Modulation of Brain Hyperexcitability: Potential New Therapeutic Approaches in Alzheimer’s Disease


Abstract
:1. Introduction
2. Network Dysfunction and Hyperexcitability
3. Targeting Shared Molecular Pathways between Epilepsy and AD
3.1. Amyloid Aβ and Neurodegeneration through Epileptogenesis
3.2. Tau and Neurodegeneration through Epileptogenesis
4. Pro-Epileptogenic Neurotransmitters and Role of Antiseizure Medications
5. Contribution of Vascular Mechanisms and Neuroinflammation to Epileptogenesis
5.1. Cerebrovascular Risk Factors
5.2. Neuroinflammation
6. Who, When, and How to Treat Brain Hyperexcitability: Diagnostic and Therapeutic Challenges
6.1. Diagnostic Tools
6.2. Therapeutic Tools
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ASM | Antiseizure medication |
| MRI | Magnetic resonance imaging |
| EEG | Electroencephalogram |
| E/I | Excitation and inhibition |
| APP | Amyloid precursor protein |
| PSEN1 | Presenilin-1 (gene mutation) |
| PS1 | Presenilin-1 (protein) |
| LTP | Long-term potentiation |
| LTD | Long-term depression |
| hiPSC | Human induced pluripotent stem cell |
| CSF | Cerebrospinal fluid |
| PiB | Pittsburgh B compound amyloid ligand |
| BACE-1 | β-site amyloid precursor protein cleaving enzyme 1 |
| NFT | Neurofibrillary tangles |
| NFL | Neurofilament light chain |
| OGA | O- GlcNAcase |
| MCI | Mild cognitive impairment |
| PDE4 | Phosphodiesterase E4 |
| BBB | Blood–brain barrier |
| ASOs | Antisense oligonucleotides |
| MTBR | Microtubule binding region |
| GSK-3β | Glycogen synthase kinase 3 beta |
| GLT-1 | Glutamate transporter-1 |
| VGLT-1 | Vesicular glutamate transporter 1 |
| SV2A | Synaptic vesicle glycoprotein 2A |
| PSEN2 | Presenilin-2 |
| fMRI | Functional magnetic resonance imaging |
| GlyT-1 | Glycine transporter 1 |
| GlyT-2 | Glycine transporter 2 |
| KD | Ketogenic diet |
| SVD | Small vessel cerebrovascular disease |
| SGLT-2 | Sodium-glucose cotransporter-2 |
| DM | Diabetes mellitus |
| AAV | Adeno-associated virus |
| TMS | Transcranial magnetic stimulation |
| tDCS | Transcranial direct current stimulation |
| tACS | Transcranial alternating current stimulation |
References
- Harris, S.S.; Wolf, F.; De Strooper, B.; Busche, M.A. Tipping the Scales: Peptide-Dependent Dysregulation of Neural Circuit Dynamics in Alzheimer’s Disease. Neuron 2020, 107, 417–435. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Capelli, V.; Husain, M. Cognition and dementia in older patients with epilepsy. Brain 2018, 141, 1592–1608. [Google Scholar] [CrossRef]
- Palop, J.J.; Mucke, L. Epilepsy and cognitive impairments in alzheimer disease. Arch. Neurol. 2009, 66, 435–440. [Google Scholar] [CrossRef] [Green Version]
- Vossel, K.A.; Tartaglia, M.C.; Nygaard, H.B.; Zeman, A.Z.; Miller, B.L. Epileptic activity in Alzheimer’s disease: Causes and clinical relevance. Lancet Neurol. 2017, 16, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Edwards, M.; Robertson, N.P. Seizures in Alzheimer’s disease: Is there more beneath the surface? J. Neurol. 2018, 265, 226–228. [Google Scholar] [CrossRef] [Green Version]
- Vossel, K.A.; Ranasinghe, K.G.; Beagle, A.J.; Mizuiri, D.; Honma, S.M.; Dowling, A.F.; Darwish, S.M.; Van Berlo, V.; Barnes, D.E.; Mantle, M.; et al. Incidence and impact of subclinical epileptiform activity in Alzheimer’s disease. Ann. Neurol. 2016, 80, 858–870. [Google Scholar] [CrossRef] [PubMed]
- Hesdorffer, D.C.; Hauser, W.A.; Annegers, J.F.; Kokmen, E.; Rocca, W.A. Dementia and adult-onset unprovoked seizures. Neurology 1996, 46, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Hauser, W.A.; Morris, M.L.; Heston, L.L.; Anderson, V.E. Seizures and myoclonus in patients with Alzheimer’s disease. Neurology 1986, 36, 1226–1230. [Google Scholar] [CrossRef]
- Difrancesco, J.C.; Tremolizzo, L.; Polonia, V.; Giussani, G.; Bianchi, E.; Franchi, C.; Nobili, A.; Appollonio, I.; Beghi, E.; Ferrarese, C. Adult-Onset Epilepsy in Presymptomatic Alzheimer’s Disease: A Retrospective Study. J. Alzheimer’s Dis. 2017, 60, 1267–1274. [Google Scholar] [CrossRef]
- Pandis, D.; Scarmeas, N. Seizures in alzheimer disease: Clinical and epidemiological data. Epilepsy Curr. 2012, 12, 184–187. [Google Scholar] [CrossRef]
- Scarmeas, N.; Honig, L.S.; Choi, H.; Cantero, J.; Brandt, J.; Blacker, D.; Albert, M.; Amatniek, J.C.; Marder, K.; Bell, K.; et al. Seizures in Alzheimer disease: Who, when, and how common? Arch. Neurol. 2009, 66, 992–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, A.D.; Deck, G.; Goldman, A.; Eskandar, E.N.; Noebels, J.; Cole, A.J. Silent hippocampal seizures and spikes identified by foramen ovale electrodes in Alzheimer’s disease. Nat. Med. 2017, 23, 678–680. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.; Libretto, T.; Henley, W.; Zeman, A. The prevalence and clinical features of epileptic seizures in a memory clinic population. Seizure 2019, 71, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Chin, J.; Roberson, E.D.; Wang, J.; Thwin, M.T.; Bien-Ly, N.; Yoo, J.; Ho, K.O.; Yu, G.Q.; Kreitzer, A.; et al. Aberrant Excitatory Neuronal Activity and Compensatory Remodeling of Inhibitory Hippocampal Circuits in Mouse Models of Alzheimer’s Disease. Neuron 2007, 55, 697–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandratavicius, L.; Alves Balista, P.; Lopes-Aguiar, C.; Ruggiero, R.N.; Umeoka, E.H.; Garcia-Cairasco, N.; Bueno-Junior, L.S.; Leite, J.P. Animal models of epilepsy: Use and limitations. Neuropsychiatr. Dis. Treat. 2014, 10, 1693–1705. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Muller, R.U.; Huang, L.T.; Kubie, J.L.; Rotenberg, A.; Rivard, B.; Cilio, M.R.; Holmes, G.L. Seizure-Induced Changes in Place Cell Physiology: Relationship to Spatial Memory. J. Neurosci. 2003, 23, 11505–11515. [Google Scholar] [CrossRef]
- Shuman, T.; Aharoni, D.; Cai, D.J.; Lee, C.R.; Chavlis, S.; Page-Harley, L.; Vetere, L.M.; Feng, Y.; Yang, C.Y.; Mollinedo-Gajate, I.; et al. Breakdown of spatial coding and interneuron synchronization in epileptic mice. Nat. Neurosci. 2020, 23, 229–238. [Google Scholar] [CrossRef]
- Holmes, G.L. Cognitive impairment in epilepsy: The role of network abnormalities. Epileptic Disord. 2015, 17, 101–116. [Google Scholar] [CrossRef]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 31, 750–754. [Google Scholar] [CrossRef] [Green Version]
- Sierksma, A.; Escott-Price, V.; De Strooper, B. Translating genetic risk of Alzheimer’s disease into mechanistic insight and drug targets. Science 2020, 370, 61–66. [Google Scholar] [CrossRef]
- Sala Frigerio, C.; De Strooper, B. Alzheimer’s Disease Mechanisms and Emerging Roads to Novel Therapeutics. Annu. Rev. Neurosci. 2016, 39, 57–79. [Google Scholar] [CrossRef] [PubMed]
- Bakker, A.; Krauss, G.L.; Albert, M.S.; Speck, C.L.; Jones, L.R.; Stark, C.E.; Yassa, M.A.; Bassett, S.S.; Shelton, A.L.; Gallagher, M. Reduction of Hippocampal Hyperactivity Improves Cognition in Amnestic Mild Cognitive Impairment. Neuron 2012, 74, 467–474. [Google Scholar] [CrossRef] [Green Version]
- Dickerson, B.C.; Salat, D.H.; Greve, D.N.; Chua, E.F.; Rand-Giovannetti, E.; Rentz, D.M.; Bertram, L.; Mullin, K.; Tanzi, R.E.; Blacker, D.; et al. Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD. Neurology 2005, 65, 404–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Leal, S.L.; Landau, S.M.; Bell, R.K.; Jagust, W.J. Hippocampal activation is associated with longitudinal amyloid accumulation and cognitive decline. eLife 2017, 6, e22978. [Google Scholar] [CrossRef] [PubMed]
- Buzsáki, G.; Logothetis, N.; Singer, W. Scaling brain size, keeping timing: Evolutionary preservation of brain rhythms. Neuron 2013, 80, 751–764. [Google Scholar] [CrossRef] [Green Version]
- Ung, H.; Cazares, C.; Nanivadekar, A.; Kini, L.; Wagenaar, J.; Becker, D.; Krieger, A.; Lucas, T.; Litt, B.; Davis, K.A. Interictal epileptiform activity outside the seizure onset zone impacts cognition. Brain 2017, 140, 2157–2168. [Google Scholar] [CrossRef] [Green Version]
- Gelinas, J.N.; Khodagholy, D.; Thesen, T.; Devinsky, O.; Buzsáki, G. Interictal epileptiform discharges induce hippocampal-cortical coupling in temporal lobe epilepsy. Nat. Med. 2016, 22, 641–648. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, U.J.; Centeno, M.; Carmichael, D.W.; Vollmar, C.; Rodionov, R.; Bonelli, S.; Stretton, J.; Pressler, R.; Eriksson, S.H.; Sisodiya, S.; et al. Imaging the interaction: Epileptic discharges, working memory, and behavior. Hum. Brain Mapp. 2013, 34, 2910–2917. [Google Scholar] [CrossRef]
- Englot, D.J.; Konrad, P.E.; Morgan, V.L. Regional and global connectivity disturbances in focal epilepsy, related neurocognitive sequelae, and potential mechanistic underpinnings. Epilepsia 2016, 57, 1546–1557. [Google Scholar] [CrossRef] [Green Version]
- Brier, M.R.; Thomas, J.B.; Ances, B.M. Network dysfunction in Alzheimer’s disease: Refining the disconnection hypothesis. Brain Connect. 2014, 4, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Delbeuck, X.; Van Der Linden, M.; Collette, F. Alzheimer’s Disease as a Disconnection Syndrome? Neuropsychol. Rev. 2003, 13, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Urrestarazu, E.; Jirsch, J.D.; LeVan, P.; Hall, J.; Gotman, J. High-frequency intracerebral EEG activity (100–500 Hz) following interictal spikes. Epilepsia 2006, 47, 1465–1476. [Google Scholar] [CrossRef] [PubMed]
- Busche, M.A.; Kekuš, M.; Adelsberger, H.; Noda, T.; Förstl, H.; Nelken, I.; Konnerth, A. Rescue of long-range circuit dysfunction in Alzheimer’s disease models. Nat. Neurosci. 2015, 18, 1623–1630. [Google Scholar] [CrossRef]
- De Gennaro, L.; Gorgoni, M.; Reda, F.; Lauri, G.; Truglia, I.; Cordone, S.; Scarpelli, S.; Mangiaruga, A.; D’Atri, A.; Lacidogna, G.; et al. The Fall of Sleep K-Complex in Alzheimer Disease. Sci. Rep. 2017, 7, 39688. [Google Scholar] [CrossRef] [Green Version]
- Kastanenka, K.V.; Hou, S.S.; Shakerdge, N.; Logan, R.; Feng, D.; Wegmann, S.; Chopra, V.; Hawkes, J.M.; Chen, X.; Bacskai, B.J. Optogenetic restoration of disrupted slow oscillations halts amyloid deposition and restores calcium homeostasis in an animal model of Alzheimer’s disease. PLoS ONE 2017, 12, e0170275. [Google Scholar] [CrossRef]
- Lucey, B.P.; McCullough, A.; Landsness, E.C.; Toedebusch, C.D.; McLeland, J.S.; Zaza, A.M.; Fagan, A.M.; McCue, L.; Xiong, C.; Morris, J.C.; et al. Reduced non–rapid eye movement sleep is associated with tau pathology in early Alzheimer’s disease. Sci. Transl. Med. 2019, 11, eaau6550. [Google Scholar] [CrossRef]
- Ranasinghe, K.G.; Cha, J.; Iaccarino, L.; Hinkley, L.B.; Beagle, A.J.; Pham, J.; Jagust, W.J.; Miller, B.L.; Rankin, K.P.; Rabinovici, G.D.; et al. Neurophysiological signatures in Alzheimer’s disease are distinctly associated with TAU, amyloid-β accumulation, and cognitive decline. Sci. Transl. Med. 2020, 12, eaaz4069. [Google Scholar] [CrossRef]
- Kam, K.; Parekh, A.; Sharma, R.A.; Andrade, A.; Lewin, M.; Castillo, B.; Bubu, O.M.; Chua, N.J.; Miller, M.D.; Mullins, A.E.; et al. Sleep oscillation-specific associations with Alzheimer’s disease CSF biomarkers: Novel roles for sleep spindles and tau. Mol. Neurodegener. 2019, 14, 10. [Google Scholar] [CrossRef] [Green Version]
- Fritschy, J.M. Epilepsy, E/I balance and GABAA receptor plasticity. Front. Mol. Neurosci. 2008, 1, 5. [Google Scholar] [CrossRef] [Green Version]
- Busche, M.A.; Konnerth, A. Impairments of neural circuit function in Alzheimer’s disease. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20150429. [Google Scholar] [CrossRef] [PubMed]
- Minkeviciene, R.; Rheims, S.; Dobszay, M.B.; Zilberter, M.; Hartikainen, J.; Fülöp, L.; Penke, B.; Zilberter, Y.; Harkany, T.; Pitkänen, A.; et al. Amyloid β-induced neuronal hyperexcitability triggers progressive epilepsy. J. Neurosci. 2009, 29, 3453–3462. [Google Scholar] [CrossRef] [PubMed]
- Gschwind, T.; Lafourcade, C.; Gfeller, T.; Zaichuk, M.; Rambousek, L.; Knuesel, I.; Fritschy, J.M. Contribution of early Alzheimer’s disease-related pathophysiology to the development of acquired epilepsy. Eur. J. Neurosci. 2018, 47, 1534–1562. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Mucke, L. Amyloid-Β-induced neuronal dysfunction in Alzheimer’s disease: From synapses toward neural networks. Nat. Neurosci. 2010. [Google Scholar] [CrossRef] [PubMed]
