Adipokines and Metabolic Regulators in Human and Experimental Pulmonary Arterial Hypertension

 ,
, 
Abstract
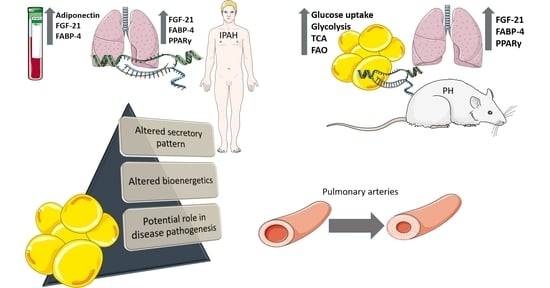
:
1. Introduction
2. Results
2.1. Circulating Concentrations of FABP-4, FGF-21, and Adiponectin Are Significantly Elevated in Patients with Idiopathic Pulmonary Arterial Hypertension (IPAH) Compared to Healthy Controls
2.2. Lung Levels of FGF-21, FABP-4, and PPARγ mRNA Are Significantly Elevated in Patients with IPAH Compared to Healthy Controls
2.3. Pulmonary Hypertension Was Induced in All Three PH-Experimental Models
2.4. Lung Levels of FGF-21, FABP-4, and PPARγ mRNA Are Significantly Elevated in Experimental Models of PH
2.5. Altered Expression of PPARγ and FABP-4 in Adipose Tissue in Experimental PH
2.6. Metabolic Pathway Gene Expression Is Altered in Rat Adipose Tissue in Experimental PH
3. Discussion
4. Materials and Methods
4.1. Human IPAH and Control Samples
4.2. Animals and Experimental Models of Pulmonary Hypertension
4.3. Hemodynamic Measurements.
4.4. Right Ventricular Weight and FI
4.5. Rat Tissue Isolation
4.6. Gene Expression Analysis
4.7. Measurement of Circulating Levels of Adipokines
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACC2 | Acetyl-CoA carboxylase-2 |
| ACADS | AcylCoA dehydrogenases (ACAD) for short fatty acid substrates |
| ACADAM | AcylCoA dehydrogenases (ACAD) for medium fatty acid substrates |
| ACADVL | AcylCoA dehydrogenases (ACAD) for very long-chain fatty acid substrates |
| ADIPOR1 | Adiponectin receptor 1 |
| ADIPOR2 | Adiponectin receptor 2 |
| AND | Adiponectin |
| CS | Citrate Synthase |
| FABP-4 | Fatty Acid Binding Protein-4 |
| FGF-21 | Fibroblast Growth Factor-21 |
| FAO | Fatty Acid Oxidation |
| FI | Fulton’s Index |
| GLUT-1 | Glucose transporter-1 |
| GLUT-4 | Glucose transported-4 |
| IPAH | Idiopathic Pulmonary Arterial Hypertension |
| MCT | Monocrotaline |
| LVSP | Left Ventricular Systolic Pressure |
| PAH | Pulmonary Arterial Hypertension |
| PH | Pulmonary Hypertension |
| PDHb | Pyruvate dehydrogenase beta subunit |
| PDK | Pyruvate Dehydrogenase Kinase |
| PFK-1 | Phosphofructokinase-1 |
| PPARα | Peroxisome proliferator-activated receptor α |
| PPARγ | Peroxisome proliferator-activated receptor γ |
| RVSP | Right Ventricular Systolic Pressure |
| TCA | Tricarboxylic Acid Cycle |
References
- Sutendra, G.; Michelakis, E.D. The metabolic basis of pulmonary arterial hypertension. Cell Metab. 2014, 19, 558–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassoun, P.M.; Mouthon, L.; Barberà, J.A.; Eddahibi, S.; Flores, S.C.; Grimminger, F.; Jones, P.L.; Maitland, M.L.; Michelakis, E.D.; Morrell, N.W.; et al. Inflammation, Growth Factors, and Pulmonary Vascular Remodeling. J. Am. Coll. Cardiol. 2009, 54, S10–S19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabinovitch, M.; Guignabert, C.; Humbert, M.; Nicolls, M.R. Inflammation and Immunity in the Pathogenesis of Pulmonary Arterial Hypertension. Circ. Res. 2014, 115, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Zamanian, R.T.; Hansmann, G.; Snook, S.; Lilienfeld, D.; Rappaport, K.M.; Reaven, G.M.; Rabinovitch, M.; Doyle, R.L. Insulin resistance in pulmonary arterial hypertension. Eur. Respir. J. 2009, 33, 318–324. [Google Scholar] [CrossRef]
