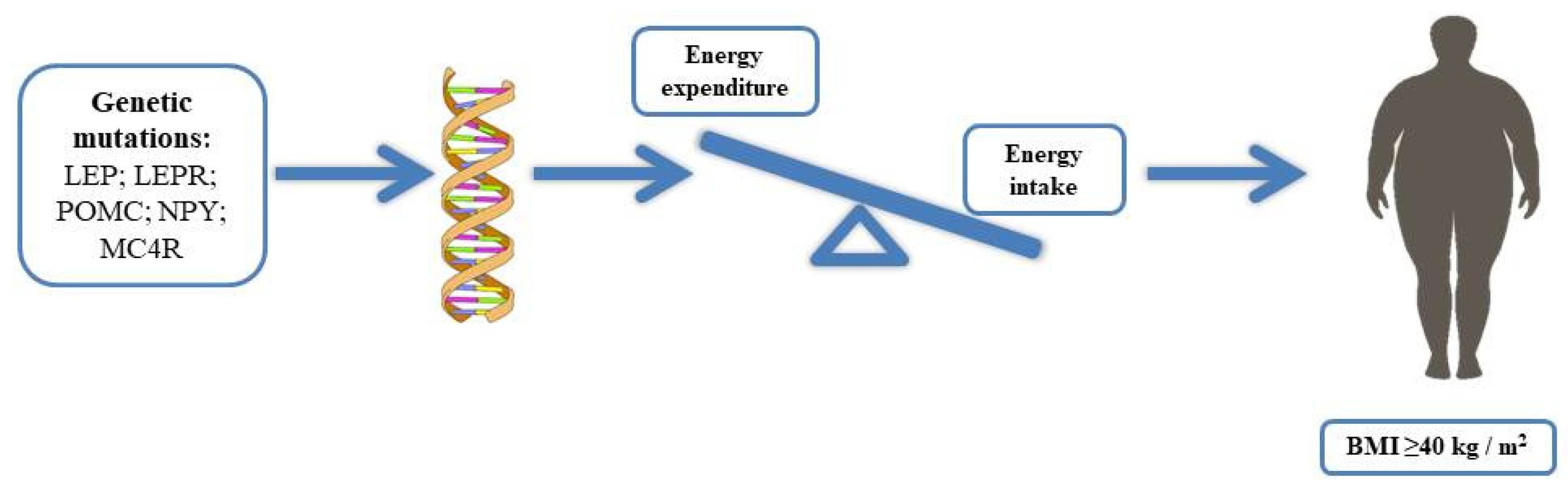
3. Monogenic Obesity
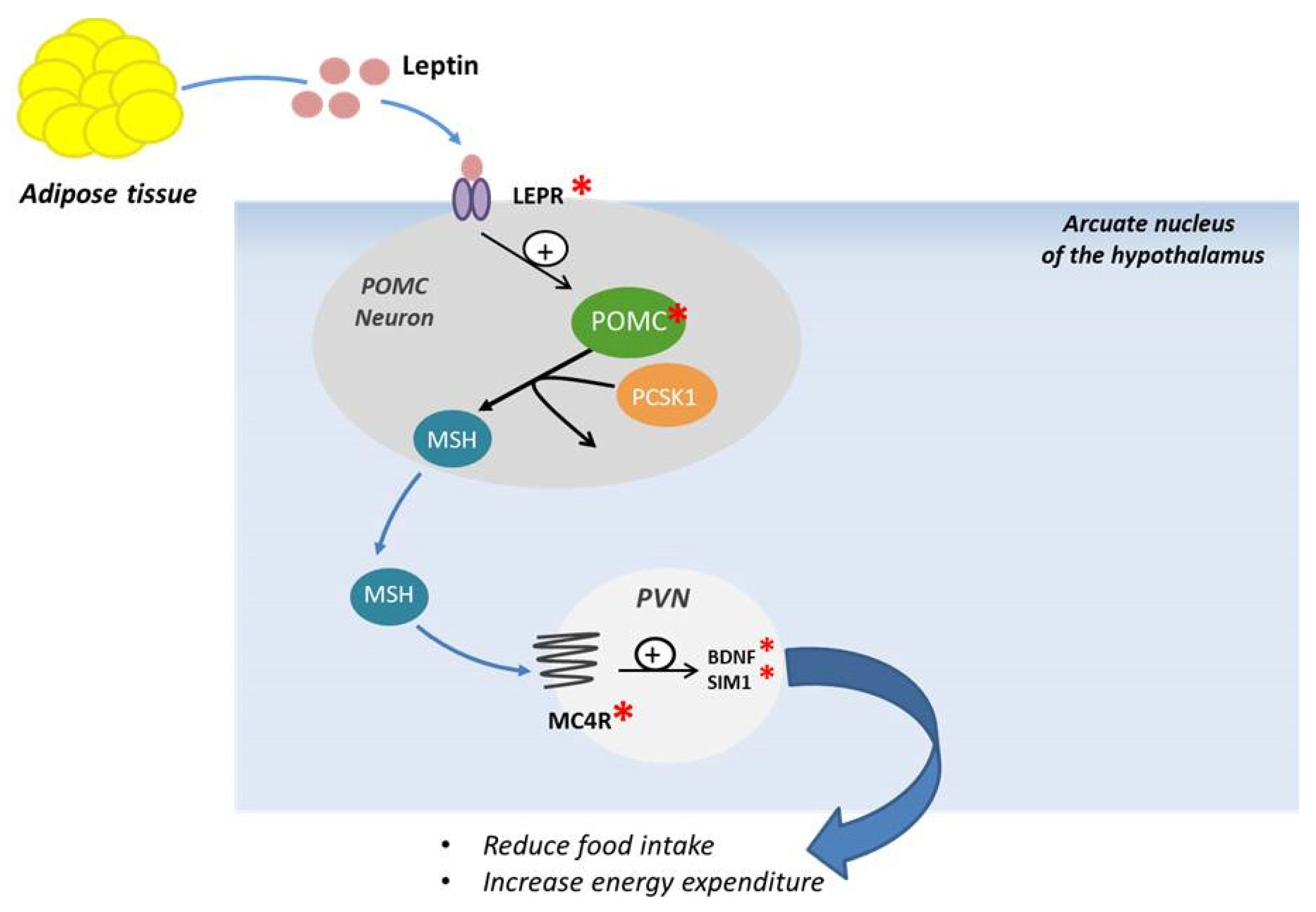
3.1. Leptin and Leptin Receptor
3.2. Proopiomelanocortin (POMC)
3.3. Prohormone Convertase 1/3 and 2 (PC1/3 and PC2)
3.4. Melanocortin Receptor (MC4R)
3.5. SIM1, BDNF and TRKB
4. Syndromic Obesity
5. Polygenic Obesity
6. Epigenetic Mechanisms and Obesity Risk
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACTH | Adrenocorticotrophic Hormone |
| AGRP | Agouti Related Protein |
| ARC | Arcuate Nucleus |
| BDNF | Brain-derived Neurotrophic Factor |
| BMI | Body Mass Index |
| cAMP | cyclic Adenosine Monophosphate |
| CCK | Cholecystokinin |
| CNS | Central Nervous System |
| FTO | Fat mass and obesity |
| GHSR | Growth Hormone Receptor |
| GLP1 | Glucagon-like peptide 1 |
| INSIG2 | Insulin-induced Gene 2 |
| IRS | Insulin Receptor Substrate |
| LEP | Leptin |
| LEPR | Leptin Receptor |
| MC4R | Melanocortin Receptor |
| ncRNA | non-coding RNA |
| NPY | Neuropeptide Y |
| PC | Prohormone Convertase |
| PI3K | Phosphatidylinositide-3 Kinase |
| POMC | Proopiomelanocortin |
| PYY | Tyrosine- Tyrosine Polypeptide |
| SIM1 | Sim BHLH Transcription Factor1 |
| TRKB | Tropomyosin-Related Kinase B |
| WHO | World Health Organization |
| α-MSH | α-melanocyte stimulating hormone |
References
- Hruby, A.; Hu, F.B. The Epidemiology of Obesity: A Big Picture. Pharmacoeconomics 2015, 33, 673–689. [Google Scholar] [CrossRef] [PubMed]
- De Lorenzo, A.; Gratteri, S.; Gualtieri, P.; Cammarano, A.; Bertucci, P.; Di Renzo, L. Why primary obesity is a disease? J. Transl. Med. 2019, 17, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pi-Sunyer, X. The Medical Risks of Obesity. Postgrad. Med. 2009, 121, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Alasmari, H.D.; Al-Shehri, A.D.; Aljuaid, T.A.; Alzaidi, B.A.; Alswat, K.A. Relationship Between Body Mass Index and Obesity Awareness in School Students. J. Clin. Med. Res. 2017, 9, 520–524. [Google Scholar] [CrossRef] [Green Version]
- Xia, Q.; Grant, S.F.A. The genetics of human obesity. Ann. New York Acad. Sci. 2013, 1281, 178–190. [Google Scholar] [CrossRef]
- Romieu, I.; On behalf of the IARC working group on Energy Balance and Obesity; Dossus, L.; Barquera, S.; Blottière, H.M.; Franks, P.W.; Gunter, M.; Hwalla, N.; Hursting, S.D.; Leitzmann, M.; et al. Energy balance and obesity: What are the main drivers? Cancer Causes Control. 2017, 28, 247–258. [Google Scholar] [CrossRef] [Green Version]
- Hill, J.; Wyatt, H.R.; Peters, J.C. Energy Balance and Obesity. Circulation 2012, 126, 126–132. [Google Scholar] [CrossRef]