- Busche, M.A.; Wegmann, S.; Dujardin, S.; Commins, C.; Schiantarelli, J.; Klickstein, N.; Kamath, T.V.; Carlson, G.A.; Nelken, I.; Hyman, B.T. Tau impairs neural circuits, dominating amyloid-β effects, in Alzheimer models in vivo. Nat. Neurosci. 2019, 22, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Pooler, A.M.; Phillips, E.C.; Lau, D.H.W.; Noble, W.; Hanger, D.P. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013, 14, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.W.; Hussaini, S.A.; Bastille, I.M.; Rodriguez, G.A.; Mrejeru, A.; Rilett, K.; Sanders, D.W.; Cook, C.; Fu, H.; Boonen, R.A.C.M.; et al. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat. Neurosci. 2016, 19, 1085–1092. [Google Scholar] [CrossRef]
- Tai, X.Y.; Koepp, M.; Duncan, J.S.; Fox, N.; Thompson, P.; Baxendale, S.; Liu, J.Y.W.; Reeves, C.; Michalak, Z.; Thom, M. Hyperphosphorylated tau in patients with refractory epilepsy correlates with cognitive decline: A study of temporal lobe resections. Brain 2016, 139, 2441–2455. [Google Scholar] [CrossRef]
- Joutsa, J.; Rinne, J.O.; Hermann, B.; Karrasch, M.; Anttinen, A.; Shinnar, S.; Sillanpaa, M. Association between childhood-onset epilepsy and amyloid burden 5 decades later. JAMA Neurol. 2017, 74, 583–590. [Google Scholar] [CrossRef]
- Busche, M.A.; Konnerth, A. Neuronal hyperactivity—A key defect in Alzheimer’s disease? BioEssays 2015, 37, 624–632. [Google Scholar] [CrossRef]
- Powell, G.; Ziso, B.; Larner, A.J. The overlap between epilepsy and Alzheimer’s disease and the consequences for treatment. Expert Rev. Neurother. 2019, 19, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewerenz, J.; Maher, P. Chronic glutamate toxicity in neurodegenerative diseases-What is the evidence? Front. Neurosci. 2015, 9, 469. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mejia, R.O.; Newman, J.W.; Toh, S.; Yu, G.Q.; Zhou, Y.; Halabisky, B.; Cissé, M.; Scearce-Levie, K.; Cheng, I.H.; Gan, L.; et al. Phospholipase A2 reduction ameliorates cognitive deficits in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2008, 11, 1311–1318. [Google Scholar] [CrossRef]
- Busche, M.A.; Chen, X.; Henning, H.A.; Reichwald, J.; Staufenbiel, M.; Sakmann, B.; Konnerth, A. Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2012, 109, 8740–8745. [Google Scholar] [CrossRef] [Green Version]
- Zott, B.; Simon, M.M.; Hong, W.; Unger, F.; Chen-Engerer, H.J.; Frosch, M.P.; Sakmann, B.; Walsh, D.M.; Konnerth, A. A vicious cycle of β amyloid−dependent neuronal hyperactivation. Science 2019, 365, 559–565. [Google Scholar] [CrossRef]
- Abramov, E.; Dolev, I.; Fogel, H.; Ciccotosto, G.D.; Ruff, E.; Slutsky, I. Amyloid-Β as a positive endogenous regulator of release probability at hippocampal synapses. Nat. Neurosci. 2009, 12, 1567–1576. [Google Scholar] [CrossRef]
- Keskin, A.D.; Kekuš, M.; Adelsberger, H.; Neumann, U.; Shimshek, D.R.; Song, B.; Zott, B.; Peng, T.; Förstl, H.; Staufenbiel, M.; et al. BACE inhibition-dependent repair of Alzheimer’s pathophysiology. Proc. Natl. Acad. Sci. USA 2017, 114, 8631–8636. [Google Scholar] [CrossRef] [Green Version]
- Busche, M.A.; Eichhoff, G.; Adelsberger, H.; Abramowski, D.; Wiederhold, K.H.; Haass, C.; Staufenbiel, M.; Konnerth, A.; Garaschuk, O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 2008, 321, 1686–1689. [Google Scholar] [CrossRef] [Green Version]
- Koffie, R.M.; Meyer-Luehmann, M.; Hashimoto, T.; Adams, K.W.; Mielke, M.L.; Garcia-Alloza, M.; Micheva, K.D.; Smith, S.J.; Kim, M.L.; Lee, V.M.; et al. Oligomeric amyloid β associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc. Natl. Acad. Sci. USA 2009, 106, 4012–4017. [Google Scholar] [CrossRef] [Green Version]
- Bero, A.W.; Yan, P.; Roh, J.H.; Cirrito, J.R.; Stewart, F.R.; Raichle, M.E.; Lee, J.M.; Holtzman, D.M. Neuronal activity regulates the regional vulnerability to amyloid-β 2 deposition. Nat. Neurosci. 2011, 14, 750–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.Y.; Hudry, E.; Hashimoto, T.; Kuchibhotla, K.; Rozkalne, A.; Fan, Z.; Spires-Jones, T.; Xie, H.; Arbel-Ornath, M.; Grosskreutz, C.L.; et al. Amyloid β induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J. Neurosci. 2010, 30, 2636–2649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, E.M.; Nong, Y.; Almeida, C.G.; Paul, S.; Moran, T.; Choi, E.Y.; Nairn, A.C.; Salter, M.W.; Lombroso, P.J.; Gouras, G.K.; et al. Regulation of NMDA receptor trafficking by amyloid-β. Nat. Neurosci. 2005, 8, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Ghatak, S.; Dolatabadi, N.; Trudler, D.; Zhang, X.; Wu, Y.; Mohata, M.; Ambasudhan, R.; Talantova, M.; Lipton, S.A. Mechanisms of hyperexcitability in alzheimer’s disease hiPSC-derived neurons and cerebral organoids vs. Isogenic control. eLife 2019, 8, e50333. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Marin, V.; Blazquez-Llorca, L.; Rodriguez, J.R.; Boluda, S.; Muntane, G.; Ferrer, I.; DeFelipe, J. Diminished perisomatic GABAergic terminals on cortical neurons adjacent to amyloid plaques. Front. Neuroanat. 2009, 3, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Meng, X.; Zhang, J.; Li, Y.; Wang, L.; Qin, X.; Sui, N.; Zhang, Y. GABA attenuates amyloid toxicity by downregulating its endocytosis and improves cognitive impairment. J. Alzheimer’s Dis. 2012, 31, 635–649. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Losa, M.; Tracy, T.E.; Ma, K.; Verret, L.; Clemente-Perez, A.; Khan, A.S.; Cobos, I.; Ho, K.; Gan, L.; Mucke, L.; et al. Nav1.1-Overexpressing Interneuron Transplants Restore Brain Rhythms and Cognition in a Mouse Model of Alzheimer’s Disease. Neuron 2018, 98, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Verret, L.; Mann, E.O.; Hang, G.B.; Barth, A.M.I.; Cobos, I.; Ho, K.; Devidze, N.; Masliah, E.; Kreitzer, A.C.; Mody, I.; et al. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in alzheimer model. Cell 2012, 149, 708–721. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Zhao, M.; Han, Y.; Zhang, H. GABAergic Inhibitory Interneuron Deficits in Alzheimer’s Disease: Implications for Treatment. Front. Neurosci. 2020. [Google Scholar] [CrossRef]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Romoli, M.; Liguori, C.; Farotti, L.; Eusebi, P.; Bedetti, C.; Siliquini, S.; Cesarini, E.N.; Romigi, A.; Mercuri, N.B.; et al. Alzheimer’s disease and late-onset epilepsy of unknown origin: Two faces of beta amyloid pathology. Neurobiol. Aging 2019, 73, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.P.; Xie, Y.; Meng, X.Y.; Kang, J.S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target. Ther. 2019, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef] [PubMed]
- Hitt, B.D.; Jaramillo, T.C.; Chetkovich, D.M.; Vassar, R. BACE1-/- mice exhibit seizure activity that does not correlate with sodium channel level or axonal localization. Mol. Neurodegener. 2010, 5, 31. [Google Scholar] [CrossRef] [Green Version]
- Moussa-Pacha, N.M.; Abdin, S.M.; Omar, H.A.; Alniss, H.; Al-Tel, T.H. BACE1 inhibitors: Current status and future directions in treating Alzheimer’s disease. Med. Res. Rev. 2020, 40, 339–384. [Google Scholar] [CrossRef]
- Egan, M.F.; Kost, J.; Tariot, P.N.; Aisen, P.S.; Cummings, J.L.; Vellas, B.; Sur, C.; Mukai, Y.; Voss, T.; Furtek, C.; et al. Randomized trial of verubecestat for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2018, 378, 1691–1703. [Google Scholar] [CrossRef]
- Satir, T.M.; Agholme, L.; Karlsson, A.; Karlsson, M.; Karila, P.; Illes, S.; Bergström, P.; Zetterberg, H. Partial reduction of amyloid β production by β-secretase inhibitors does not decrease synaptic transmission. Alzheimer’s Res. Ther. 2020, 147, 256–274. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Marinković, P.; Blumenstock, S.; Goltstein, P.M.; Korzhova, V.; Peters, F.; Knebl, A.; Herms, J. In vivo imaging reveals reduced activity of neuronal circuits in a mouse tauopathy model. Brain 2019, 142, 1051–1062. [Google Scholar] [CrossRef] [PubMed]
- Green, C.; Sydow, A.; Vogel, S.; Anglada-Huguet, M.; Wiedermann, D.; Mandelkow, E.; Mandelkow, E.M.; Hoehn, M. Functional networks are impaired by elevated tau-protein but reversible in a regulatable Alzheimer’s disease mouse model. Mol. Neurodegener. 2019, 14, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menkes-Caspi, N.; Yamin, H.G.; Kellner, V.; Spires-Jones, T.L.; Cohen, D.; Stern, E.A. Pathological tau disrupts ongoing network activity. Neuron 2015, 85, 959–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, H.; Rodriguez, G.A.; Herman, M.; Emrani, S.; Nahmani, E.; Barrett, G.; Figueroa, H.Y.; Goldberg, E.; Hussaini, S.A.; Duff, K.E. Tau Pathology Induces Excitatory Neuron Loss, Grid Cell Dysfunction, and Spatial Memory Deficits Reminiscent of Early Alzheimer’s Disease. Neuron 2017, 93, 533–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatch, R.J.; Wei, Y.; Xia, D.; Götz, J. Hyperphosphorylated tau causes reduced hippocampal CA1 excitability by relocating the axon initial segment. Acta Neuropathol. 2017, 133, 717–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller-Thomsen, L.; Borgmann, D.; Morcinek, K.; Schröder, S.; Dengler, B.; Moser, N.; Neumaier, F.; Schneider, T.; Schröder, H.; Huggenberger, S. Consequences of hyperphosphorylated tau on the morphology and excitability of hippocampal neurons in aged tau transgenic mice. Neurobiol. Aging 2020, 93, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Pickett, E.K.; Herrmann, A.G.; McQueen, J.; Abt, K.; Dando, O.; Tulloch, J.; Jain, P.; Dunnett, S.; Sohrabi, S.; Fjeldstad, M.P.; et al. Amyloid Beta and Tau Cooperate to Cause Reversible Behavioral and Transcriptional Deficits in a Model of Alzheimer’s Disease. Cell Rep. 2019, 29, 3592–3604. [Google Scholar] [CrossRef] [Green Version]
- Roberson, E.D.; Halabisky, B.; Yoo, J.W.; Yao, J.; Chin, J.; Yan, F.; Wu, T.; Hamto, P.; Devidze, N.; Yu, G.Q.; et al. Amyloid-β/fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of alzheimer’s disease. J. Neurosci. 2011, 31, 700–711. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Thangavel, R.; Rysted, J.; Kim, Y.; Francis, M.B.; Adams, E.; Lin, Z.; Taugher, R.J.; Wemmie, J.A.; Usachev, Y.M.; et al. Loss of tau and Fyn reduces compensatory effects of MAP2 for tau and reveals a Fyn-independent effect of tau on calcium. J. Neurosci. Res. 2019, 97, 1393–1413. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-β toxicity in alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Hall, A.M.; Kelinske, M.; Roberson, E.D. Seizure resistance without parkinsonism in aged mice after tau reduction. Neurobiol. Aging 2014, 35, 2617–2624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeVos, S.L.; Goncharoff, D.K.; Chen, G.; Kebodeaux, C.S.; Yamada, K.; Stewart, F.R.; Schuler, D.R.; Maloney, S.E.; Wozniak, D.F.; Rigo, F.; et al. Antisense reduction of tau in adult mice protects against seizures. J. Neurosci. 2013, 33, 12887–12897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calhoun, M.E.; Wiederhold, K.H.; Abramowski, D.; Phinney, A.L.; Probst, A.; Sturchler-Pierrat, C.; Staufenbiel, M.; Sommer, B.; Jucker, M. Neuron loss in APP transgenic mice. Nature 1998, 395, 755–756. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Thom, M.; Martinian, L.; Harding, B.; Cross, J.H.; Nikolic, M.; Sisodiya, S.M. Pathological tau tangles localize to focal cortical dysplasia in older patients. Epilepsia 2007, 48, 1447–1454. [Google Scholar] [CrossRef]
- Keller, C.J.; Truccolo, W.; Gale, J.T.; Eskandar, E.; Thesen, T.; Carlson, C.; Devinsky, O.; Kuzniecky, R.; Doyle, W.K.; Madsen, J.R.; et al. Heterogeneous neuronal firing patterns during interictal epileptiform discharges in the human cortex. Brain 2010, 133, 668–681. [Google Scholar] [CrossRef] [Green Version]
- Congdon, E.E.; Sigurdsson, E.M. Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef]
- Yanamandra, K.; Kfoury, N.; Jiang, H.; Mahan, T.E.; Ma, S.; Maloney, S.E.; Wozniak, D.F.; Diamond, M.I.; Holtzman, D.M. Anti-tau antibodies that block tau aggregate seeding invitro markedly decrease pathology and improve cognition in vivo. Neuron 2013, 80, 402–414. [Google Scholar] [CrossRef] [Green Version]
