Therapeutic Targeting Strategies for Early- to Late-Staged Alzheimer’s Disease


Abstract
:1. Introduction
2. Central Hypothesis for AD Pathogenesis
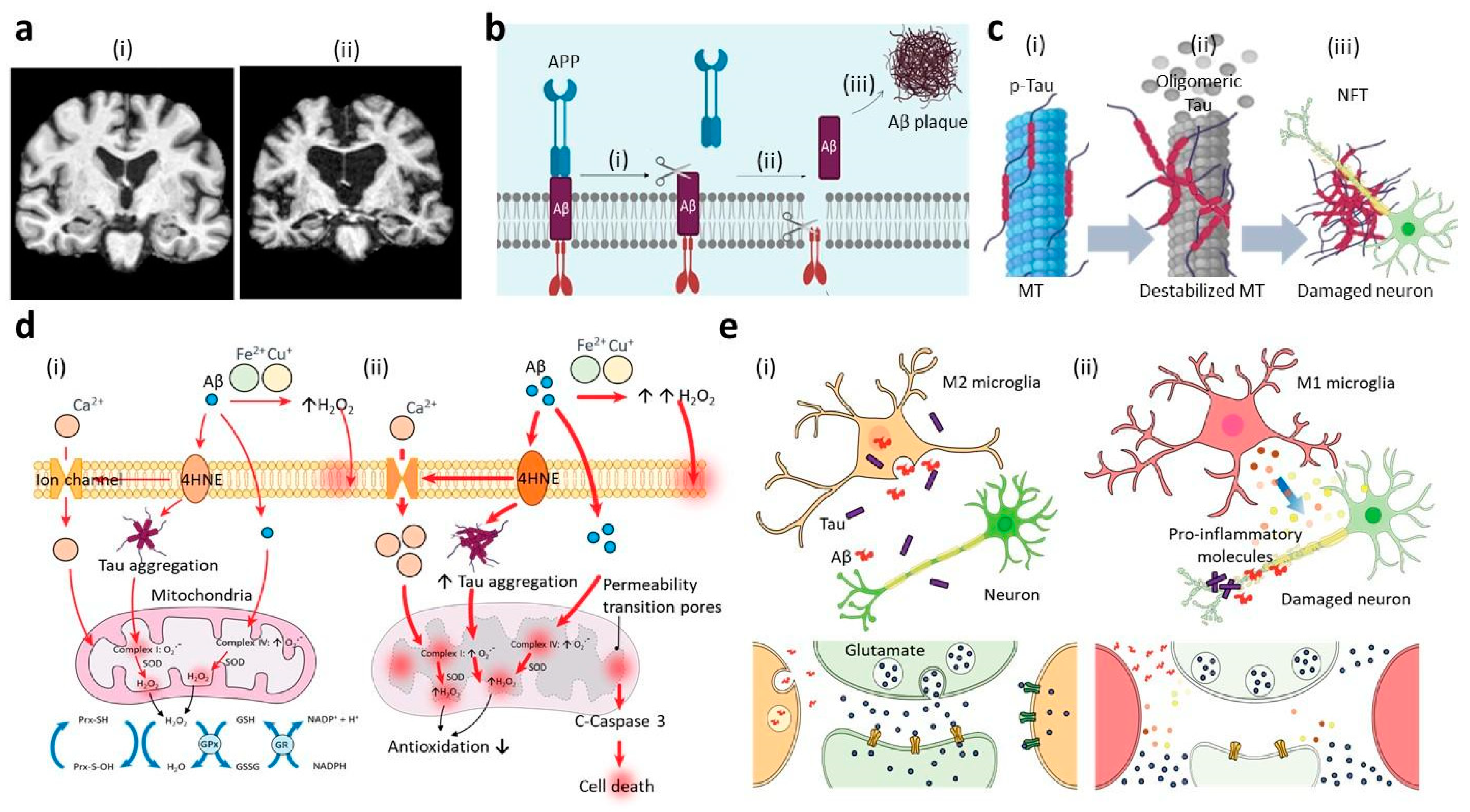
2.1. Aβ Hypothesis

2.2. Tau Hypothesis
2.3. Oxidative Stress
2.4. Neuroinflammation
3. Promising Strategies for Targeting Each Stage of AD Development
3.1. Conventional Strategies Targeting Aβ
3.1.1. Inhibition of Aβ Cascade
3.1.2. Passive Immunotherapy Targeting Aβ
3.1.3. Inhibition of Aβ Aggregation
3.1.4. Challenges
3.2. Inhibition of Tauopathy
3.2.1. Inhibition of Hyperphosphorylation in Tau
3.2.2. Inhibition of Tau Aggregation
3.2.3. Inhibition of Tau Activity
3.2.4. Challenges
3.3. Neuroinflammatory Modulation
3.3.1. Inhibition of JAK2/STAT3 Pathway
3.3.2. Inhibition of NF-κB/NLRP3 Pathway
3.3.3. Inhibition of p38 MAPK Pathway
3.3.4. Calcium/Calcineurin/NFAT Pathway
3.3.5. TREM2 Pathway
3.3.6. TLR Pathway
3.3.7. RAGE/CSF1R/P2Y1R Pathway
3.3.8. Anti-Inflammatory Therapy Targeting Adaptive Immune System
3.3.9. Challenges
4. Neuroregeneration Restoring Cognitive Impairment
4.1. Supplement of Neurotrophic Factors
4.1.1. Supplement of Neurotrophins
4.1.2. Increase of Neurotrophic Effects by Peptide Mimetics
4.2. Supplement of Neuronal Cells
4.2.1. Conversion of Glial Cells to Neuronal Cells
4.2.2. Transplanting Stem Cells into Brains
4.2.3. Challenges
5. Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 7,8-DHF | 7,8-dihydroxyflavone |
| AAV2 | Adeno-associated virus serotype 2 |
| AD | Alzheimer’s Disease |
| ALS | Amyotrophic lateral sclerosis |
| APOE | Apolipoprotein E |
| APP | Amyloid-beta precursor protein |
| ATP | Adenosine Triphosphate |
| Aβ | Amyloid beta |
| APLP1 | Amyloid-like protein 1 |
| BAC | Bacterial Artificial Chromosome |
| BACE-1 | Beta-site APP Cleaving Enzyme 1 |
| BDNF | Brain-Derived Neurotrophic Factor |
| CAMK II | Ca2+/calmodulin-dependent protein kinase II |
| CAT | Catalase |
| CBD | Corticobasal Degeneration |
| CDK5 | Cycle-dependent Kinase 5 |
| CHL1 | Close homolog of L1 |
| CNS | Central Nervous System |
| CNTFs | Ciliary Neurotrophic Factors |
| COX | Cyclooxygenase |
| CSF | Cerebrospinal Fluid |
| CSF1R | Colony-Stimulating Factor-1 Receptor |
| DAM | Disease-Associated Microglia |
| DAPT | N-(N-(3,5-difluorophenacetyl)-L-alanyl)-S-phenylglycine t-butyl ester |
| DNA | Deoxyribonucleic acid |
| ERK | Extracellular signal Regulated Kinase |
| FAD | Familial Alzheimer’s Disease |
| FTDP-17 | Frontotemporal dementia with parkinsonism-17 |
| GDNFs | Glial cell line-Derived Neurotrophic Factors |
| GABAergic | γ-Aminobutyric Acid-producing |