- Herrera, B.M.; Lindgren, C.M. The Genetics of Obesity. Curr. Diabetes Rep. 2010, 10, 498–505. [Google Scholar] [CrossRef] [Green Version]
- McPherson, R. Genetic contributors to obesity. Can. J. Cardiol. 2007, 23, 23A–27A. [Google Scholar] [CrossRef] [Green Version]
- Thaker, V.V. Genetic and epigenetic causes of obesity. Adolesc. Med. State art Rev. 2017, 28, 379–405. [Google Scholar]
- Rao, K.R.; Lal, N.; Giridharan, N. Genetic & epigenetic approach to human obesity. Indian J. Med Res. 2014, 140, 589–603. [Google Scholar] [PubMed]
- Hinney, A.; Vogel, C.I.G.; Hebebrand, J. From monogenic to polygenic obesity: Recent advances. Eur. Child Adolesc. Psychiatry 2010, 19, 297–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Djik, S.; Tellam, R.L.; Morrison, J.L.; Mühlhäusler, B.S.; Molloy, P. Recent developments on the role of epigenetics in obesity and metabolic disease. Clin. Epigenetics 2015, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzika, E.; Dreker, T.; Imhof, A. Epigenetics and Metabolism in Health and Disease. Front. Genet. 2018, 9, 361. [Google Scholar] [CrossRef] [Green Version]
- Chiurazzi, M.; Di Maro, M.; Cozzolino, M.; Colantuoni, A. Mitochondrial Dynamics and Microglia as New Targets in Metabolism Regulation. Int. J. Mol. Sci. 2020, 21, 3450. [Google Scholar] [CrossRef]
- Perry, B.G.; Wang, Y. Appetite regulation and weight control: The role of gut hormones. Nutr. Diabetes 2012, 2, e26. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Zhang, C.; Borgquist, A.; Nestor, C.C.; Smith, A.W.; Bosch, M.A.; Ku, S.; Wagner, E.J.; Rønnekleiv, O.K.; Kelly, M.J. Insulin Excites Anorexigenic Proopiomelanocortin Neurons via Activation of Canonical Transient Receptor Potential Channels. Cell Metab. 2014, 19, 682–693. [Google Scholar] [CrossRef] [Green Version]
- Batterham, R.L.; Bloom, S.R. The Gut Hormone Peptide YY Regulates Appetite. Ann. N. Y. Acad. Sci. 2003, 994, 162–168. [Google Scholar] [CrossRef]
- Rowlands, J.; Heng, J.; Newsholme, P.; Carlessi, R. Pleiotropic Effects of GLP-1 and Analogs on Cell Signaling, Metabolism, and Function. Front. Endocrinol. 2018, 9, 672. [Google Scholar] [CrossRef] [Green Version]
- Huvenne, H.; Dubern, B.; Clément, K.; Poitou, C. Rare Genetic Forms of Obesity: Clinical Approach and Current Treatments in 2016. Obes. Facts 2016, 9, 158–173. [Google Scholar] [CrossRef]
- Ranadive, S.A.; Vaisse, C. Lessons from extreme human obesity: Monogenic disorders. Endocrinol. Metab. Clin. N. Am. 2008, 37, 733–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.S. The role of leptin-melanocortin system and human weight regulation: Lessons from experiments of nature. Ann. Acad. Med. Singapore 2009, 38, 34–44. [Google Scholar] [PubMed]