- Kontsekova, E.; Zilka, N.; Kovacech, B.; Skrabana, R.; Novak, M. Identification of structural determinants on tau protein essential for its pathological function: Novel therapeutic target for tau immunotherapy in Alzheimer’s disease. Alzheimer’s Res. Ther. 2014, 6, 45. [Google Scholar] [CrossRef] [Green Version]
- Ondrus, M.; Novak, P. Design of the phase II clinical study of the tau vaccine AADvac1 in patients with mild Alzheimer’s disease. Neurobiol. Aging 2016, 39, 26. [Google Scholar] [CrossRef]
- Theunis, C.; Crespo-Biel, N.; Gafner, V.; Pihlgren, M.; López-Deber, M.P.; Reis, P.; Hickman, D.T.; Adolfsson, O.; Chuard, N.; Ndao, D.M.; et al. Efficacy and safety of a liposome-based vaccine against protein Tau, assessed in Tau.P301L mice that model tauopathy. PLoS ONE 2013, 8, e72301. [Google Scholar] [CrossRef] [Green Version]
- AXON Neuroscience SE. Axon Presented Positive Phase II Trial Results of AADvac1 at AAT-AD/PD 2020; Biospace: Vienna, Austria, 2020. [Google Scholar]
- Gauthier, S.; Feldman, H.H.; Schneider, L.S.; Wilcock, G.K.; Frisoni, G.B.; Hardlund, J.H.; Moebius, H.J.; Bentham, P.; Kook, K.A.; Wischik, D.J.; et al. Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: A randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet 2016, 388, 2873–2884. [Google Scholar] [CrossRef] [Green Version]
- Wilcock, G.K.; Gauthier, S.; Frisoni, G.B.; Jia, J.; Hardlund, J.H.; Moebius, H.J.; Bentham, P.; Kook, K.A.; Schelter, B.O.; Wischik, D.J.; et al. Potential of Low Dose Leuco-Methylthioninium Bis(Hydromethanesulphonate) (LMTM) Monotherapy for Treatment of Mild Alzheimer’s Disease: Cohort Analysis as Modified Primary Outcome in a Phase III Clinical Trial. J. Alzheimer’s Dis. 2018, 61, 435–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuzwa, S.A.; Shan, X.; MacAuley, M.S.; Clark, T.; Skorobogatko, Y.; Vosseller, K.; Vocadlo, D.J. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat. Chem. Biol. 2012, 8, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, B.H.; Schmechel, D.; Hirman, J.; Blackwell, A.; Keith, J.; Gold, M.; Schmechel, D.; Kirby, L.; Huszar, L.; Walling, D.; et al. A double-blind, placebo-controlled, ascending-dose, randomized study to evaluate the safety, tolerability and effects on cognition of AL-108 after 12 weeks of intranasal administration in subjects with mild cognitive impairment. Dement. Geriatr. Cogn. Disord. 2013, 35, 325–336. [Google Scholar] [CrossRef]
- Gurney, M.E.; D’Amato, E.C.; Burgin, A.B. Phosphodiesterase-4 (PDE4) Molecular Pharmacology and Alzheimer’s Disease. Neurotherapeutics 2015, 12, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, B.; Vitolo, O.V.; Trinchese, F.; Liu, S.; Shelanski, M.; Arancio, O. Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J. Clin. Investig. 2004, 114, 1624–1634. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.B.; Farr, S.A.; Flood, J.F.; Kamlesh, V.; Franko, M.; Banks, W.A.; Morley, J.E. Site-directed antisense oligonucleotide decreases the expression of amyloid precursor protein and reverses deficits in learning and memory in aged SAMP8 mice. Peptides 2000, 21, 1769–1775. [Google Scholar] [CrossRef]
- Schoch, K.M.; Miller, T.M. Antisense Oligonucleotides: Translation from Mouse Models to Human Neurodegenerative Diseases. Neuron 2017, 94, 1056–1070. [Google Scholar] [CrossRef] [Green Version]
- Wurster, C.D.; Ludolph, A.C. Antisense oligonucleotides in neurological disorders. Ther. Adv. Neurol. Disord. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Muyllaert, D.; Kremer, A.; Jaworski, T.; Borghgraef, P.; Devijver, H.; Croes, S.; Dewachter, I.; Van Leuven, F. Glycogen synthase kinase-3β, or a link between amyloid and tau pathology? Genes Brain Behav. 2008, 7, 57–66. [Google Scholar] [CrossRef]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leroy, K.; Boutajangout, A.; Authelet, M.; Woodgett, J.R.; Anderton, B.H.; Brion, J.P. The active form of glycogen synthase kinase-3β is associated with granulovacuolar degeneration in neurons in Alzheimers’s disease. Acta Neuropathol. 2002, 103, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Avila, J. Tau phosphorylation and aggregation in Alzheimer’s disease pathology. FEBS Lett. 2006, 580, 2922–2927. [Google Scholar] [CrossRef] [Green Version]
- ue Barreda, E.G.; Pérez, M.; Ramos, P.G.; de Cristobal, J.; Martín-Maestro, P.; Morán, A.; Dawson, H.N.; Vitek, M.P.; Lucas, J.J.; Hernández, F.; et al. Tau-knockout mice show reduced GSK3-induced hippocampal degeneration and learning deficits. Neurobiol. Dis. 2010, 37, 622–629. [Google Scholar] [CrossRef]
- Toral-Rios, D.; Pichardo-Rojas, P.S.; Alonso-Vanegas, M.; Campos-Peña, V. GSK3β and Tau Protein in Alzheimer’s Disease and Epilepsy. Front. Cell. Neurosci. 2020, 14, 19. [Google Scholar] [CrossRef]
- Takashima, A.; Murayama, M.; Murayama, O.; Kohno, T.; Honda, T.; Yasutake, K.; Nihonmatsu, N.; Mercken, M.; Yamaguchi, H.; Sugihara, S.; et al. Presenilin 1 associates with glycogen synthase kinase-3β and its substrate tau. Proc. Natl. Acad. Sci. USA 1998, 95, 9637–9641. [Google Scholar] [CrossRef] [Green Version]
- Peineau, S.; Taghibiglou, C.; Bradley, C.; Wong, T.P.; Liu, L.; Lu, J.; Lo, E.; Wu, D.; Saule, E.; Bouschet, T.; et al. LTP Inhibits LTD in the Hippocampus via Regulation of GSK3β. Neuron 2007, 53, 703–717. [Google Scholar] [CrossRef] [Green Version]
- Collingridge, G.L.; Isaac, J.T.R.; Yu, T.W. Receptor trafficking and synaptic plasticity. Nat. Rev. Neurosci. 2004, 5, 952–962. [Google Scholar] [CrossRef]
- Abraham, W.C.; Bear, M.F. Metaplasticity: The plasticity of synaptic plasticity. Trends Neurosci. 1996, 19, 126–130. [Google Scholar] [CrossRef]
- Rockenstein, E.; Torrance, M.; Adame, A.; Mante, M.; Bar-on, P.; Rose, J.B.; Crews, L.; Masliah, E. Neuroprotective effects of regulators of the glycogen synthase kinase-3β signaling pathway in a transgenic model of Alzheimer’s disease are associated with reduced amyloid precursor protein phosphorylation. J. Neurosci. 2007, 27, 1981–1991. [Google Scholar] [CrossRef]
- Phiel, C.J.; Wilson, C.A.; Lee, V.M.Y.; Klein, P.S. GSK-3α regulates production of Alzheimer’s disease amyloid-β peptides. Nature 2003, 423, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, H.; Ishihara, T.; Suguimoto, P.; Yokota, O.; Oshima, E.; Kugo, A.; Terada, S.; Hamamura, T.; Trojanowski, J.Q.; Lee, V.M.Y.; et al. Chronic lithium treatment decreases tau lesions by promoting ubiquitination in a mouse model of tauopathies. Acta Neuropathol. 2005, 110, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Aourz, N.; Serruys, A.S.K.; Chabwine, J.N.; Balegamire, P.B.; Afrikanova, T.; Edrada-Ebel, R.; Grey, A.I.; Kamuhabwa, A.R.; Walrave, L.; Esguerra, C.V.; et al. Identification of GSK-3 as a Potential Therapeutic Entry Point for Epilepsy. ACS Chem. Neurosci. 2019, 10, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- Eldar-Finkelman, H.; Martinez, A. GSK-3 Inhibitors: Preclinical and Clinical Focus on CNS. Front. Mol. Neurosci. 2011, 4, 32. [Google Scholar] [CrossRef] [Green Version]
- Serenó, L.; Coma, M.; Rodríguez, M.; Sánchez-Ferrer, P.; Sánchez, M.B.; Gich, I.; Agulló, J.M.; Pérez, M.; Avila, J.; Guardia-Laguarta, C.; et al. A novel GSK-3β inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol. Dis. 2009, 35, 359–367. [Google Scholar] [CrossRef]
- Del Ser, T.; Steinwachs, K.C.; Gertz, H.J.; Andrés, M.V.; Gómez-Carrillo, B.; Medina, M.; Vericat, J.A.; Redondo, P.; Fleet, D.; León, T. Treatment of Alzheimer’s disease with the GSK-3 inhibitor tideglusib: A pilot study. J. Alzheimer’s Dis. 2013, 33, 205–215. [Google Scholar] [CrossRef]
- Lovestone, S.; Boada, M.; Dubois, B.; Hüll, M.; Rinne, J.O.; Huppertz, H.J.; Calero, M.; Andrés, M.V.; Gómez-Carrillo, B.; León, T.; et al. A phase II trial of tideglusib in alzheimer’s disease. J. Alzheimer’s Dis. 2015, 45, 75–88. [Google Scholar] [CrossRef]
- Liu, X.; Chen, L.; Chen, Y. N-methyl-D-aspartate receptors mediate epilepsy-induced axonal impairment and tau phosphorylation via activating glycogen synthase kinase-3ß and cyclin-dependent kinase 5. Discov. Med. 2017, 23, 221–234. [Google Scholar]
- Esposito, Z.; Belli, L.; Toniolo, S.; Sancesario, G.; Bianconi, C.; Martorana, A. Amyloid β, glutamate, excitotoxicity in alzheimer’s disease: Are we on the right track? CNS Neurosci. Ther. 2013, 19, 549–555. [Google Scholar] [CrossRef]
- Klyubin, I.; Wang, Q.; Reed, M.N.; Irving, E.A.; Upton, N.; Hofmeister, J.; Cleary, J.P.; Anwyl, R.; Rowan, M.J. Protection against Aβ-mediated rapid disruption of synaptic plasticity and memory by memantine. Neurobiol. Aging 2011, 32, 614–623. [Google Scholar] [CrossRef]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Aβ1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrent, L.; Ferrer, I. PP2A and Alzheimer Disease. Curr. Alzheimer Res. 2012, 9, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Chohan, M.O.; Khatoon, S.; Iqbal, I.G.; Iqbal, K. Involvement of I2PP2A in the abnormal hyperphosphorylation of tau and its reversal by Memantine. FEBS Lett. 2006, 580, 3973–3979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nygaard, H.B.; Van Dyck, C.H.; Strittmatter, S.M. Fyn kinase inhibition as a novel therapy for Alzheimer’s disease. Alzheimer’s Res. Ther. 2014, 6, 8. [Google Scholar] [CrossRef] [Green Version]
- Nygaard, H.B. Targeting Fyn Kinase in Alzheimer’s Disease. Biol. Psychiatry 2018, 83, 369–376. [Google Scholar] [CrossRef]
- Kaufman, A.C.; Salazar, S.V.; Haas, L.T.; Yang, J.; Kostylev, M.A.; Jeng, A.T.; Robinson, S.A.; Gunther, E.C.; Van Dyck, C.H.; Nygaard, H.B.; et al. Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Ann. Neurol. 2015, 77, 953–971. [Google Scholar] [CrossRef] [Green Version]
- Toyonaga, T.; Smith, L.M.; Finnema, S.J.; Gallezot, J.D.; Naganawa, M.; Bini, J.; Mulnix, T.; Cai, Z.; Ropchan, J.; Huang, Y.; et al. In vivo synaptic density imaging with 11C-UCB-J detects treatment effects of saracatinib in a mouse model of Alzheimer disease. J. Nucl. Med. 2019, 60, 1780–1786. [Google Scholar] [CrossRef]
- Van Dyck, C.H.; Nygaard, H.B.; Chen, K.; Donohue, M.C.; Raman, R.; Rissman, R.A.; Brewer, J.B.; Koeppe, R.A.; Chow, T.W.; Rafii, M.S.; et al. Effect of AZD0530 on Cerebral Metabolic Decline in Alzheimer Disease: A Randomized Clinical Trial. JAMA Neurol. 2019, 76, 1219–1229. [Google Scholar] [CrossRef] [Green Version]
- Piette, F.; Belmin, J.; Vincent, H.; Schmidt, N.; Pariel, S.; Verny, M.; Marquis, C.; Mely, J.; Hugonot-Diener, L.; Kinet, J.P.; et al. Masitinib as an adjunct therapy for mild-to-moderate Alzheimer’s disease: A randomised, placebo-controlled phase 2 trial. Alzheimer’s Res. Ther. 2011, 3, 16. [Google Scholar] [CrossRef]
- Science, A. AB Science Reports the Outcome from the Interim Analysis of Study AB09004 in Alzheimer’s Disease; AB Science: Paris, France, 2019. [Google Scholar]
- Vandenberghe, R.; Rinne, J.O.; Boada, M.; Katayama, S.; Scheltens, P.; Vellas, B.; Tuchman, M.; Gass, A.; Fiebach, J.B.; Hill, D.; et al. Bapineuzumab for mild to moderate Alzheimer’s disease in two global, randomized, phase 3 trials. Alzheimer’s Res. Ther. 2016, 8, 18. [Google Scholar] [CrossRef] [Green Version]
- Landen, J.W.; Cohen, S.; Billing, C.B.; Cronenberger, C.; Styren, S.; Burstein, A.H.; Sattler, C.; Lee, J.H.; Jack, C.R.; Kantarci, K.; et al. Multiple-dose ponezumab for mild-to-moderate Alzheimer’s disease: Safety and efficacy. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2017, 3, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Henley, D.; Raghavan, N.; Sperling, R.; Aisen, P.; Raman, R.; Romano, G. Preliminary Results of a Trial of Atabecestat in Preclinical Alzheimer’s Disease. N. Engl. J. Med. 2019, 380, 1483–1485. [Google Scholar] [CrossRef] [PubMed]
- Novak, G.; Streffer, J.R.; Timmers, M.; Henley, D.; Brashear, H.R.; Bogert, J.; Russu, A.; Janssens, L.; Tesseur, I.; Tritsmans, L.; et al. Long-term safety and tolerability of atabecestat (JNJ-54861911), an oral BACE1 inhibitor, in early Alzheimer’s disease spectrum patients: A randomized, double-blind, placebo-controlled study and a two-period extension study. Alzheimer’s Res. Ther. 2020, 12, 58. [Google Scholar] [CrossRef] [PubMed]
- Jacob, C.P.; Koutsilieri, E.; Bartl, J.; Neuen-Jacob, E.; Arzberger, T.; Zander, N.; Ravid, R.; Roggendorf, W.; Riederer, P.; Grünblatt, E. Alterations in expression of glutamatergic transporters and receptors in sporadic Alzheimer’s disease. J. Alzheimer’s Dis. 2007, 11, 97–116. [Google Scholar] [CrossRef]