- Mair, K.M.; Gaw, R.; MacLean, M.R. Obesity, estrogens and adipose tissue dysfunction—Implications for pulmonary arterial hypertension. Pulm. Circ. 2020, 10, 2045894020952019. [Google Scholar] [CrossRef]
- Poms, A.D.; Turner, M.; Farber, H.W.; Meltzer, L.A.; McGoon, M.D. Comorbid conditions and outcomes in patients with pulmonary arterial hypertension: A REVEAL registry analysis. Chest 2013, 144, 169–176. [Google Scholar] [CrossRef]
- Assad, T.R.; Hemnes, A.R. Metabolic Dysfunction in Pulmonary Arterial Hypertension. Curr. Hypertens. Rep. 2015, 17, 20. [Google Scholar] [CrossRef] [Green Version]
- Friedman, S.E.; Andrus, B.W. Obesity and Pulmonary Hypertension: A Review of Pathophysiologic Mechanisms. J. Obes. 2012, 2012, 505274. [Google Scholar] [CrossRef] [Green Version]
- Ranchoux, B.; Nadeau, V.; Bourgeois, A.; Provencher, S.; Tremblay, É.; Omura, J.; Coté, N.; Abu-Alhayja’a, R.; Dumais, V.; Nachbar, R.T.; et al. Metabolic Syndrome Exacerbates Pulmonary Hypertension due to Left Heart Disease. Circ. Res. 2019, 125, 449–466. [Google Scholar] [CrossRef]
- Li, C.; Xu, M.M.; Wang, K.; Adler, A.J.; Vella, A.T.; Zhou, B. Macrophage polarization and meta-inflammation. Transl. Res. 2018, 191, 29–44. [Google Scholar] [CrossRef]
- Luo, L.; Liu, M. Adipose tissue in control of metabolism. J. Endocrinol. 2016, 231, R77–R99. [Google Scholar] [CrossRef] [Green Version]
- Perrotta, F.; Nigro, E.; Mollica, M.; Costigliola, A.; D’Agnano, V.; Daniele, A.; Bianco, A.; Guerra, G. Pulmonary Hypertension and Obesity: Focus on Adiponectin. Int. J. Mol. Sci. 2019, 20, 912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Q.; Johns, R.A. Resistin family proteins in pulmonary diseases. Am. J. Physiol. Cell. Mol. Physiol. 2020, 319, L422–L434. [Google Scholar] [CrossRef] [PubMed]
- Paulin, R.; Michelakis, E.D. The Metabolic Theory of Pulmonary Arterial Hypertension. Circ. Res. 2014, 115, 148–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucherat, O.; Peterlini, T.; Bourgeois, A.; Nadeau, V.; Breuils-Bonnet, S.; Boilet-Molez, S.; Potus, F.; Meloche, J.; Chabot, S.; Lambert, C.; et al. Mitochondrial HSP90 accumulation promotes vascular remodelin in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2018, 198, 90. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Calvo, R.; Girona, J.; Alegret, J.M.; Bosquet, A.; Ibarretxe, D.; Masana, L. Role of the fatty acid-binding protein 4 in heart failure and cardiovascular disease. J. Endocrinol. 2017, 233, R173–R184. [Google Scholar] [CrossRef] [PubMed]