- Funcke, J.-B.; Von Schnurbein, J.; Lennerz, B.S.; Lahr, G.; Debatin, K.-M.; Fischer, S.; Wabitsch, M. Monogenic forms of childhood obesity due to mutations in the leptin gene. Mol. Cell. Pediatr. 2014, 1, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahima, R.S.; Antwi, D.A. Brain Regulation of Appetite and Satiety. Endocrinol. Metab. Clin. N. Am. 2008, 37, 811–823. [Google Scholar] [CrossRef] [Green Version]
- Klok, M.D.; Jakobsdottir, S.; Drent, M.L. The role of leptin and ghrelin in the regulation of food intake and body weight in humans: A review. Obes. Rev. 2007, 8, 21–34. [Google Scholar] [CrossRef]
- Montague, C.T.; Farooqi, I.S.; Whitehead, J.P.; Soos, M.A.; Rau, H.; Wareham, N.J.; Sewter, C.P.; Digby, J.E.; Mohammed, S.N.; Hurst, J.A.; et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nat. Cell Biol. 1997, 387, 903–908. [Google Scholar] [CrossRef]
- Farooqi, I.S.; O’Rahilly, S. Genetics of Obesity in Humans. Endocr. Rev. 2006, 27, 710–718. [Google Scholar] [CrossRef]
- Yupanqui-Lozno, H.; Bastarrachea, R.A.; Yupanqui-Velazco, M.E.; Alvarez-Jaramillo, M.; Medina-Méndez, E.; Giraldo-Peña, A.P.; Arias-Serrano, A.; Torres-Forero, C.; Garcia-Ordoñez, A.M.; Mastronardi, C.A.; et al. Congenital Leptin Deficiency and Leptin Gene Missense Mutation Found in Two Colombian Sisters with Severe Obesity. Genes 2019, 10, 342. [Google Scholar] [CrossRef] [Green Version]
- Blüher, S.; Shah, S.; Mantzoros, C.S. Leptin Deficiency: Clinical Implications and Opportunities for therapeutic interventions. J. Investig. Med. 2009, 57, 784–788. [Google Scholar] [CrossRef]
- Fairbrother, U.; Kidd, E.; Malagamuwa, T.; Walley, A. Genetics of Severe Obesity. Curr. Diabetes Rep. 2018, 18, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Clément, K.; Vaisse, C.; Lahlou, N.; Cabrol, S.; Pelloux, V.; Cassuto, D.; Gourmelen, M.; Dina, C.; Chambaz, J.; Lacorte, J.-M.; et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nat. Cell Biol. 1998, 392, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Di Blasio, A.M. Faculty Opinions recommendation of Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor. N. Engl. J. Med. 2007, 356, 237–247. [Google Scholar] [CrossRef]
- Millington, G.W. The role of proopiomelanocortin (POMC) neurones in feeding behaviour. Nutr. Metab. 2007, 4, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harno, E.; Ramamoorthy, T.G.; Coll, A.P.; White, A. POMC: The Physiological Power of Hormone Processing. Physiol. Rev. 2018, 98, 2381–2430. [Google Scholar] [CrossRef] [PubMed]