- Texidó, L.; Martín-Satué, M.; Alberdi, E.; Solsona, C.; Matute, C. Amyloid β peptide oligomers directly activate NMDA receptors. Cell Calcium 2011, 49, 184–190. [Google Scholar] [CrossRef]
- Papouin, T.; Ladépêche, L.; Ruel, J.; Sacchi, S.; Labasque, M.; Hanini, M.; Groc, L.; Pollegioni, L.; Mothet, J.P.; Oliet, S.H.R. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell 2012, 150, 633–646. [Google Scholar] [CrossRef] [Green Version]
- Benarroch, E.E. Glutamatergic synaptic plasticity and dysfunction in Alzheimer disease: Emerging mechanisms. Neurology 2018, 91, 125–132. [Google Scholar] [CrossRef]
- Liu, Q.; Huang, Y.; Xue, F.; Simard, A.; DeChon, J.; Li, G.; Zhang, J.; Lucero, L.; Wang, M.; Sierks, M.; et al. A novel nicotinic acetylcholine receptor subtype in basal forebrain cholinergic neurons with high sensitivity to amyloid peptides. J. Neurosci. 2009, 29, 918–929. [Google Scholar] [CrossRef]
- Ménard, C.; Quirion, R.; Vigneault, E.; Bouchard, S.; Ferland, G.; El Mestikawy, S.; Gaudreau, P. Glutamate presynaptic vesicular transporter and postsynaptic receptor levels correlate with spatial memory status in aging rat models. Neurobiol. Aging 2015, 36, 1471–1482. [Google Scholar] [CrossRef] [Green Version]
- Kashani, A.; Lepicard, È.; Poirel, O.; Videau, C.; David, J.P.; Fallet-Bianco, C.; Simon, A.; Delacourte, A.; Giros, B.; Epelbaum, J.; et al. Loss of VGLUT1 and VGLUT2 in the prefrontal cortex is correlated with cognitive decline in Alzheimer disease. Neurobiol. Aging 2008, 29, 1619–1630. [Google Scholar] [CrossRef] [Green Version]
- Cretin, B. Pharmacotherapeutic strategies for treating epilepsy in patients with Alzheimer’s disease. Expert Opin. Pharmacother. 2018, 19, 1201–1209. [Google Scholar] [CrossRef] [PubMed]
- Mula, M.; Trimble, M.R. Antiepileptic drug-induced cognitive adverse effects: Potential mechanisms and contributing factors. CNS Drugs 2009, 23, 121–137. [Google Scholar] [CrossRef] [PubMed]
- Ziyatdinova, S.; Gurevicius, K.; Kutchiashvili, N.; Bolkvadze, T.; Nissinen, J.; Tanila, H.; Pitkänen, A. Spontaneous epileptiform discharges in a mouse model of Alzheimer’s disease are suppressed by antiepileptic drugs that block sodium channels. Epilepsy Res. 2011, 94, 75–85. [Google Scholar] [CrossRef]
- Chen, G.; Huang, L.D.; Jiang, Y.M.; Manji, H.K. The mood-stabilizing agent valproate inhibits the activity of glycogen synthase kinase-3. J. Neurochem. 2000, 72, 1327–1330. [Google Scholar] [CrossRef] [PubMed]
- Qing, H.; He, G.; Ly, P.T.T.; Fox, C.J.; Staufenbiel, M.; Cai, F.; Zhang, Z.; Wei, S.; Sun, X.; Chen, C.H.; et al. Valproic acid inhibits aβ production, neuritic plaque formation, and behavioral deficits in alzheimer’s disease mouse models. J. Exp. Med. 2008, 205, 2781–2789. [Google Scholar] [CrossRef] [PubMed]
- Tariot, P.N.; Schneider, L.S.; Cummings, J.; Thomas, R.G.; Raman, R.; Jakimovich, L.J.; Loy, R.; Bartocci, B.; Fleisher, A.; Ismail, M.S.; et al. Chronic divalproex sodium to attenuate agitation and clinical progression of Alzheimer disease. Arch. Gen. Psychiatry 2011, 68, 853–861. [Google Scholar] [CrossRef] [Green Version]
- Fleisher, A.S.; Truran, D.; Mai, J.T.; Langbaum, J.B.S.; Aisen, P.S.; Cummings, J.L.; Jack, C.R.; Weiner, M.W.; Thomas, R.G.; Schneider, L.S.; et al. Chronic divalproex sodium use and brain atrophy in Alzheimer disease. Neurology 2011, 77, 1263–1271. [Google Scholar] [CrossRef] [Green Version]
- Galiana, G.L.; Gauthier, A.C.; Mattson, R.H. Eslicarbazepine Acetate: A New Improvement on a Classic Drug Family for the Treatment of Partial-Onset Seizures. Drugs R D 2017, 17, 329–339. [Google Scholar] [CrossRef] [Green Version]
- Meador, K.J.; Seliger, J.; Boyd, A.; Razavi, B.; Falco-Walter, J.; Le, S.; Loring, D.W. Comparative neuropsychological effects of carbamazepine and eslicarbazepine acetate. Epilepsy Behav. 2019, 94, 151–157. [Google Scholar] [CrossRef]
- Koch, H.J.; Szecsey, A.; Vogel, M. Sedation caused by primidone may exacerbate dementia. Epilepsy Behav. 2003, 4, 592. [Google Scholar] [CrossRef]
- Defrancesco, M.; Marksteiner, J.; Wolfgang Fleischhacker, W.; Blasko, I. Use of benzodiazepines in Alzheimer’s disease: A systematic review of literature. Int. J. Neuropsychopharmacol. 2015, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supasitthumrong, T.; Bolea-Alamanac, B.M.; Asmer, S.; Woo, V.L.; Abdool, P.S.; Davies, S.J.C. Gabapentin and pregabalin to treat aggressivity in dementia: A systematic review and illustrative case report. Br. J. Clin. Pharmacol. 2019, 85, 690–703. [Google Scholar] [CrossRef] [PubMed]
- Salinsky, M.; Storzbach, D.; Munoz, S. Cognitive effects of pregabalin in healthy volunteers: A double-blind, placebo-controlled trial. Neurology 2010, 74, 755–761. [Google Scholar] [CrossRef]
- Shi, J.Q.; Wang, B.R.; Tian, Y.Y.; Xu, J.; Gao, L.; Zhao, S.L.; Jiang, T.; Xie, H.G.; Zhang, Y.D. Antiepileptics topiramate and levetiracetam alleviate behavioral deficits and reduce neuropathology in APPswe/PS1dE9 transgenic mice. CNS Neurosci. Ther. 2013, 19, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Wandschneider, B.; Burdett, J.; Townsend, L.; Hill, A.; Thompson, P.J.; Duncan, J.S.; Koepp, M.J. Effect of topiramate and zonisamide on fMRI cognitive networks. Neurology 2017, 88, 1165–1171. [Google Scholar] [CrossRef] [Green Version]
- Leach, M.J.; Marden, C.M.; Miller, A.A. Pharmacological Studies on Lamotrigine, A Novel Potential Antiepileptic Drug: II. Neurochemical Studies on the Mechanism of Action. Epilepsia 1986, 27, 490–497. [Google Scholar] [CrossRef]
- Zhang, M.Y.; Zheng, C.Y.; Zou, M.M.; Zhu, J.W.; Zhang, Y.; Wang, J.; Liu, C.F.; Li, Q.F.; Xiao, Z.C.; Li, S.; et al. Lamotrigine attenuates deficits in synaptic plasticity and accumulation of amyloid plaques in APP/PS1 transgenic mice. Neurobiol. Aging 2014, 35, 2713–2725. [Google Scholar] [CrossRef]
- Vossel, K.A.; Beagle, A.J.; Rabinovici, G.D.; Shu, H.; Lee, S.E.; Naasan, G.; Hegde, M.; Cornes, S.B.; Henry, M.L.; Nelson, A.B.; et al. Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. 2013, 70, 1158–1166. [Google Scholar] [CrossRef]
- Tekin, S.; Aykut-Bingöl, C.; Tanridaǧ, T.; Aktan, S. Antiglutamatergic therapy in Alzheimer’s disease—Effects of lamotrigine. J. Neural Transm. 1998, 105, 295–303. [Google Scholar] [CrossRef]
- Larner, A.J. Presenilin-1 mutations in alzheimer’s disease: An update on genotype-phenotype relationships. J. Alzheimer’s Dis. 2013, 37, 653–659. [Google Scholar] [CrossRef]
- Lancman, M.E.; Fertig, E.J.; Trobliger, R.W.; Perrine, K.; Myers, L.; Iyengar, S.S.; Malik, M. The effects of lacosamide on cognition, quality-of-life measures, and quality of life in patients with refractory partial epilepsy. Epilepsy Behav. 2016, 61, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Meador, K.J.; Loring, D.W.; Boyd, A.; Echauz, J.; LaRoche, S.; Velez-Ruiz, N.; Korb, P.; Byrnes, W.; Dilley, D.; Borghs, S.; et al. Randomized double-blind comparison of cognitive and EEG effects of lacosamide and carbamazepine. Epilepsy Behav. 2016, 62, 267–275. [Google Scholar] [CrossRef]
- Sarkis, R.A.; Nicolas, J.; Lee, J.W. Tolerability of lacosamide or zonisamide in elderly patients with seizures. Seizure 2017, 49, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Lorenzo, F.; Motta, C.; Caltagirone, C.; Koch, G.; Mercuri, N.B.; Martorana, A. Lacosamide in the management of behavioral symptoms in frontotemporal dementia. Alzheimer Dis. Assoc. Disord. 2018, 32, 364–365. [Google Scholar] [CrossRef] [PubMed]
- Toniolo, S.; Di Lorenzo, F.; Bozzali, M.; Yogarajah, M. The impact of lacosamide on mood disorders in adult patients with epilepsy: A systematic review. Epilepsy Behav. 2020, 111, 107179. [Google Scholar] [CrossRef] [PubMed]
- Toledo, M. Effects of adjunctive perampanel on sleep quality, daytime somnolence and cognition in refractory focal epilepsy: Further data. Author’s response. Epilepsy Behav. 2017, 68, 238. [Google Scholar] [CrossRef] [PubMed]
- Meador, K.J.; Yang, H.; Piña-Garza, J.E.; Laurenza, A.; Kumar, D.; Wesnes, K.A. Cognitive effects of adjunctive perampanel for partial-onset seizures: A randomized trial. Epilepsia 2016, 57, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Abou-Khalil, B. Levetiracetam in the treatment of epilepsy. Neuropsychiatr. Dis. Treat. 2008, 4, 507–523. [Google Scholar] [CrossRef] [Green Version]
- Koh, M.T.; Haberman, R.P.; Foti, S.; McCown, T.J.; Gallagher, M. Treatment strategies targeting excess hippocampal activity benefit aged rats with cognitive impairment. Neuropsychopharmacology 2010, 35, 1016–1025. [Google Scholar] [CrossRef]
- Nygaard, H.B.; Kaufman, A.C.; Sekine-Konno, T.; Huh, L.L.; Going, H.; Feldman, S.J.; Kostylev, M.A.; Strittmatter, S.M. Brivaracetam, but not ethosuximide, reverses memory impairments in an Alzheimer’s disease mouse model. Alzheimer’s Res. Ther. 2015, 7, 25. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, P.E.; Zhu, L.; Verret, L.; Vossel, K.A.; Orr, A.G.; Cirrito, J.R.; Devidze, N.; Ho, K.; Yu, G.Q.; Palop, J.J.; et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc. Natl. Acad. Sci. USA 2012, 109, E2895–E2903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, I.A.; Gallagher, M.; Eichenbaum, H.; Tanila, H. Neurocognitive aging: Prior memories hinder new hippocampal encoding. Trends Neurosci. 2006, 29, 662–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakker, A.; Albert, M.S.; Krauss, G.; Speck, C.L.; Gallagher, M. Response of the medial temporal lobe network in amnestic mild cognitive impairment to therapeutic intervention assessed by fMRI and memory task performance. NeuroImage Clin. 2015, 7, 688–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lippa, C.F.; Rosso, A.; Hepler, M.; Jenssen, S.; Pillai, J.; Irwin, D. Levetiracetam: A practical option for seizure management in elderly patients with cognitive impairment. Am. J. Alzheimer’s Dis. Other Demen. 2010, 25, 149–154. [Google Scholar] [CrossRef] [Green Version]
- Cumbo, E.; Ligori, L.D. Levetiracetam, lamotrigine, and phenobarbital in patients with epileptic seizures and Alzheimer’s disease. Epilepsy Behav. 2010, 17, 461–466. [Google Scholar] [CrossRef]
- Klein, P.; Diaz, A.; Gasalla, T.; Whitesides, J. A review of the pharmacology and clinical efficacy of brivaracetam. Clin. Pharmacol. Adv. Appl. 2018, 10, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Glauser, T.A.; Cnaan, A.; Shinnar, S.; Hirtz, D.G.; Dlugos, D.; Masur, D.; Clark, P.O.; Capparelli, E.V.; Adamson, P.C. Ethosuximide, valproic acid, and lamotrigine in childhood absence epilepsy. N. Engl. J. Med. 2010, 362, 790–799. [Google Scholar] [CrossRef] [Green Version]
- Löscher, W.; Schmidt, D. Epilepsy: Perampanel—New promise for refractory epilepsy? Nat. Rev. Neurol. 2012, 8, 661–662. [Google Scholar] [CrossRef]
- Zhang, X.; Heng, X.; Li, T.; Li, L.; Yang, D.; Zhang, X.; Du, Y.; Doody, R.S.; Le, W. Long-term treatment with lithium alleviates memory deficits and reduces amyloid-β production in an aged Alzheimer’s disease transgenic mouse model. J. Alzheimer’s Dis. 2011, 24, 739–749. [Google Scholar] [CrossRef]
- Matsunaga, S.; Kishi, T.; Annas, P.; Basun, H.; Hampel, H.; Iwata, N. Lithium as a Treatment for Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Alzheimer’s Dis. 2015, 48, 403–410. [Google Scholar] [CrossRef]
- Ware, K.; Tillery, E.; Linder, L. General pharmacokinetic/pharmacodynamic concepts of mood stabilizers in the treatment of bipolar disorder. Ment. Health Clin. 2016, 6, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Javitt, D.C. Glycine transport inhibitors in the treatment of schizophrenia. Handb. Exp. Pharmacol. 2012, 367–399. [Google Scholar] [CrossRef]
- Li, Y.; Krupa, B.; Kang, J.S.; Bolshakov, V.Y.; Liu, G. Glycine site of NMDA receptor serves as a spatiotemporal detector of synaptic activity patterns. J. Neurophysiol. 2009, 102, 578–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingelheim, B. Boehringer Ingelheim Refocuses Gly-T1 Inhibition Brain Research on Schizophrenia. 2020. Available online: https://www.boehringer-ingelheim.us/media-statements/media-statements/boehringer-ingelheim-refocuses-gly-t1-inhibition-brain-research (accessed on 7 December 2020).