- Sunaga, H.; Koitabashi, N.; Iso, T.; Matsui, H.; Obokata, M.; Kawakami, R.; Murakami, M.; Yokoyama, T.; Kurabayashi, M. Activation of cardiac AMPK-FGF21 feed-forward loop in acute myocardial infarction: Role of adrenergic overdrive and lipolysis byproducts. Sci. Rep. 2019, 9, 11841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciarka, A.; Doan, V.; Velez-Roa, S.; Naeije, R.; van de Borne, P. Prognostic significance of sympathetic nervous system acti-vation in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2010, 181, 1269–1275. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Bernlohr, D.A. Metabolic functions of FABPs—Mechanisms and therapeutic implications. Nat. Rev. Endocrinol. 2015, 11, 592–605. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.E.; Samocha-Bonet, D.; Whitworth, P.T.; Fazakerley, D.J.; Turner, N.; Biden, T.J.; James, D.E.; Cantley, J. Identification of fatty acid binding protein 4 as an adipokine that regulates insulin secretion during obesity. Mol. Metab. 2014, 3, 465–473. [Google Scholar] [CrossRef]
- Wang, Y.; Mak, J.C.; Lee, M.Y.K.; Xu, A.; Ip, M.S. Low-Frequency Intermittent Hypoxia Promotes Subcutaneous Adipogenic Differentiation. Oxid. Med. Cell. Longev. 2018, 2018, 4501757. [Google Scholar] [CrossRef] [PubMed]
- Lam, D.C.-L.; Xu, A.; Lam, K.S.-L.; Lam, B.; Lam, J.C.-M.; Lui, M.M.-S.; Ip, M.S.-M. Serum adipocyte-fatty acid binding protein level is elevated in severe OSA and correlates with insulin resistance. Eur. Respir. J. 2008, 33, 346–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rich, S.; Dantzker, D.R.; Ayres, S.M.; Bergofsky, E.H.; Brundage, B.H.; Detre, K.M.; Fishman, A.P.; Goldring, R.M.; Groves, B.M.; Koerner, S.K.; et al. Primary Pulmonary Hypertension. Ann. Intern. Med. 1987, 107, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Shields, K.J.; Verdelis, K.; Passineau, M.J.; Faight, E.M.; Zourelias, L.; Wu, C.; Chong, R.; Benza, R.L. Three-Dimensional Micro Computed Tomography Analysis of the Lung Vasculature and Differential Adipose Proteomics in the Sugen/Hypoxia Rat Model of Pulmonary Arterial Hypertension. Pulm. Circ. 2016, 6, 586–596. [Google Scholar] [CrossRef]
- Summer, R.; Fiack, C.A.; Ikeda, Y.; Sato, K.; Dwyer, D.; Ouchi, N.; Fine, A.; Farber, H.W.; Walsh, K. Adiponectin deficiency: A model of pulmonary hypertension associated with pulmonary vascular disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L432–L438. [Google Scholar] [CrossRef] [Green Version]
- Weng, M.; Raher, M.J.; Leyton, P.; Combs, T.P.; Scherer, P.E.; Bloch, K.D.; Medoff, B.D. Adiponectin Decreases Pulmonary Arterial Remodeling in Murine Models of Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2011, 45, 340–347. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhang, B.; Hao, C.; Huang, X.; Li, X.; Huang, Y.; Luo, Z.-Q. Omentin-A Novel Adipokine in Respiratory Diseases. Int. J. Mol. Sci. 2017, 19, 73. [Google Scholar] [CrossRef] [Green Version]
- Santos, M.; Reis, A.; Gonçalves, F.; Ferreira-Pinto, M.J.; Cabral, S.; Torres, S.; Leite-Moreira, A.F.; Henriques-Coelho, T. Adiponectin Levels Are Elevated in Patients with Pulmonary Arterial Hypertension. Clin. Cardiol. 2013, 37, 21–25. [Google Scholar] [CrossRef]
- Menzaghi, C.; Trischitta, V. The Adiponectin Paradox for All-Cause and Cardiovascular Mortality. Diabetes 2017, 67, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ma, X.L.; Lau, W.B. Cardiovascular Adiponectin Resistance: The Critical Role of Adiponectin Receptor Modification. Trends Endocrinol. Metab. 2017, 28, 519–530. [Google Scholar] [CrossRef]