- Krude, H.; Grüters, A. Implications of Proopiomelanocortin (POMC) Mutations in Humans: The POMC Deficiency Syndrome. Trends Endocrinol. Metab. 2000, 11, 15–22. [Google Scholar] [CrossRef]
- Wardlaw, S.L. Hypothalamic proopiomelanocortin processing and the regulation of energy balance. Eur. J. Pharmacol. 2011, 660, 213–219. [Google Scholar] [CrossRef] [Green Version]
- D’Agostino, G.; Diano, S. alpha-Melanocyte stimulating hormone: Production and degradation. J. Mol. Med. 2010, 88, 1195–1201. [Google Scholar] [CrossRef] [Green Version]
- Krude, H.; Biebermann, H.; Luck, W.; Horn, R.; Brabant, G.; Grüters, A. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat. Genet. 1998, 19, 155–157. [Google Scholar] [CrossRef]
- Creemers, J.W.M.; Lee, Y.S.; Oliver, R.L.; Bahceci, M.; Tuzcu, A.; Gokalp, D.; Keogh, J.; Herber, S.; White, A.; O’Rahilly, S.; et al. Mutations in the Amino-Terminal Region of Proopiomelanocortin (POMC) in Patients with Early-Onset Obesity Impair POMC Sorting to the Regulated Secretory Pathway. J. Clin. Endocrinol. Metab. 2008, 93, 4494–4499. [Google Scholar] [CrossRef] [Green Version]
- Hook, V.; Funkelstein, L.; Lu, D.; Bark, S.; Wegrzyn, J.; Hwang, S.-R. Proteases for Processing Proneuropeptides into Peptide Neurotransmitters and Hormones. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 393–423. [Google Scholar] [CrossRef] [Green Version]
- Farooqi, I.S.; Volders, K.; Stanhope, R.; Heuschkel, R.; White, A.; Lank, E.; Keogh, J.; O’Rahilly, S.; Creemers, J.W.M. Hyperphagia and Early-Onset Obesity due to a Novel Homozygous Missense Mutation in Prohormone Convertase 1/3. J. Clin. Endocrinol. Metab. 2007, 92, 3369–3373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Rahilly, S.; Gray, H.; Humphreys, P.J.; Krook, A.; Polonsky, K.S.; White, A.; Gibson, S.; Taylor, K.; Carr, C. Impaired Processing of Prohormones Associated with Abnormalities of Glucose Homeostasis and Adrenal Function. New Engl. J. Med. 1995, 333, 1386–1391. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Molina, B.; Martin, M.G.; Lindberg, I. PCSK1 Variants and Human Obesity. Prog. Mol. Biol. Transl. Sci. 2016, 140, 47–74. [Google Scholar] [CrossRef]
- Krashes, M.J.; Lowell, B.B.; Garfield, A.S. Melanocortin-4 receptor–regulated energy homeostasis. Nat. Neurosci. 2016, 19, 206–219. [Google Scholar] [CrossRef] [Green Version]
- Begriche, K.; Girardet, C.; McDonald, P.; Butler, A.A. Melanocortin-3 Receptors and Metabolic Homeostasis. Prog. Mol. Bio.l Transl. Sci. 2013, 114, 109–146. [Google Scholar] [CrossRef] [Green Version]