- Avanir Pharmaceuticals Avanir Pharmaceuticals, Inc. Reports Phase 3 Data Evaluating Investigational AVP-786 for the Treatment of Moderate-to-Severe Agitation in Patients with Alzheimer’s Dementia; PRNewswire: Aliso Viejo, CA, USA, 2019. [Google Scholar]
- Okamoto, M.; Gray, J.D.; Larson, C.S.; Kazim, S.F.; Soya, H.; McEwen, B.S.; Pereira, A.C. Riluzole reduces amyloid beta pathology, improves memory, and restores gene expression changes in a transgenic mouse model of early-onset Alzheimer’s disease. Transl. Psychiatry 2018, 8, 153. [Google Scholar] [CrossRef]
- Gutierres, J.M.; Carvalho, F.B.; Schetinger, M.R.C.; Marisco, P.; Agostinho, P.; Rodrigues, M.; Rubin, M.A.; Schmatz, R.; Da Silva, C.R.; De Cognato, G.P.; et al. Anthocyanins restore behavioral and biochemical changes caused by streptozotocin-induced sporadic dementia of Alzheimer’s type. Life Sci. 2014, 96, 7–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, H.I.; Lin, C.C.; Tu, Y.F.; Chang, C.M.; Hsu, H.C.; Chi, C.H.; Kao, C.H. An increased risk of reversible dementia may occur after zolpidem derivative use in the elderly population a population-based case-control study. Medicine 2015, 94, e809. [Google Scholar] [CrossRef] [PubMed]
- Caltagirone, C.; Ferrannini, L.; Marchionni, N.; Nappi, G.; Scapagnini, G.; Trabucchi, M. The potential protective effect of tramiprosate (homotaurine) against Alzheimer’s disease: A review. Aging Clin. Exp. Res. 2012, 24, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Gervais, F.; Paquette, J.; Morissette, C.; Krzywkowski, P.; Yu, M.; Azzi, M.; Lacombe, D.; Kong, X.; Aman, A.; Laurin, J.; et al. Targeting soluble Aβ peptide with Tramiprosate for the treatment of brain amyloidosis. Neurobiol. Aging 2007, 28, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Aisen, P.S.; Gauthier, S.; Ferris, S.H.; Saumier, D.; Haine, D.; Garceau, D.; Duong, A.; Suhy, J.; Oh, J.; Lau, W.C.; et al. Tramiprosate in mild-to-moderate Alzheimer’s disease—A randomized, double-blind, placebo-controlled, multi-centre study (the alphase study). Arch. Med. Sci. 2011, 7, 102–111. [Google Scholar] [CrossRef] [Green Version]
- Abushakra, S.; Porsteinsson, A.; Scheltens, P.; Sadowsky, C.; Vellas, B.; Cummings, J.; Gauthier, S.; Hey, J.A.; Power, A.; Wang, P.; et al. Clinical Effects of Tramiprosate in APOE4/4 Homozygous Patients with Mild Alzheimer’s Disease Suggest Disease Modification Potential. J. Prev. Alzheimer’s Dis. 2017, 4, 149–156. [Google Scholar] [CrossRef]
- Abushakra, S.; Bracoud, L.; Schaerer, J.; Power, A.; Hey, J.; Scott, D.; Suhy, J.; Tolar, M. ADNI. APOE4/4 early to mild AD subjects show high rates of hippocampal atrophy and cognitive decline in ADNI-1 and tramiprosate datasets. J. Prev. Alzheimer’s Dis. 2018. [Google Scholar] [CrossRef]
- Froestl, W.; Gallagher, M.; Jenkins, H.; Madrid, A.; Melcher, T.; Teichman, S.; Mondadori, C.G.; Pearlman, R. SGS742: The first GABAB receptor antagonist in clinical trials. Biochem. Pharmacol. 2004, 68, 1479–1487. [Google Scholar] [CrossRef] [PubMed]
- Rice, H.C.; de Malmazet, D.; Schreurs, A.; Frere, S.; van Molle, I.; Volkov, A.N.; Creemers, E.; Vertkin, I.; Nys, J.; Ranaivoson, F.M.; et al. Secreted amyloid-b precursor protein functions as a GABA B R1a ligand to modulate synaptic transmission. Science 2019, 363. [Google Scholar] [CrossRef] [PubMed]
- Lilamand, M.; Porte, B.; Cognat, E.; Hugon, J.; Mouton-Liger, F.; Paquet, C. Are ketogenic diets promising for Alzheimer’s disease? A translational review. Alzheimer’s Res. Ther. 2020, 12, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Mcgill, K.J.; Jackson, C.F.; Bresnahan, R.; Levy, R.G.; Cooper, P.N. Ketogenic diets for drug-resistant epilepsy. Cochrane Database Syst. Rev. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Devivo, D.C.; Leckie, M.P.; Ferrendelli, J.S.; McDougal, D.B. Chronic ketosis and cerebral metabolism. Ann. Neurol. 1978, 3, 331–337. [Google Scholar] [CrossRef]
- Kashiwaya, Y.; Takeshima, T.; Mori, N.; Nakashima, K.; Clarke, K.; Veech, R.L. D-β-hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 5440–5444. [Google Scholar] [CrossRef] [Green Version]
- Torosyan, N.; Sethanandha, C.; Grill, J.D.; Dilley, M.L.; Lee, J.; Cummings, J.L.; Ossinalde, C.; Silverman, D.H. Changes in regional cerebral blood flow associated with a 45 day course of the ketogenic agent, caprylidene, in patients with mild to moderate Alzheimer’s disease: Results of a randomized, double-blinded, pilot study. Exp. Gerontol. 2018, 111, 118–121. [Google Scholar] [CrossRef]
- Taylor, M.K.; Sullivan, D.K.; Mahnken, J.D.; Burns, J.M.; Swerdlow, R.H. Feasibility and efficacy data from a ketogenic diet intervention in Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 28–36. [Google Scholar] [CrossRef]
- Rusek, M.; Pluta, R.; Ułamek-Kozioł, M.; Czuczwar, S.J. Ketogenic diet in alzheimer’s disease. Int. J. Mol. Sci. 2019, 20, 3892. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.; Chanda, S.; Marro, S.; Ng, Y.H.; Janas, J.A.; Haag, D.; Ang, C.E.; Tang, Y.; Flores, Q.; Mall, M.; et al. Generation of pure GABAergic neurons by transcription factor programming. Nat. Methods 2017, 14, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Lee, S.R.; Kim, S.U.; Lee, H.J. Alzheimer’s Disease and Stem Cell Therapy. Exp. Neurobiol. 2014, 23, 45–52. [Google Scholar] [CrossRef]
- Tong, L.M.; Fong, H.; Huang, Y. Stem cell therapy for Alzheimer’s disease and related disorders: Current status and future perspectives. Exp. Mol. Med. 2015, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, L.M.; Djukic, B.; Arnold, C.; Gillespie, A.K.; Yoon, S.Y.; Wang, M.M.; Zhang, O.; Knoferle, J.; Rubenstein, J.L.R.; Alvarez-Buylla, A.; et al. Inhibitory interneuron progenitor transplantation restores normal learning and memory in ApoE4 knock-in mice without or with Aβ accumulation. J. Neurosci. 2014, 34, 9506–9515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fouad, G.I. Stem cells as a promising therapeutic approach for Alzheimer’s disease: A review. Bull. Natl. Res. Cent. 2019, 43. [Google Scholar] [CrossRef]
- Hamm, V.; Héraud, C.; Bott, J.B.; Herbeaux, K.; Strittmatter, C.; Mathis, C.; Goutagny, R. Differential contribution of APP metabolites to early cognitive deficits in a TgCRND8 mouse model of Alzheimer’s disease. Sci. Adv. 2017, 3, e1601068. [Google Scholar] [CrossRef] [Green Version]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Imfeld, P.; Bodmer, M.; Schuerch, M.; Jick, S.S.; Meier, C.R. Seizures in patients with Alzheimer’s disease or vascular dementia: A population-based nested case-control analysis. Epilepsia 2013, 54, 700–707. [Google Scholar] [CrossRef]
- Sen, A.; Jette, N.; Husain, M.; Sander, J.W. Epilepsy in older people. Lancet 2020, 395, 735–748. [Google Scholar] [CrossRef]
- Sillanpää, M.; Anttinen, A.; Rinne, J.O.; Joutsa, J.; Sonninen, P.; Erkinjuntti, M.; Hermann, B.; Karrasch, M.; Saarinen, M.; Tiitta, P.; et al. Childhood-onset epilepsy five decades later. A prospective population-based cohort study. Epilepsia 2015, 56, 1774–1783. [Google Scholar] [CrossRef]
- Gottesman, R.F.; Schneider, A.L.C.; Zhou, Y.; Coresh, J.; Green, E.; Gupta, N.; Knopman, D.S.; Mintz, A.; Rahmim, A.; Sharrett, A.R.; et al. Association between midlife vascular risk factors and estimated brain amyloid deposition. J. Am. Med. Assoc. 2017, 317, 1443–1450. [Google Scholar] [CrossRef] [PubMed]
- Gibson, L.M.; Hanby, M.F.; Al-Bachari, S.M.; Parkes, L.M.; Allan, S.M.; Emsley, H.C.A. Late-onset epilepsy and occult cerebrovascular disease. J. Cereb. Blood Flow Metab. 2014, 34, 564–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, W.L. Aβ toxicity in Alzheimer’s disease: Globular oligomers (ADDLs) as new vaccine and drug targets. Neurochem. Int. 2002, 41, 345–352. [Google Scholar] [CrossRef]
- Wang, J.; Ho, L.; Chen, L.; Zhao, Z.; Zhao, W.; Qian, X.; Humala, N.; Seror, I.; Bartholomew, S.; Rosendorff, C.; et al. Valsartan lowers brain β-amyloid protein levels and improves spatial learning in a mouse model of Alzheimer disease. J. Clin. Investig. 2007, 117, 3393–3402. [Google Scholar] [CrossRef]
- Sa-nguanmoo, P.; Tanajak, P.; Kerdphoo, S.; Jaiwongkam, T.; Pratchayasakul, W.; Chattipakorn, N.; Chattipakorn, S.C. SGLT2-inhibitor and DPP-4 inhibitor improve brain function via attenuating mitochondrial dysfunction, insulin resistance, inflammation, and apoptosis in HFD-induced obese rats. Toxicol. Appl. Pharmacol. 2017, 333, 43–50. [Google Scholar] [CrossRef]
- Wium-Andersen, I.K.; Osler, M.; Jørgensen, M.B.; Rungby, J.; Wium-Andersen, M.K. Antidiabetic medication and risk of dementia in patients with type 2 diabetes: A nested case-control study. Eur. J. Endocrinol. 2019, 181, 499–507. [Google Scholar] [CrossRef]
- Sood, S.; Jain, K.; Gowthamarajan, K. Intranasal therapeutic strategies for management of Alzheimer’s disease. J. Drug Target. 2014, 22, 279–294. [Google Scholar] [CrossRef]
- Li, R.; Huang, Y.; Chen, L.; Zhou, H.; Zhang, M.; Chang, L.; Shen, H.; Zhou, M.; Su, P.; Zhu, D. Targeted delivery of intranasally administered nanoparticles-mediated neuroprotective peptide NR2B9c to brain and neuron for treatment of ischemic stroke. Nanomed. Nanotechnol. Biol. Med. 2019, 18, 380–390. [Google Scholar] [CrossRef]
- González-Nieto, D.; Fernández-Serra, R.; Pérez-Rigueiro, J.; Panetsos, F.; Martinez-Murillo, R.; Guinea, G.V. Biomaterials to Neuroprotect the Stroke Brain: A Large Opportunity for Narrow Time Windows. Cells 2020, 9, 1074. [Google Scholar] [CrossRef]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Wilkinson, C.W.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Breitner, J.C.S.; DeGroodt, W.; et al. Intranasal insulin improves cognition and modulates β-amyloid in early AD. Neurology 2008, 70, 440–448. [Google Scholar] [CrossRef]