- Hui, X.; Feng, T.; Liu, Q.; Gao, Y.; Xu, A. The FGF21–adiponectin axis in controlling energy and vascular homeostasis. J. Mol. Cell Biol. 2016, 8, 110–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, W.L.; Adams, A.C.; Brozinick, J.T.; Bui, H.H.; Miyauchi, Y.; Kusminski, C.M.; Bauer, S.M.; Wade, M.; Singhal, E.; Cheng, C.C.; et al. An FGF21-adiponectin-ceramide axis con-trols energy expenditure and insulin action in mice. Cell Metab. 2013, 17, 790–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tezze, C.; Romanello, V.; Sandri, M. FGF21 as Modulator of Metabolism in Health and Disease. Front. Physiol. 2019, 10, 419. [Google Scholar] [CrossRef] [PubMed]
- Suomalainen, A.; Elo, J.M.; Pietiläinen, K.H.; Hakonen, A.H.; Sevastianova, K.; Korpela, M.; Isohanni, P.; Marjavaara, S.K.; Tyni, T.; Kiuru-Enari, S.; et al. FGF-21 as a biomarker for mus-cle-manifesting mitochondrial respiratory chain deficiencies: A diagnostic study. Lancet Neurol. 2011, 10, 806–818. [Google Scholar] [CrossRef]
- Markan, K.R.; Naber, M.C.; Ameka, M.K.; Anderegg, M.D.; Mangelsdorf, D.J.; Kliewer, S.A.; Mohammadi, M.; Potthoff, M.J. Circulating FGF21 Is Liver Derived and Enhances Glucose Uptake During Refeeding and Overfeeding. Diabetes 2014, 63, 4057–4063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.H.; Jeong, Y.T.; Oh, H.; Kim, S.-H.; Cho, J.M.; Kim, Y.-N.; Kim, S.S.; Kim, D.H.; Hur, K.Y.; Kim, H.K.; et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat. Med. 2013, 19, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izumiya, Y.; Bina, H.A.; Ouchi, N.; Akasaki, Y.; Kharitonenkov, A.; Walsh, K. FGF21 is an Akt-regulated myokine. FEBS Lett. 2008, 582, 3805–3810. [Google Scholar] [CrossRef] [Green Version]
- Ruan, C.-C.; Kong, L.-R.; Chen, X.-H.; Ma, Y.; Pan, X.-X.; Zhang, Z.-B.; Gao, P. A2A Receptor Activation Attenuates Hypertensive Cardiac Remodeling via Promoting Brown Adipose Tissue-Derived FGF21. Cell Metab. 2018, 28, 476–489.e5. [Google Scholar] [CrossRef] [Green Version]
- Cai, G.; Liu, J.; Wang, M.; Su, L.; Cai, M.; Huang, K.; Li, X.; Li, M.; Wang, L.; Huang, X. Mutual promotion of FGF21 and PPARγ attenuates hypoxia-induced pulmonary hypertension. Exp. Biol. Med. 2019, 244, 252–261. [Google Scholar] [CrossRef]
- Liu, J.; Cai, G.; Li, M.; Fan, S.; Yao, B.; Ping, W.; Huang, Z.; Cai, H.; Dai, Y.; Wang, L.; et al. Fibroblast growth factor 21 attenuates hypoxia-induced pulmonary hypertension by upregulating PPARγ expression and suppressing inflammatory cytokine levels. Biochem. Biophys. Res. Commun. 2018, 504, 478–484. [Google Scholar] [CrossRef]
- Chen, A.; Liu, J.; Zhu, J.; Wang, X.; Xu, Z.; Cui, Z.; Yao, D.; Huang, Z.; Xu, M.; Chen, M.; et al. FGF21 attenuates hypoxia-induced dysfunction and apoptosis in HPAECs through alleviating endoplasmic reticulum stress. Int. J. Mol. Med. 2018, 42, 1684–1694. [Google Scholar] [CrossRef] [PubMed]
- Trayhurn, P. Hypoxia and Adipocyte Physiology: Implications for Adipose Tissue Dysfunction in Obesity. Annu. Rev. Nutr. 2014, 34, 207–236. [Google Scholar] [CrossRef] [PubMed]