- Valette, M.; Bellisle, F.; Carette, C.; Poitou, C.; Dubern, B.; Paradis, G.; Hercberg, S.; Muzard, L.; Clement, K.; Czernichow, S. Eating behaviour in obese patients with melanocortin-4 receptor mutations: A literature review. Int. J. Obes. 2013, 37, 1027–1035. [Google Scholar] [CrossRef] [Green Version]
- Eneli, I.U.; Xu, J.; Webster, M.; McCagg, A.; Van Der Ploeg, L.; Garfield, A.S.; Estrada, E. Tracing the effect of the melanocortin-4 receptor pathway in obesity: Study design and methodology of the TEMPO registry. Appl. Clin. Genet. 2019, 12, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Farooqi, I.S.; Yeo, G.S.H.; Keogh, J.M.; Aminian, S.; Jebb, S.A.; Butler, G.; Cheetham, T.; O’Rahilly, S. Dominant and recessive inheritance of morbid obesity associated with melanocortin 4 receptor deficiency. J. Clin. Investig. 2000, 106, 271–279. [Google Scholar] [CrossRef] [Green Version]
- Vaisse, C.; Clement, K.; Guy-Grand, B.; Froguel, P. A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat. Genet. 1998, 20, 113–114. [Google Scholar] [CrossRef]
- Collet, T.-H.; Dubern, B.; Mokrosinski, J.; Connors, H.; Keogh, J.M.; De Oliveira, E.M.; Henning, E.; Poitou-Bernert, C.; Oppert, J.-M.; Tounian, P.; et al. Evaluation of a melanocortin-4 receptor (MC4R) agonist (Setmelanotide) in MC4R deficiency. Mol. Metab. 2017, 6, 1321–1329. [Google Scholar] [CrossRef]
- Kühnen, P.; Clément, K.; Wiegand, S.; Blankenstein, O.; Gottesdiener, K.; Martini, L.L.; Mai, K.; Blume-Peytavi, U.; Grüters, A.; Krude, H. Proopiomelanocortin Deficiency Treated with a Melanocortin-4 Receptor Agonist. New Engl. J. Med. 2016, 375, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.; Zhang, J.; Thomsen, H.S.; Hansen, E.L.; Hollensted, M.; Madsbad, S.; Hansen, T.; Holst, J.J.; Holm, J.-C.; Torekov, S.S. Patients with Obesity Caused by Melanocortin-4 Receptor Mutations Can Be Treated with a Glucagon-like Peptide-1 Receptor Agonist. Cell Metab. 2018, 28, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Michaud, J.L.; Boucher, F.; Melnyk, A.; Gauthier, F.; Goshu, E.; Lévy, E.; Mitchell, G.A.; Himms-Hagen, J.; Fan, C.-M. Sim1 haploinsufficiency causes hyperphagia, obesity and reduction of the paraventricular nucleus of the hypothalamus. Hum. Mol. Genet. 2001, 10, 1465–1473. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Xie, X. Neurotrophic factor control of satiety and body weight. Nat. Rev. Neurosci. 2016, 17, 282–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stagi, S.; Bianconi, M.; Sammarco, R.A.M.A.; Giglio, S.; De Martino, M. New Thoughts on Pediatric Genetic Obesity: Pathogenesis, Clinical Characteristics and Treatment Approach; IntechOpen: London, UK, 2017; p. 213. [Google Scholar]