- Morris, J.K.; Burns, J.M. Insulin: An emerging treatment for alzheimer’s disease dementia? Curr. Neurol. Neurosci. Rep. 2012, 12, 520–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De La Monte, S.M. Early intranasal insulin therapy halts progression of neurodegeneration: Progress in Alzheimer’s disease therapeutics. Aging Health 2012, 8, 61–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craft, S.; Claxton, A.; Baker, L.D.; Hanson, A.J.; Cholerton, B.; Trittschuh, E.H.; Dahl, D.; Caulder, E.; Neth, B.; Montine, T.J.; et al. Effects of Regular and Long-Acting Insulin on Cognition and Alzheimer’s Disease Biomarkers: A Pilot Clinical Trial. J. Alzheimer’s Dis. 2017, 57, 1325–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craft, S.; Raman, R.; Chow, T.W.; Rafii, M.S.; Sun, C.K.; Rissman, R.A.; Donohue, M.C.; Brewer, J.B.; Jenkins, C.; Harless, K.; et al. Safety, Efficacy, and Feasibility of Intranasal Insulin for the Treatment of Mild Cognitive Impairment and Alzheimer Disease Dementia: A Randomized Clinical Trial. JAMA Neurol. 2020, 77, 1099–1109. [Google Scholar] [CrossRef] [PubMed]
- Di Paolo, G.; Kim, T.W. Linking lipids to Alzheimer’s disease: Cholesterol and beyond. Nat. Rev. Neurosci. 2011, 12, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Di Battista, A.; Heinsinger, N.M.; William Rebeck, G. Alzheimer’s Disease Genetic Risk Factor APOE-ε4 Also Affects Normal Brain Function. Curr. Alzheimer Res. 2016, 13, 1200–1207. [Google Scholar] [CrossRef]
- Thalman, C.; Horta, G.; Qiao, L.; Endle, H.; Tegeder, I.; Cheng, H.; Laube, G.; Sigrudsson, T.; Hauser, M.J.; Tenzer, S.; et al. Synaptic phospholipids as a new target for cortical hyperexcitability and E/I balance in psychiatric disorders. Mol. Psychiatry 2018, 23, 1699–1710. [Google Scholar] [CrossRef]
- Shi, J.; Dong, Y.; Cui, M.Z.; Xu, X. Lysophosphatidic acid induces increased BACE1 expression and Aβ formation. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Sayas, C.L.; Moreno-Flores, M.T.; Avila, J.; Wandosell, F. The neurite retraction induced by lysophosphatidic acid increases Alzheimer’s disease-like Tau phosphorylation. J. Biol. Chem. 1999, 274, 37046–37052. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Kim, N.H.; Yang, H.; Kim, S.H.; Huh, S.O. Lysophosphatidic acid induces neurite retraction in differentiated neuroblastoma cells via GSK-3β activation. Mol. Cells 2011, 31, 483–489. [Google Scholar] [CrossRef] [Green Version]
- Castilla-Ortega, E.; Sánchez-López, J.; Hoyo-Becerra, C.; Matas-Rico, E.; Zambrana-Infantes, E.; Chun, J.; De Fonseca, F.R.; Pedraza, C.; Estivill-Torrús, G.; Santin, L.J. Exploratory, anxiety and spatial memory impairments are dissociated in mice lacking the LPA1 receptor. Neurobiol. Learn. Mem. 2010, 94, 73–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLimans, K.E.; Willette, A.A. Autotaxin is related to metabolic dysfunction and predicts Alzheimer’s disease outcomes. J. Alzheimer’s Dis. 2017, 56, 403–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramesh, S.; Govindarajulu, M.; Suppiramaniam, V.; Moore, T.; Dhanasekaran, M. Autotaxin–lysophosphatidic acid signaling in alzheimer’s disease. Int. J. Mol. Sci. 2018, 9, 1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardura-Fabregat, A.; Boddeke, E.W.G.M.; Boza-Serrano, A.; Brioschi, S.; Castro-Gomez, S.; Ceyzériat, K.; Dansokho, C.; Dierkes, T.; Gelders, G.; Heneka, M.T.; et al. Targeting Neuroinflammation to Treat Alzheimer’s Disease. CNS Drugs 2017, 31, 1057–1082. [Google Scholar] [CrossRef] [Green Version]
- Vezzani, A.; Baram, T.Z. New Roles for Interleukin-1 Beta in the Mechanisms of Epilepsy. Epilepsy Curr. 2007, 7, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Mrak, R.E.; Griffin, W.S.T. Interleukin-1 and the immunogenetics of Alzheimer disease. J. Neuropathol. Exp. Neurol. 2000, 59, 471–476. [Google Scholar] [CrossRef] [Green Version]
- Mrak, R.E.; Griffin, W.S.T. Interleukin-1, neuroinflammation, and Alzheimer’s disease. Neurobiol. Aging 2001, 22, 903–908. [Google Scholar] [CrossRef]
- O’Connor, J.J.; Coogan, A.N. Actions of the pro-inflammatory cytokine IL-1β on central synaptic transmission. Exp. Physiol. 1999, 84, 601–614. [Google Scholar] [CrossRef]
- Tanaka, S.; Ide, M.; Shibutani, T.; Ohtaki, H.; Numazawa, S.; Shioda, S.; Yoshida, T. Lipopolysaccharide-induced microglial activation induces learning and memory deficits without neuronal cell death in rats. J. Neurosci. Res. 2006, 83, 557–566. [Google Scholar] [CrossRef]
- Vezzani, A.; French, J.; Bartfai, T.; Baram, T.Z. The role of inflammation in epilepsy. Nat. Rev. Neurol. 2011, 7, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Depino, A.M.; Alonso, M.; Ferrari, C.; del Rey, A.; Anthony, D.; Besedovsky, H.; Medina, J.H.; Pitossi, F. Learning modulation by endogenous hippocampal IL-1: Blockade of endogenous IL-1 facilitates memory formation. Hippocampus 2004, 14, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Simon, A.; Van Der Meer, J.W.M. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 2012, 11, 633–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaturapatporn, D.; Isaac, M.G.E.K.N.; McCleery, J.; Tabet, N. Aspirin, steroidal and non-steroidal anti-inflammatory drugs for the treatment of Alzheimer’s disease. Cochrane Database Syst. Rev. 2012. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; Prins, N.; Lammertsma, A.; Yaqub, M.; Gouw, A.; Wink, A.M.; Chu, H.M.; van Berckel, B.N.M.; Alam, J. An exploratory clinical study of p38α kinase inhibition in Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2018, 5, 464–473. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.Y.; Lee, K.C.; Pei, Z.; Khan, A.; Bakshi, K.; Burns, L.H. PTI-125 binds and reverses an altered conformation of filamin A to reduce Alzheimer’s disease pathogenesis. Neurobiol. Aging 2017, 55, 99–114. [Google Scholar] [CrossRef]
- Wang, H.Y.; Pei, Z.; Lee, K.C.; Lopez-Brignoni, E.; Nikolov, B.; Crowley, C.A.; Marsman, M.R.; Barbier, R.; Friedmann, N.; Burns, L.H. PTI-125 Reduces Biomarkers of Alzheimer’s Disease in Patients. J. Prev. Alzheimers Dis. 2020, 7, 256–264. [Google Scholar] [CrossRef]
- Zhang, L.; Huang, T.; Teaw, S.; Nguyen, L.H.; Hsieh, L.S.; Gong, X.; Burns, L.H.; Bordey, A. Filamin A inhibition reduces seizure activity in a mouse model of focal cortical malformations. Sci. Transl. Med. 2020, 12, eaay0289. [Google Scholar] [CrossRef]
- Andrews-Zwilling, Y.; Bien-Ly, N.; Xu, Q.; Li, G.; Bernardo, A.; Yoon, S.Y.; Zwilling, D.; Yan, T.X.; Chen, L.; Huang, Y. Apolipoprotein E4 causes age- and Tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J. Neurosci. 2010, 30, 13707–13717. [Google Scholar] [CrossRef]
- Aboud, O.; Mrak, R.E.; Boop, F.A.; Griffin, W.S.T. Epilepsy: Neuroinflammation, neurodegeneration, and APOE genotype. Acta Neuropathol. Commun. 2014, 1, 41. [Google Scholar] [CrossRef] [Green Version]
- Amatniek, J.C.; Hauser, W.A.; DelCastillo-Castaneda, C.; Jacobs, D.M.; Marder, K.; Bell, K.; Albert, M.; Brandt, J.; Stern, Y. Incidence and predictors of seizures in patients with Alzheimer’s disease. Epilepsia 2006, 47, 867–872. [Google Scholar] [CrossRef]
- Nuriel, T.; Angulo, S.L.; Khan, U.; Ashok, A.; Chen, Q.; Figueroa, H.Y.; Emrani, S.; Liu, L.; Herman, M.; Barrett, G.; et al. Neuronal hyperactivity due to loss of inhibitory tone in APOE4 mice lacking Alzheimer’s disease-like pathology. Nat. Commun. 2017, 8, 1464. [Google Scholar] [CrossRef] [PubMed]
- Kunz, L.; Schröder, T.N.; Lee, H.; Montag, C.; Lachmann, B.; Sariyska, R.; Reuter, M.; Stirnberg, R.; Stöcker, T.; Messing-Floeter, P.C.; et al. Reduced grid-cell-like representations in adults at genetic risk for Alzheimer’s disease. Science 2015, 35, 430–433. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Gottesdiener, A.J.; Parmar, M.; Li, M.; Kaminsky, S.M.; Chiuchiolo, M.J.; Sondhi, D.; Sullivan, P.M.; Holtzman, D.M.; Crystal, R.G.; et al. Intracerebral adeno-associated virus gene delivery of apolipoprotein E2 markedly reduces brain amyloid pathology in Alzheimer’s disease mouse models. Neurobiol. Aging 2016, 44, 159–172. [Google Scholar] [CrossRef]
- Rosenberg, J.B.; Kaplitt, M.G.; De, B.P.; Chen, A.; Flagiello, T.; Salami, C.; Pey, E.; Zhao, L.; Ricart Arbona, R.J.; Monette, S.; et al. AAVrh.10-Mediated APOE2 Central Nervous System Gene Therapy for APOE4-Associated Alzheimer’s Disease. Hum. Gene Ther. Clin. Dev. 2018, 29, 24–47. [Google Scholar] [CrossRef] [PubMed]
- Hämäläinen, A.; Pihlajamäki, M.; Tanila, H.; Hänninen, T.; Niskanen, E.; Tervo, S.; Karjalainen, P.A.; Vanninen, R.L.; Soininen, H. Increased fMRI responses during encoding in mild cognitive impairment. Neurobiol. Aging 2007, 28, 1889–1903. [Google Scholar] [CrossRef] [PubMed]
- Yassa, M.A.; Stark, S.M.; Bakker, A.; Albert, M.S.; Gallagher, M.; Stark, C.E.L. High-resolution structural and functional MRI of hippocampal CA3 and dentate gyrus in patients with amnestic Mild Cognitive Impairment. Neuroimage 2010, 51, 1242–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celone, K.A.; Calhoun, V.D.; Dickerson, B.C.; Atri, A.; Chua, E.F.; Miller, S.L.; DePeau, K.; Rentz, D.M.; Selkoe, D.J.; Blacker, D.; et al. Alterations in memory networks in mild cognitive impairment and Alzheimer’s disease: An independent component analysis. J. Neurosci. 2006, 26, 10222–10231. [Google Scholar] [CrossRef] [Green Version]
- Quiroz, Y.T.; Budson, A.E.; Celone, K.; Ruiz, A.; Newmark, R.; Castrillõn, G.; Lopera, F.; Stern, C.E. Hippocampal hyperactivation in presymptomatic familial Alzheimer’s disease. Ann. Neurol. 2010, 68, 865–875. [Google Scholar] [CrossRef] [Green Version]
- Bondi, M.W.; Houston, W.S.; Eyler, L.T.; Brown, G.G. fMRI evidence of compensatory mechanisms in older adults at genetic risk for Alzheimer disease. Neurology 2005, 64, 501–508. [Google Scholar] [CrossRef] [Green Version]
- Bassett, S.S.; Yousem, D.M.; Cristinzio, C.; Kusevic, I.; Yassa, M.A.; Caffo, B.S.; Zeger, S.L. Familial risk for Alzheimer’s disease alters fMRI activation patterns. Brain 2006, 129, 1229–1239. [Google Scholar] [CrossRef] [Green Version]
- Putcha, D.; Brickhouse, M.; O’Keefe, K.; Sullivan, C.; Rentz, D.; Marshall, G.; Dickerson, B.; Sperling, R. Hippocampal hyperactivation associated with cortical thinning in Alzheimer’s disease signature regions in non-demented elderly adults. J. Neurosci. 2011, 31, 17680–17688. [Google Scholar] [CrossRef]
- Huijbers, W.; Mormino, E.C.; Schultz, A.P.; Wigman, S.; Ward, A.M.; Larvie, M.; Amariglio, R.E.; Marshall, G.A.; Rentz, D.M.; Johnson, K.A.; et al. Amyloid-β deposition in mild cognitive impairment is associated with increased hippocampal activity, atrophy and clinical progression. Brain 2015, 138, 1023–1035. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J. EEG dynamics in patients with Alzheimer’s disease. Clin. Neurophysiol. 2004, 115, 1490–1505. [Google Scholar] [CrossRef]