- Broderick, T.L.; King, T.M. Upregulation of GLUT-4 in right ventricle of rats with monocrotaline-induced pulmonary hypertension. Med. Sci. Monit. 2008, 14, Br261–Br264. [Google Scholar] [PubMed]
- Calvier, L.; Chouvarine, P.; Legchenko, E.; Hoffmann, N.; Geldner, J.; Borchert, P.; Jonigk, D.; Mozes, M.M.; Hansmann, G. PPARγ Links BMP2 and TGFβ1 Pathways in Vascular Smooth Muscle Cells, Regulating Cell Proliferation and Glucose Metabolism. Cell Metab. 2017, 25, 1118–1134.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.D.; Peng, J.; Lu, C.; Hsin, M.; Mura, M.; Wu, L.; Chu, L.; Zamel, R.; Machuca, T.; Waddell, T.; et al. Metabolomic Heterogeneity of Pulmonary Arterial Hypertension. PLoS ONE 2014, 9, e88727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bujak, R.; Mateo, J.; Blanco, I.; Izquierdo-Garcia, J.L.; Dudzik, D.; Markuszewski, M.J.; Peinado, V.I.; Laclaustra, M.; Barberà, J.A.; Barbas, C.; et al. New Biochemical Insights into the Mechanisms of Pulmonary Arterial Hypertension in Humans. PLoS ONE 2016, 11, e0160505. [Google Scholar] [CrossRef] [PubMed]
- Talati, M.H.; Brittain, E.L.; Fessel, J.P.; Penner, N.; Atkinson, J.; Funke, M.; Greuter, C.; Jerome, W.G.; Freeman, M.; Newman, J.H.; et al. Mechanisms of Lipid Accumulation in the Bone Mor-phogenetic Protein Receptor Type 2 Mutant Right Ventricle. Am. J. Respir. Crit. Care Med. 2016, 194, 719–728. [Google Scholar] [CrossRef] [Green Version]
- Legchenko, E.; Chouvarine, P.; Borchert, P.; Fernandez-Gonzalez, A.; Snay, E.; Meier, M.; Maegel, L.; Mitsialis, S.A.; Rog-Zielinska, E.A.; kourembanas, S.; et al. PPARgamma agonist pioglitazone reverses pulmonary hypertension and prevents right heart failure via fatty acid oxidation. Sci. Transl. Med. 2018, 10, 438. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.-H.; Piao, L.; Hong, Z.; Toth, P.T.; Marsboom, G.; Bache-Wiig, P.; Rehman, J.; Archer, S.L. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: Exploiting Randle’s cycle. J. Mol. Med. 2011, 90, 31–43. [Google Scholar] [CrossRef] [Green Version]
- Hue, L.; Taegtmeyer, H. The Randle cycle revisited: A new head for an old hat. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E578–E591. [Google Scholar] [CrossRef] [Green Version]
- Daval, M.; Foufelle, F.; Ferré, P. Functions of AMP-activated protein kinase in adipose tissue. J. Physiol. 2006, 574, 55–62. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. FGF21 activates AMPK signaling: Impact on metabolic regulation and the aging process. J. Mol. Med. 2016, 95, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Dutchak, P.A.; Katafuchi, T.; Bookout, A.L.; Choi, J.H.; Yu, R.T.; Mangelsdorf, D.J.; Kliewer, S.A. Fibroblast Growth Factor-21 Regulates PPARγ Activity and the Antidiabetic Actions of Thiazolidinediones. Cell 2012, 148, 556–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astapova, O.; Leff, T. Adiponectin and PPARγ: Cooperative and interdependent actions of two key regulators of metabolism. Vitam. Horm. 2012, 90, 143–162. [Google Scholar] [PubMed]