- Forsythe, E.; Beales, P.L. Bardet–Biedl syndrome. Eur. J. Hum. Genet. 2013, 21, 8–13. [Google Scholar] [CrossRef]
- Manara, E.; Paolacci, S.; D’Esposito, F.; Abeshi, A.; Ziccardi, L.; Falsini, B.; Colombo, L.; Iarossi, G.; Pilotta, A.; Boccone, L.; et al. Mutation profile of BBS genes in patients with Bardet–Biedl syndrome: An Italian study. Ital. J. Pediatr. 2019, 45, 1–8. [Google Scholar] [CrossRef]
- Butler, M.G.; Manzardo, A.M.; Forster, J.L. Prader-Willi Syndrome: Clinical Genetics and Diagnostic Aspects with Treatment Approaches. Curr. Pediatr. Rev. 2016, 12, 136–166. [Google Scholar] [CrossRef]
- Marshall, J.D.; Maffei, P.; Collin, G.B.; Naggert, J.K. Alstrom Syndrome: Genetics and Clinical Overview. Curr. Genom. 2011, 12, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Alaimo, J.T.; Barton, L.V.; Mullegama, S.V.; Wills, R.D.; Foster, R.H.; Elsea, S.H. Individuals with Smith-Magenis syndrome display profound neurodevelopmental behavioral deficiencies and exhibit food-related behaviors equivalent to Prader-Willi syndrome. Res. Dev. Disabil. 2015, 47, 27–38. [Google Scholar] [CrossRef]
- Chen, L.; Mullegama, S.V.; Alaimo, J.T.; Elsea, S.H. Smith-Magenis syndrome and its circadian influence on development, behavior, and obesity - own experience. Dev. Period Med. 2015, 19, 149–156. [Google Scholar]
- Hinney, A.; Hebebrand, J. Polygenic obesity in humans. Obes. Facts 2008, 1, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Hess, M.E.; Brüning, J.C. The fat mass and obesity-associated (FTO) gene: Obesity and beyond? Biochim. Biophys. Acta 2014, 1842, 2039–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harbron, J.; Van Der Merwe, L.L.; Zaahl, M.G.; Kotze, M.; Senekal, M. Fat Mass and Obesity-Associated (FTO) Gene Polymorphisms Are Associated with Physical Activity, Food Intake, Eating Behaviors, Psychological Health, and Modeled Change in Body Mass Index in Overweight/Obese Caucasian Adults. Nutrients 2014, 6, 3130–3152. [Google Scholar] [CrossRef] [Green Version]
- Malzahn, D.; Müller-Nurasyid, M.; Heid, I.M.; Wichmann, H.-E.; Bickeböller, H. Controversial association results for INSIG2 on body mass index may be explained by interactions with age and with MC4R. Eur. J. Hum. Genet. 2014, 22, 1217–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prakash, J.; Mittal, B.; Srivastava, A.; Awasthi, S.; Srivastava, P.; Srivastava, N. Common Genetic Variant of INSIG2 Gene rs7566605 Polymorphism Is Associated with Severe Obesity in North India. Iran. Biomed. J. 2017, 21, 261–269. [Google Scholar] [CrossRef] [Green Version]