- Smailovic, U.; Jelic, V. Neurophysiological Markers of Alzheimer’s Disease: Quantitative EEG Approach. Neurol. Ther. 2019, 8, 37–55. [Google Scholar] [CrossRef] [Green Version]
- Zamrini, E.; Maestu, F.; Pekkonen, E.; Funke, M.; Makela, J.; Riley, M.; Bajo, R.; Sudre, G.; Fernandez, A.; Castellanos, N.; et al. Magnetoencephalography as a putative biomarker for Alzheimer’s disease. Int. J. Alzheimer’s Dis. 2011, 2011, 280289. [Google Scholar] [CrossRef] [Green Version]
- Stam, C.J.; Jones, B.F.; Manshanden, I.; van Cappellen van Walsum, A.M.; Montez, T.; Verbunt, J.P.A.; de Munck, J.C.; van Dijk, B.W.; Berendse, H.W.; Scheltens, P. Magnetoencephalographic evaluation of resting-state functional connectivity in Alzheimer’s disease. Neuroimage 2006, 32, 1335–1344. [Google Scholar] [CrossRef]
- Maestú, F.; Campo, P.; Gil-Gregorio, P.; Fernández, S.; Fernández, A.; Ortiz, T. Medial temporal lobe neuromagnetic hypoactivation and risk for developing cognitive decline in elderly population: A 2-year follow-up study. Neurobiol. Aging 2006, 27, 32–37. [Google Scholar] [CrossRef]
- López-Sanz, D.; Brunã, R.; Garcés, P.; Camara, C.; Serrano, N.; Rodríguez-Rojo, I.C.; Delgado, M.L.; Montenegro, M.; López-Higes, R.; Yus, M.; et al. Alpha band disruption in the AD-continuum starts in the Subjective Cognitive Decline stage: A MEG study. Sci. Rep. 2016, 6, 37685. [Google Scholar] [CrossRef]
- Bajo, R.; Maestú, F.; Nevado, A.; Sancho, M.; Gutiérrez, R.; Campo, P.; Castellanos, N.P.; Gil, P.; Moratti, S.; Pereda, E.; et al. Functional connectivity in mild cognitive impairment during a memory task: Implications for the disconnection hypothesis. J. Alzheimer’s Dis. 2010, 22, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Mandal, P.K.; Banerjee, A.; Tripathi, M.; Sharma, A. A comprehensive review of magnetoencephalography (MEG) studies for brain functionality in healthy aging and Alzheimer’s disease (AD). Front. Comput. Neurosci. 2018, 12, 60. [Google Scholar] [CrossRef]
- Nakamura, A.; Cuesta, P.; Fernández, A.; Arahata, Y.; Iwata, K.; Kuratsubo, I.; Bundo, M.; Hattori, H.; Sakurai, T.; Fukuda, K.; et al. Electromagnetic signatures of the preclinical and prodromal stages of Alzheimer’s disease. Brain 2018, 141, 1470–1485. [Google Scholar] [CrossRef] [Green Version]
- Ruzich, E.; Crespo-García, M.; Dalal, S.S.; Schneiderman, J.F. Characterizing hippocampal dynamics with MEG: A systematic review and evidence-based guidelines. Hum. Brain Mapp. 2019, 40, 1353–1375. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.H.; Lane, H.Y.; Lin, C.H. Brain stimulation in Alzheimer’s disease. Front. Psychiatry 2018, 9, 201. [Google Scholar] [CrossRef] [Green Version]
- Di Lorenzo, F.; Motta, C.; Casula, E.P.; Bonnì, S.; Assogna, M.; Caltagirone, C.; Martorana, A.; Koch, G. LTP-like cortical plasticity predicts conversion to dementia in patients with memory impairment. Brain Stimul. 2020, 13, 1175–1182. [Google Scholar] [CrossRef]
- Chou, Y.H.; Ton That, V.; Sundman, M. A systematic review and meta-analysis of rTMS effects on cognitive enhancement in mild cognitive impairment and Alzheimer’s disease. Neurobiol. Aging 2020, 86, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Z.; Edwards, M.J.; Rounis, E.; Bhatia, K.P.; Rothwell, J.C. Theta burst stimulation of the human motor cortex. Neuron 2005, 45, 201–206. [Google Scholar] [CrossRef] [Green Version]
- Di Lorenzo, F.; Motta, C.; Bonnì, S.; Mercuri, N.B.; Caltagirone, C.; Martorana, A.; Koch, G. LTP-like cortical plasticity is associated with verbal memory impairment in Alzheimer’s disease patients. Brain Stimul. 2019, 12, 148–151. [Google Scholar] [CrossRef]
- Di Lorenzo, F.; Ponzo, V.; Bonnì, S.; Motta, C.; Negrão Serra, P.C.; Bozzali, M.; Caltagirone, C.; Martorana, A.; Koch, G. Long-term potentiation–like cortical plasticity is disrupted in Alzheimer’s disease patients independently from age of onset. Ann. Neurol. 2016, 80, 202–210. [Google Scholar] [CrossRef]
- Ferreri, F.; Vecchio, F.; Vollero, L.; Guerra, A.; Petrichella, S.; Ponzo, D.; Määtta, S.; Mervaala, E.; Könönen, M.; Ursini, F.; et al. Sensorimotor cortex excitability and connectivity in Alzheimer’s disease: A TMS-EEG Co-registration study. Hum. Brain Mapp. 2016, 37, 2083–2096. [Google Scholar] [CrossRef]
- Pennisi, G.; Ferri, R.; Lanza, G.; Cantone, M.; Pennisi, M.; Puglisi, V.; Malaguarnera, G.; Bella, R. Transcranial magnetic stimulation in Alzheimer’s disease: A neurophysiological marker of cortical hyperexcitability. J. Neural Transm. 2011, 118, 587–598. [Google Scholar] [CrossRef]
- Koch, G.; Bonnì, S.; Pellicciari, M.C.; Casula, E.P.; Mancini, M.; Esposito, R.; Ponzo, V.; Picazio, S.; Di Lorenzo, F.; Serra, L.; et al. Transcranial magnetic stimulation of the precuneus enhances memory and neural activity in prodromal Alzheimer’s disease. Neuroimage 2018, 169, 302–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrucci, R.; Mameli, F.; Guidi, I.; Mrakic-Sposta, S.; Vergari, M.; Marceglia, S.; Cogiamanian, F.; Barbieri, S.; Scarpini, E.; Priori, A. Transcranial direct current stimulation improves recognition memory in Alzheimer disease. Neurology 2008, 71, 493–498. [Google Scholar] [CrossRef] [Green Version]
- Boggio, P.S.; Khoury, L.P.; Martins, D.C.S.; Martins, O.E.M.S.; De Macedo, E.C.; Fregni, F. Temporal cortex direct current stimulation enhances performance on a Visual recognition memory task in Alzheimer disease. J. Neurol. Neurosurg. Psychiatry 2009, 80, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Khedr, E.M.; El Gamal, N.F.; El-Fetoh, N.A.; Khalifa, H.; Ahmed, E.M.; Ali, A.M.; Noaman, M.; El-Baki, A.A.; Karim, A.A. A double-blind randomized clinical trial on the efficacy of cortical direct current stimulation for the treatment of Alzheimer’s disease. Front. Aging Neurosci. 2014, 6, 275. [Google Scholar] [CrossRef] [PubMed]
- Bystad, M.; Grønli, O.; Rasmussen, I.D.; Gundersen, N.; Nordvang, L.; Wang-Iversen, H.; Aslaksen, P.M. Transcranial direct current stimulation as a memory enhancer in patients with Alzheimer’s disease: A randomized, placebo-controlled trial. Alzheimer’s Res. Ther. 2016, 8, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, Y.; Wei, P.; Wang, C.; Shan, Y.; Yu, Y.; Qiao, Y.; Xie, B.; Shi, X.; Zhu, Z.; Lu, J.; et al. TRanscranial AlterNating current Stimulation FOR patients with Mild Alzheimer’s Disease (TRANSFORM-AD study): Protocol for a randomized controlled clinical trial. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2020, 6, e12005. [Google Scholar] [CrossRef]
- Iaccarino, H.F.; Singer, A.C.; Martorell, A.J.; Rudenko, A.; Gao, F.; Gillingham, T.Z.; Mathys, H.; Seo, J.; Kritskiy, O.; Abdurrob, F.; et al. Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature 2016, 540, 230–235. [Google Scholar] [CrossRef] [Green Version]
- Adaikkan, C.; Middleton, S.J.; Marco, A.; Pao, P.C.; Mathys, H.; Kim, D.N.W.; Gao, F.; Young, J.Z.; Suk, H.J.; Boyden, E.S.; et al. Gamma Entrainment Binds Higher-Order Brain Regions and Offers Neuroprotection. Neuron 2019, 102, 929–943. [Google Scholar] [CrossRef]
- Martorell, A.J.; Paulson, A.L.; Suk, H.J.; Abdurrob, F.; Drummond, G.T.; Guan, W.; Young, J.Z.; Kim, D.N.W.; Kritskiy, O.; Barker, S.J.; et al. Multi-sensory Gamma Stimulation Ameliorates Alzheimer’s-Associated Pathology and Improves Cognition. Cell 2019, 177, 256–271. [Google Scholar] [CrossRef] [Green Version]
- Ismail, R.; Hansen, A.K.; Parbo, P.; Brændgaard, H.; Gottrup, H.; Brooks, D.J.; Borghammer, P. The Effect of 40-Hz Light Therapy on Amyloid Load in Patients with Prodromal and Clinical Alzheimer’s Disease. Int. J. Alzheimer’s Dis. 2018, 6852303. [Google Scholar] [CrossRef] [Green Version]
- Sabbagh, M.; Sadowsky, C.; Tousi, B.; Agronin, M.E.; Alva, G.; Armon, C.; Bernick, C.; Keegan, A.P.; Karantzoulis, S.; Baror, E.; et al. Effects of a combined transcranial magnetic stimulation (TMS) and cognitive training intervention in patients with Alzheimer’s disease. Alzheimer’s Dement. 2020, 16, 641–650. [Google Scholar] [CrossRef]
- Andrade, S.M.; de Oliveira, E.A.; Alves, N.T.; dos Santos, A.C.G.; de Mendonça, C.T.P.L.; Sampaio, D.D.A.; da Silva, E.E.Q.C.; da Fonsêca, É.K.G.; de Almeida Rodrigues, E.T.; de Lima, G.N.S.; et al. Neurostimulation Combined with Cognitive Intervention in Alzheimer’s Disease (NeuroAD): Study Protocol of Double-Blind, Randomized, Factorial Clinical Trial. Front. Aging Neurosci. 2018, 10, 334. [Google Scholar] [CrossRef]
- Arendash, G.; Cao, C.; Abulaban, H.; Baranowski, R.; Wisniewski, G.; Becerra, L.; Andel, R.; Lin, X.; Zhang, X.; Wittwer, D.; et al. A Clinical Trial of Transcranial Electromagnetic Treatment in Alzheimer’s Disease: Cognitive Enhancement and Associated Changes in Cerebrospinal Fluid, Blood, and Brain Imaging. J. Alzheimer’s Dis. 2019, 71, 57–82. [Google Scholar] [CrossRef] [Green Version]
- Grossman, N.; Bono, D.; Dedic, N.; Kodandaramaiah, S.B.; Rudenko, A.; Suk, H.J.; Cassara, A.M.; Neufeld, E.; Kuster, N.; Tsai, L.H.; et al. Noninvasive Deep Brain Stimulation via Temporally Interfering Electric Fields. Cell 2017, 169, 1029–1041. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Lee, C.; Park, J.; Im, C.H. Individually customized transcranial temporal interference stimulation for focused modulation of deep brain structures: A simulation study with different head models. Sci. Rep. 2020, 10, 11730. [Google Scholar] [CrossRef]
- Chao, L.L. Effects of Home Photobiomodulation Treatments on Cognitive and Behavioral Function, Cerebral Perfusion, and Resting-State Functional Connectivity in Patients with Dementia: A Pilot Trial. Photobiomodul. Photomed. Laser Surg. 2019, 37, 133–141. [Google Scholar] [CrossRef]
- Machii, K.; Cohen, D.; Ramos-Estebanez, C.; Pascual-Leone, A. Safety of rTMS to non-motor cortical areas in healthy participants and patients. Clin. Neurophysiol. 2006, 117, 455–471. [Google Scholar] [CrossRef]
- Khedr, E.M.; Salama, R.H.; Abdel Hameed, M.; Abo Elfetoh, N.; Seif, P. Therapeutic Role of Transcranial Direct Current Stimulation in Alzheimer Disease Patients: Double-Blind, Placebo-Controlled Clinical Trial. Neurorehabil. Neural Repair 2019, 33, 384–394. [Google Scholar] [CrossRef]



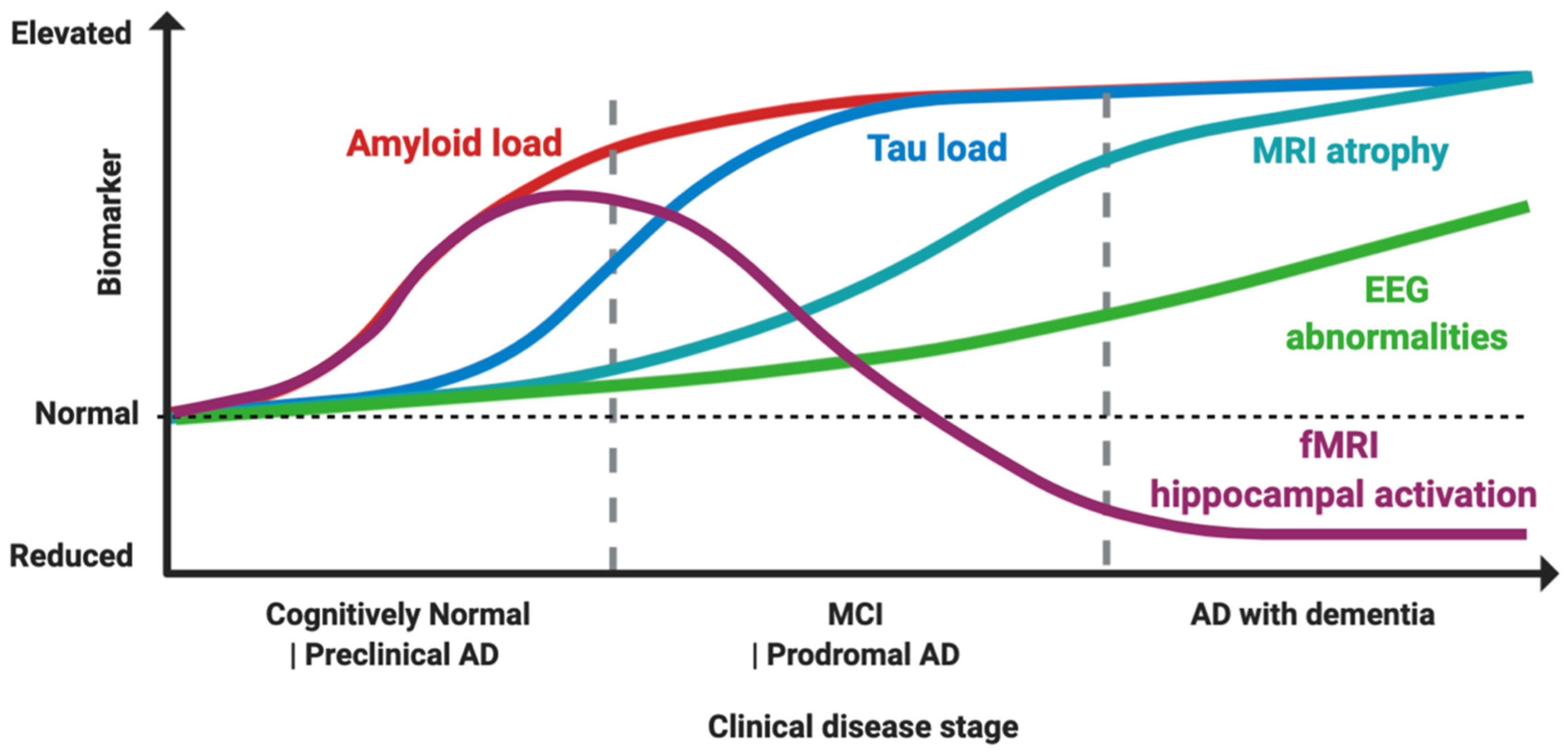{kind=link}
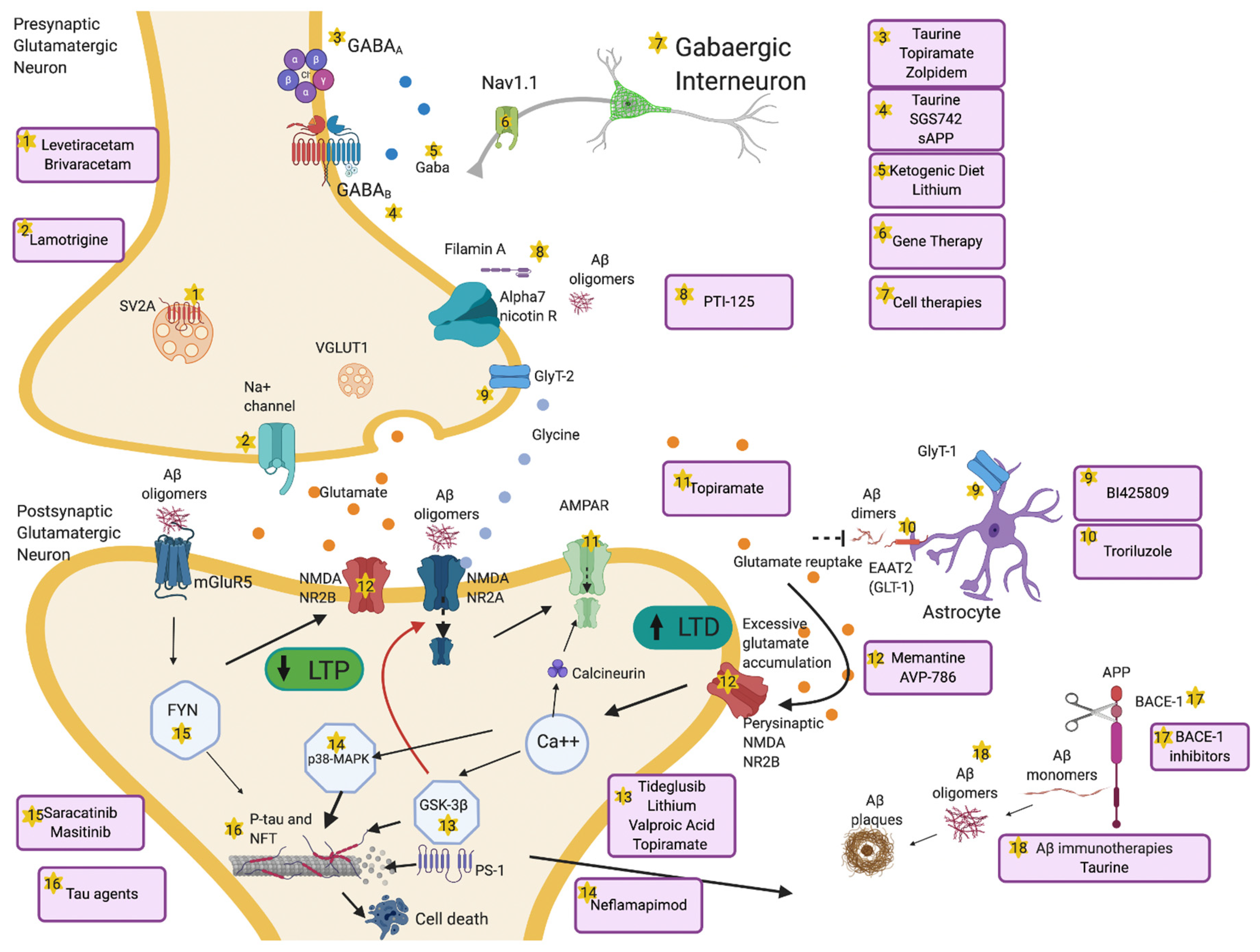{kind=link}