- Dubois, V.; Eeckhoute, J.; Lefebvre, P.; Staels, B. Distinct but complementary contributions of PPAR isotypes to energy homeostasis. J. Clin. Investig. 2017, 127, 1202–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansmann, G.; Zamanian, R.T. PPARgamma activation: A potential treatment for pulmonary hypertension. Sci. Transl. Med. 2009, 1, 12ps4. [Google Scholar] [CrossRef]
- Ameshima, S.; Golpon, H.; Cool, C.D.; Chan, D.; Vandivier, R.W.; Gardai, S.J.; Wick, M.; Namenoff, R.A.; Geraci, M.W.; Voelkel, N.F. Peroxisome proliferator-activated receptor gamma (PPARgamma) expression is decreased in pulmonary hypertension and affects endothelial cell growth. Circ. Res. 2003, 92, 1162–1169. [Google Scholar] [CrossRef] [Green Version]
- Gong, K.; Xing, D.; Li, P.; Aksut, B.; Ambalavanan, N.; Yang, Q.; Nozell, S.E.; Oparil, S.; Chen, T.-F. Hypoxia induces downregulation of PPAR-gamma in isolated pulmonary arterial smooth muscle cells and in rat lung via transforming growth factor-beta signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 301, L899–L907. [Google Scholar] [CrossRef]
- Worley, J.R.; Baugh, M.D.; Hughes, D.A.; Edwards, D.R.; Hogan, A.; Sampson, M.J.; Gavrilovic, J. Metalloproteinase expression in PMA-stimulated THP-1 cells. Effects of peroxisome proliferator-activated receptor-gamma (PPAR gamma) agonists and 9-cis-retinoic acid. J. Biol. Chem. 2003, 278, 51340–51346. [Google Scholar] [CrossRef] [Green Version]
- Martin-Nizard, F.; Furman, C.; Delerive, P.; Kandoussi, A.; Fruchart, J.C.; Staels, B.; Duriez, P. Peroxisome Proliferator–activated Receptor Activators Inhibit Oxidized Low-density Lipoprotein–induced Endothelin-1 Secretion in Endothelial Cells. J. Cardiovasc. Pharmacol. 2002, 40, 822–831. [Google Scholar] [CrossRef]
- Lehman, A.M.; Montford, J.R.; Horita, H.; Ostriker, A.C.; Weiser-Evans, M.C.; Nemenoff, R.A.; Furgeson, S.B. Activation of the retinoid X receptor modulates angiotensin II-induced smooth muscle gene expression and inflammation in vascular smooth muscle cells. Mol. Pharmacol. 2014, 86, 570–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.K.; Lee, J.; Oh, Y.; Lee, Y.-S.; Lee, S.-D. Rosiglitazone attenuates hypoxia-induced pulmonary arterial hypertension in rats. Respirology 2010, 15, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Bauer, N.R.; Moore, T.M.; McMurtry, I.F. Rodent models of PAH: Are we there yet? Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L580–L582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenmark, K.R.; Meyrick, B.; Galie, N.; Mooi, W.J.; McMurtry, I.F. Animal models of pulmonary arterial hypertension: The hope for etiological discovery and pharmacological cure. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L1013–L1032. [Google Scholar] [CrossRef]
- Provencher, S.; Archer, S.L.; Ramirez, F.D.; Hibbert, B.; Paulin, R.; Boucherat, O.; Lacasse, Y.; Bonnet, S. Standards and Methodological Rigor in Pulmonary Arterial Hypertension Preclinical and Translational Research. Circ. Res. 2018, 122, 1021–1032. [Google Scholar] [CrossRef]