- Moosavi, A.; Ardekani, A.M. Role of Epigenetics in Biology and Human Diseases. Iran. Biomed. J. 2016, 20, 246–258. [Google Scholar]
- Peaston, A.; Whitelaw, E. Epigenetics and phenotypic variation in mammals. Mamm. Genome 2006, 17, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic Modifications. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef] [Green Version]
- Al Aboud, N.M.; Tupper, C.; Jialal, I. Genetics, Epigenetic Mechanism; StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar] [PubMed]
- Qureshi, I.A.; Mehler, M.F. Epigenetic mechanisms underlying nervous system diseases. In Handbook of Clinical Neurology; Elsevier BV: Amsterdam, The Netherlands, 2018; Volume 147, pp. 43–58. [Google Scholar]
- Dendup, T.; Feng, X.; Clingan, S.; Astell-Burt, T. Environmental Risk Factors for Developing Type 2 Diabetes Mellitus: A Systematic Review. Int. J. Environ. Res. Public Heal. 2018, 15, 78. [Google Scholar] [CrossRef] [Green Version]
- Martínez, J.A.; Milagro, F.I.; Claycombe, K.J.; Schalinske, K.J.A. Epigenetics in Adipose Tissue, Obesity, Weight Loss, and Diabetes. Adv. Nutr. 2014, 5, 71–81. [Google Scholar] [CrossRef] [Green Version]
- Youngson, N.A.; Morris, M.J. What obesity research tells us about epigenetic mechanisms. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20110337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, C.; Rönn, T. Epigenetics in Human Obesity and Type 2 Diabetes. Cell Metab. 2019, 29, 1028–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y. Epigenetic Mechanisms Link Maternal Diets and Gut Microbiome to Obesity in the Offspring. Front. Genet. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Parlee, S.D.; MacDougald, O.A. Maternal nutrition and risk of obesity in offspring: The Trojan horse of developmental plasticity. Biochim. Biophys. Acta 2014, 1842, 495–506. [Google Scholar] [CrossRef] [Green Version]
- Tiffon, C. The Impact of Nutrition and Environmental Epigenetics on Human Health and Disease. Int. J. Mol. Sci. 2018, 19, 3425. [Google Scholar] [CrossRef] [Green Version]
- Indrio, F.; Martini, S.; Francavilla, R.; Corvaglia, L.; Cristofori, F.; Mastrolia, S.A.; Neu, J.; Rautava, S.; Spena, G.R.; Raimondi, F.; et al. Epigenetic Matters: The Link between Early Nutrition, Microbiome, and Long-term Health Development. Front. Pediatr. 2017, 5, 178. [Google Scholar] [CrossRef]