| Class and Name | Mechanism | Population | Phase, NCT Number and Outcomes, Reference |
|---|---|---|---|
| Aβ amyloid agents | |||
| Aducanumab | Passive immunotherapy | MCI 1 and Mild AD 2 | Phase 3 (NCT02484547)-ongoing Phase 3 (NCT02477800)-ongoing |
| Bapineuzumab | Passive immunotherapy | (1) Mild-moderate AD ApoE4+ (2) Mild-moderate AD ApoE4− | (1) Phase 3 (NCT00667810)–failed [142] (2) Phase 3 (NCT00676143)–failed [142] |
| BAN2401 | Passive immunotherapy | MCI and Mild AD | Phase 2 (NCT01767311)-ongoing Phase 3 (NCT03887455)-ongoing |
| Crenezumab | Passive immunotherapy | Mild AD | Phase 3 (NCT02670083)-terminated for lack of efficacy |
| Donanemab | Passive immunotherapy | Mild-moderate AD | Phase 2 (NCT03367403)-ongoing |
| Gantenerumab | Passive immunotherapy | Mild AD | Phase 3 (NCT03444870)-ongoing Phase 3 (NCT03443973)-ongoing |
| Ponezumab | Passive immunotherapy | Mild-moderate AD | Phase 2 (NCT00722046)–failed [143] |
| Solanezumab | Passive immunotherapy | MCI and mild AD | Phase 3 (NCT02760602)–terminated for lack of efficacy |
| BACE-1 3inhibitors | |||
| Atabecestat | BACE-1 inhibition | (1) Amyloid+ or ApoE4+ healthy subjects (2) Mild AD | (1) Phase 2/3 (NCT02569398)–failed [144] (2) Phase 2 (NCT02406027)–failed [145] |
| Elenbecestat | BACE-1 inhibition | Mild AD | Phase 3 (NCT03036280)-ongoing |
| Lanabecestat | BACE-1 inhibition | Early AD | Phase 2/3 (NCT02245737)-ongoing |
| Umibecestat | BACE-1 inhibition | ApoE4 + healthy subjects | Phase 2/3 (NCT03131453)–terminated for adverse cognitive effects |
| Verubecestat | BACE-1 inhibition | Mild AD | Phase 3 (NCT01953601)–failed [78] |
| Tau agents | |||
| AADvac1 | Active immunotherapy | (1) Mild-moderate AD (2) PPA 4 | (1) Phase 2 (NCT0257925)–no effects on cognition, reduction NFL 5 and MRI 6 atrophy [101] (2) Phase 1 (NCT03174886)-ongoing |
| ACI-35 | Active immunotherapy | MCI and Mild AD | Phase 2A (NCT04445831)-ongoing |
| Gosuranemab | Passive immunotherapy | MCI and Mild AD | Phase 2 (NCT03352557)-ongoing |
| Tilavonemab | Passive immunotherapy | MCI and Mild AD | Phase 2 (NCT02880956)-ongoing |
| Semorinemab | Passive immunotherapy | Mild-moderate AD | Phase 2 (NCT03289143)-ongoing |
| Zagotenemab | Passive immunotherapy | Mild-moderate AD | Phase 2 (NCT03518073)-ongoing Phase 2 (NCT03828747)-ongoing |
| Salsalate | Acetylation inhibitor | Mild-moderate AD | Phase 1 (NCT03277573)-ongoing |
| LMTM | Aggregator inhibitor | MCI and Mild AD | Phase 3 (NCT03446001)-ongoing |
| LY3372689 | OGA inhibitors 7 | Healthy participants | Phase 1 (NCT04392271)-ongoing |
| Davunetide | Microtubule stabilizers | MCI | Phase 2 (NCT00422981)–failed [105] |
| Rolipram | PDE4 8 inhibitor | APP/PS1 mice | Preclinical Phase [107] |
| ASOs 9 | MAPT 10 mRNA blockage | Mild AD | Phase 1/2 (NCT03186989)-ongoing |
| GSK-3β 11 inhibitors | |||
| Tideglusib | GSK-3β inhibition | Mild-moderate AD | Phase 2a (NCT00948259)-trends for cognitive benefits [127] Phase 2b (NCT01350362)–failed [128] |
| Memantine | NMDA 12 NR2B antagonist | Moderate-severe AD | Licensed in moderate-severe AD |
| Ifenprodil | NMDA NR2B antagonist | Pentylenetetrazol (PTZ)-kindled rats | Preclinical Phase [129] |
| Kinase inhibitors | |||
| Saracatinib | Src kinase inhibitor | Mild-moderate AD | Phase 2a (NCT02167256)–failed [139] |
| Masitinib | Tyrosine kinase inhibitor | Mild-moderate AD | Phase 2 (NCT00976118)-improvement in cognitive scores [140] Phase 3 (NCT01872598)-ongoing |
| Class and Name | Mechanism | Population | Phase, NCT Number and Outcomes, Reference |
|---|---|---|---|
| ASMs1 | |||
| Levetiracetam | SV2A 2 binding | (1, 2, 3) Mild-moderate AD 3 (4, 5) Mild AD (6) MCI 4 (7) ApoE4+ healthy subjects | (1) Phase 2 (NCT04004702)-ongoing (2) Phase 2 (NCT03489044)-ongoing (3) Phase 2 (NCT02002819)-ongoing (4) Phase 2 (NCT03875638)-ongoing (5) Phase n/a (NCT01554683)-not reported (6) Phase 2 (NCT01044758)-not reported (7) Phase 2 (NCT03461861)-ongoing |
| Brivaracetam | SV2A binding | APP/PS1 mice | Preclinical Phase [182] |
| Lamotrigine | Na+ channel blocker | APP/PS1 mice | Preclinical Phase [169] |
| NMDA modulators | |||
| Lithium | Downregulation of NMDA receptors, increasing GABAergic transmission, GSK-3β inhibition | MCI | Phase 4 (NCT03185208)-ongoing |
| BI425809 | GlyT-1 and GlyT-2 5 blockage | Mild AD | Phase 2 (NCT02788513)–failed [196] |
| AVP-786 | NMDA antagonist | AD | Phase 3 (NCT04464564)-ongoing Phase 3 (NCT04408755)-ongoing |
| Troriluzole | GLT-1 6 enhancement | AD | Phase 2/3 (NCT03605667)-ongoing |
| GABAergic modulators | |||
| Zolpidem | GABAA receptor agonist | AD | Phase 3 (NCT03075241)-ongoing |
| Tramiprosate | GABAA receptor agonist and GABAB receptor antagonist | MCI and AD | Nutritional supplement [203] |
| ALZ-801 | GABAA receptor agonist and GABAB receptor antagonist | Healthy subjects | Phase 1 (NCT04585347)-not reported Phase 1 (NCT04157712)-not reported |
| SGS742 | GABAB receptor antagonist | (1) MCI (2) Mild-Moderate AD | (1) Phase 2 (NCT n/a)-improvements in memory [206] (2) Phase 2 (NCT00093951)–not reported |
| sAPP 7 | GABABR1α modulator | Thy1-GCaMP6s mice | Preclinical Phase [207] |
| KD 8 | Ketone bodies production | (1, 3) AD (2) MCI and AD | (1) Phase n/a 9 (NCT03860792)-ongoing (2) Phase n/a (NCT03472664)-ongoing (3) Phase n/a (NCT02912936)-ongoing |
| Stem cells | Increase of GABAergic tone by restoring physiological cell phenotypes | (1, 4, 5) Mild-moderate AD (2) MCI (3) Mild AD (6) AD | (1) Phase 2 (NCT02833792)–ongoing (2) Phase 2 (NCT04228666)–ongoing (3) Phase 2 (NCT04482413)–ongoing (4) Phase 1/2 (NCT04388982)–ongoing (5) Phase 1/2 (NCT02899091)–ongoing (6) Phase 1/2 (NCT02054208)-not reported |
| Semagacestat | Nav1.1 channel enhancement | AD | Phase 3 (NCT00762411)–terminated for increased rates of skin cancer and lack of efficacy Phase 3 (NCT00594568)–failed [221] Phase 3 (NCT01035138)–terminated for increased rates of skin cancer and lack of efficacy |
| Class and Name | Mechanism | Population | Phase, NCT Number and Outcomes, Reference |
|---|---|---|---|
| Anti-hypertensive medication | |||
| Valsartan | Aβ amyloid reduction | Tg2576 mouse | Preclinical Phase [228] |
| Anti-diabetic medication | |||
| Dapagliflozin | SGLT2 1 inhibition | (1) Type 2 DM 2 (2) AD 3 with or without Type 2 DM | (1) Phase n/a 4 (NCT03961659)-ongoing (2) Phase 1 (NCT03801642)-ongoing |
| Intranasal insulin | Increase of brain glucose, reduce neuroinflammation and oxidative stress | (1) MCI and AD (2) MCI and AD (3) Healthy subjects or MCI (4) Healthy subjects or MCI | (1) Phase 2/3 (NCT01767909)–failed [238] (2) Phase 2 (NCT02503501)–terminated for lack of efficacy (3) Phase 2 (NCT03857321)–ongoing (4) Phase 3 (NCT04199767)–ongoing |
| Anti-inflammatory drugs | |||
| Neflamapimod | p38-MAPK 5 kinase inhibitor | Mild AD | Phase 2 (NCT03435861)-ongoing |
| PTI-125 | Filamin A inhibitor | Mild-moderate AD | Phase 2 (NCT04388254)-ongoing |
| Gene therapy | |||
| AAVrh.10-APOE2 | AAV 6 vectors | ApoE4+ MCI 7 or AD | Phase 1 (NCT03634007)-ongoing |
| Class and Name | Mechanism | Population | Phase, NCT Number and Outcomes, Reference |
|---|---|---|---|
| TMS 1 | Coil-induced depolarizing magnetic field | (1, 10) AD 2 (2) MCI 3 or AD (3, 4, 5, 6, 7, 8, 9, 12) Mild-moderate AD (11) PPA 4, MCI, AD | (1) Phase 2 (NCT00814697)–not reported (2) Phase 2 (NCT04555941)-ongoing (3) Phase n/a 5 (NCT03778151)-ongoing (4) Phase n/a (NCT04260724)-ongoing (5) Phase n/a (NCT03121066)-ongoing (6) Phase n/a (NCT02537496)-not reported (7) Phase n/a (NCT01481961)–ongoing (8) Phase n/a (NCT04263194)-ongoing (9) Phase n/a (NCT04294888)-ongoing (10) Phase n/a (NCT04562506)-not reported (11) Phase n/a (NCT04045990)-ongoing (12) Phase 4 (NCT02190084)-not reported |
| tDCS 6 | Low direct electric currents | Mild-moderate AD | Phase n/a (NCT03288363)–ongoing Phase n/a (NCT04404153)–ongoing Phase n/a (NCT03313518)–cognitive improvement and increase in CSF 7 Aβ42 [312] |
| tACS 8 | Sinusoidal, alternating low frequency currents | (1, 3) Mild-moderate AD (2) MCI (4) MCI and AD (5, 6) AD | (1) Phase n/a (NCT03290326)-not reported (2) Phase n/a (NCT04515433)-ongoing (3) Phase n/a (NCT03412604)-ongoing (4) Phase 1/2 (NCT03880240)-ongoing (5) Phase n/a (NCT03920826)-ongoing (6) Phase n/a (NCT04088643)-ongoing |
| GammaSense stimulation | 40 Hz LED light and auditory stimuli | (1, 2) MCI and AD | (1) Phase n/a (NCT03556280)-ongoing (2) Phase n/a (NCT03661034)-ongoing |
| TMS/tDCS and cognitive stimulation | Brain stimulation and computer-based cognitive stimulation | (1, 2) Mild-moderate AD | (1) Phase n/a (NCT01825317)–not reported (2) Phase n/a (NCT01825330)–not reported |
| NeuroEM | Transcranial electromagnetic treatment (TEMT) | (1, 2) Mild-moderate AD | (1) Phase n/a (NCT03927040)-ongoing (2) Phase 1/2 (NCT04271163)-ongoing |
| Temporal interference stimulation (TI) | Two different electric fields via electrodes | Healthy subjects | Phase n/a (NCT03747601)-ongoing |
| Photobiomodulation | Intranasal delivery of near infrared light via diodes | (1) Healthy subjects at risk for AD (2) AD (3) AD | (1) Phase 2 (NCT04018092)-ongoing (2) Phase n/a (NCT03405662)-ongoing (3) Phase n/a (NCT03160027)–improvements in cognition, cerebral perfusion and brain connectivity [310] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toniolo, S.; Sen, A.; Husain, M. Modulation of Brain Hyperexcitability: Potential New Therapeutic Approaches in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 9318. https://doi.org/10.3390/ijms21239318
Toniolo S, Sen A, Husain M. Modulation of Brain Hyperexcitability: Potential New Therapeutic Approaches in Alzheimer’s Disease. International Journal of Molecular Sciences. 2020; 21(23):9318. https://doi.org/10.3390/ijms21239318
Chicago/Turabian StyleToniolo, Sofia, Arjune Sen, and Masud Husain. 2020. "Modulation of Brain Hyperexcitability: Potential New Therapeutic Approaches in Alzheimer’s Disease" International Journal of Molecular Sciences 21, no. 23: 9318. https://doi.org/10.3390/ijms21239318
APA StyleToniolo, S., Sen, A., & Husain, M. (2020). Modulation of Brain Hyperexcitability: Potential New Therapeutic Approaches in Alzheimer’s Disease. International Journal of Molecular Sciences, 21(23), 9318. https://doi.org/10.3390/ijms21239318