- Mam, V.; Tanbe, A.F.; Vitali, S.H.; Arons, E.; Christou, H.A.; Khalil, R.A. Impaired Vasoconstriction and Nitric Oxide-Mediated Relaxation in Pulmonary Arteries of Hypoxia- and Monocrotaline-Induced Pulmonary Hypertensive Rats. J. Pharmacol. Exp. Ther. 2009, 332, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Christou, H.; Hudalla, H.; Michael, Z.; Filatava, E.J.; Minglin, Z.; Zhu, M.; Possomato-Vieira, J.S.; Dias-Junior, C.; Kourembanas, S.; Khalil, R.A. Impaired Pulmonary Arterial Vasoconstriction and Nitric Oxide–Mediated Relaxation Underlie Severe Pulmonary Hypertension in the Sugen-Hypoxia Rat Model. J. Pharmacol. Exp. Ther. 2018, 364, 258–274. [Google Scholar] [CrossRef]
- Christou, H.; Reslan, O.M.; Mam, V.; Tanbe, A.F.; Vitali, S.H.; Touma, M.; Arons, E.; Mitsialis, S.A.; Kourembanas, S.; Khalil, R.A. Improved pulmonary vascular reactivity and decreased hypertrophic remodeling during nonhypercapnic acidosis in experimental pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L875–L890. [Google Scholar] [CrossRef] [Green Version]
- Hudalla, H.; Michael, Z.; Christodoulou, N.; Willis, G.R.; Fernandez-Gonzalez, A.; Filatava, E.J.; Dieffenbach, P.; Fredenburgh, L.E.; Stearman, R.S.; Geraci, M.W.; et al. Carbonic Anhydrase Inhibition Ameliorates Inflammation and Experimental Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2019, 61, 512–524. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RVSP (Mean ± SEM) mm Hg | LVSP (Mean ± SEM) Mm Hg | Fulton’s Index (Mean ± SEM) | RV/BW Ratio (Mean ± SEM) | |
|---|---|---|---|---|
| Normoxia | 21.20 ± 1.8 | 81.83 ± 6.4 | 0.24 ± 0.006 | 0.55 ± 0.02 |
| Hypoxia (Hx) | 37.03 ± 1.5 * | 84.43 ± 6.6 | 0.40 ± 0.050 * | 0.88 ± 0.09 * |
| Monocrotaline (MCT) | 52.18 ± 8.8 * | 97.11 ± 1.9 | 0.46 ± 0.035 * | 0.94 ± 0.09 * |
| SuHx | 38.91 ± 2.3 * | 69.83 ± 6.6 | 0.69 ± 0.048 * | 1.69 ± 0.14 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papathanasiou, A.E.; Spyropoulos, F.; Michael, Z.; Joung, K.E.; Briana, D.D.; Malamitsi-Puchner, A.; Mantzoros, C.S.; Christou, H. Adipokines and Metabolic Regulators in Human and Experimental Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2021, 22, 1435. https://doi.org/10.3390/ijms22031435
Papathanasiou AE, Spyropoulos F, Michael Z, Joung KE, Briana DD, Malamitsi-Puchner A, Mantzoros CS, Christou H. Adipokines and Metabolic Regulators in Human and Experimental Pulmonary Arterial Hypertension. International Journal of Molecular Sciences. 2021; 22(3):1435. https://doi.org/10.3390/ijms22031435
Chicago/Turabian StylePapathanasiou, Aimilia Eirini, Fotios Spyropoulos, Zoe Michael, Kyoung E. Joung, Despina D. Briana, Ariadne Malamitsi-Puchner, Christos S. Mantzoros, and Helen Christou. 2021. "Adipokines and Metabolic Regulators in Human and Experimental Pulmonary Arterial Hypertension" International Journal of Molecular Sciences 22, no. 3: 1435. https://doi.org/10.3390/ijms22031435
APA StylePapathanasiou, A. E., Spyropoulos, F., Michael, Z., Joung, K. E., Briana, D. D., Malamitsi-Puchner, A., Mantzoros, C. S., & Christou, H. (2021). Adipokines and Metabolic Regulators in Human and Experimental Pulmonary Arterial Hypertension. International Journal of Molecular Sciences, 22(3), 1435. https://doi.org/10.3390/ijms22031435