- Begum, G.; Stevens, A.; Smith, E.B.; Connor, K.; Challis, J.R.G.; Bloomfield, F.; White, A. Epigenetic changes in fetal hypothalamic energy regulating pathways are associated with maternal undernutrition and twinning. FASEB J. 2012, 26, 1694–1703. [Google Scholar] [CrossRef] [Green Version]
- Samblas, M.; Milagro, F.I.; Martínez, J.A. DNA methylation markers in obesity, metabolic syndrome, and weight loss. Epigenetics 2019, 14, 421–444. [Google Scholar] [CrossRef]
- Ideraabdullah, F.Y.; Zeisel, S.H. Dietary Modulation of the Epigenome. Physiol. Rev. 2018, 98, 667–695. [Google Scholar] [CrossRef] [Green Version]
- Campión, J.; Milagro, F.I.; Goyenechea, E.; Martinez, J.A. TNF-α Promoter Methylation as a Predictive Biomarker for Weight-loss Response. Obesity 2009, 17, 1293–1297. [Google Scholar] [CrossRef]
- Cordero, P.; Campión, J.; Milagro, F.; Goyenechea, E.; Steemburgo, T.; Javierre, B.M.; Martinez, J.A. Leptin and TNF-alpha promoter methylation levels measured by MSP could predict the response to a low-calorie diet. J. Physiol. Biochem. 2011, 67, 463–470. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Genetic Obesity | ||
|---|---|---|
| Non-Syndromic Forms | Syndromic Form | |
| Monogenic Obesity | Polygenic Obesity | Chromosomal or Pleiotropic Forms |
| Leptin deficiency LEPR mutation POMC deficiency PC mutation MC4R mutation SIM1, BDNF, TRKB mutations | MC4R mutation FTO mutation INSIG2 mutation | Bardet Biedl syndrome Prader Willi syndrome Alstrom syndrome Smith–Magenis syndrome |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiurazzi, M.; Cozzolino, M.; Orsini, R.C.; Di Maro, M.; Di Minno, M.N.D.; Colantuoni, A. Impact of Genetic Variations and Epigenetic Mechanisms on the Risk of Obesity. Int. J. Mol. Sci. 2020, 21, 9035. https://doi.org/10.3390/ijms21239035
Chiurazzi M, Cozzolino M, Orsini RC, Di Maro M, Di Minno MND, Colantuoni A. Impact of Genetic Variations and Epigenetic Mechanisms on the Risk of Obesity. International Journal of Molecular Sciences. 2020; 21(23):9035. https://doi.org/10.3390/ijms21239035
Chicago/Turabian StyleChiurazzi, Martina, Mauro Cozzolino, Roberta Clara Orsini, Martina Di Maro, Matteo Nicola Dario Di Minno, and Antonio Colantuoni. 2020. "Impact of Genetic Variations and Epigenetic Mechanisms on the Risk of Obesity" International Journal of Molecular Sciences 21, no. 23: 9035. https://doi.org/10.3390/ijms21239035
APA StyleChiurazzi, M., Cozzolino, M., Orsini, R. C., Di Maro, M., Di Minno, M. N. D., & Colantuoni, A. (2020). Impact of Genetic Variations and Epigenetic Mechanisms on the Risk of Obesity. International Journal of Molecular Sciences, 21(23), 9035. https://doi.org/10.3390/ijms21239035