| GFAP | Glial Fibrillary Acidic Protein |
| GLP-1 | Glucagon-Like Peptide-1 |
| GPX | Glutathione Peroxidase |
| GSH | Glutathione |
| GSK-3β | Glycogen synthase kinase 3 beta |
| hNGFp | painless NGF protein |
| IFN-γ | Interferon gamma |
| IFITM3 | Interferon-induced transmembrane protein 3 |
| IGF-I | Insulin-like Growth Factor-I |
| IgG | Immunoglobulin G |
| IL | Interleukin |
| JAK | Janus Kinase |
| LMTM | Leuco-methylthioninium bis (hydro-methanesulfonate) |
| MAPK | Mitogen-Activated Protein Kinase |
| MB | Methylene blue |
| MCI | Mild cognitive impairment |
| MPACs | Metal protein attenuating compounds |
| MSCs | Mesenchymal Stem Cells |
| NADP | Nicotinamide adenine dinucleotide phosphate |
| NRF2 | Nuclear factor erythroid 2-related factor |
| NFAT | Nuclear Factor of Activated T-cells |
| NFT | Neurofibrillary Tangles |
| NF-κB | Nuclear factor-κB |
| NGF | Nerve Growth Factor |
| NLR | Nod-Like Receptor |
| NLRP3 | NLR family Pyrin domain containing 3 |
| NO | Nitric Oxide |
| NSAIDs | Non-Steroidal Anti-Inflammatory Drugs |
| NSCs | Neural Stem Cells |
| NT | Neurotrophin |
| OKA | Okadaic acid |
| P2Y1R | P2Y1 purinoreceptor |
| PET | Positron emission tomography |
| PD | Parkinson’s Disease |
| PHF | Paired Helical Filament |
| PI3K | Phosphatidylinositol 3-Kinase |
| PiD | Pick Disease |
| PKA | cAMP-dependent protein kinase |
| PP | Protein phosphatase |
| PPZ | Perphenazine |
| PS | Presenilin |
| PSP | Progressive Supranuclear Palsy |
| RAGE | Receptor for Advanced Glycosylation End products |
| RNA | Ribonucleic acid |
| RNS | Reactive Nitrogen Species |
| ROS | Reactive Oxygen Species |
| SAD | Sporadic Alzheimer’s Disease |
| SAG | Smoothened Agonist |
| SOCS3 | Suppressor of Cytokine Signaling 3 |
| SOD | Superoxide Dismutase |
| STAT | Signal Transducer and Activator of Transcription |
| sTREM2 | Soluble TREM2 |
| TBB | 4,5,6,7-tetrabromobenzotriazole |
| TGF-β | Transforming growth factor beta |
| Th1 | Type I helper T cells |
| Th2 | Type II helper T cells |
| TLR | Toll-Like Receptor |
| TNF-α | Tumor Necrosis Factor |
| Tregs | Regulation T cells |
| TREM2 | Triggering Receptor Expressed on Myeloid cells 2 |
| Trks | Tropomyosin Receptor Kinases |
| TTNPB | 4-((E)-2-(5,6,7,8-Tetrahydro-5,5,8,8-tetramethyl-2-naphthalenyl)-1-propenyl)-benzoic acid |
| VEGF | Vascular Endothelial Growth Factor |
| VPA | Valproic Acid |
References
- Ferri, C.P.; Prince, M.; Brayne, C.; Brodaty, H.; Fratiglioni, L.; Ganguli, M.; Hall, K.; Hasegawa, K.; Hendrie, H.; Huang, Y.; et al. Global Prevalence of Dementia: A Delphi Consensus Study. Lancet 2005, 366, 2112–2117. [Google Scholar] [CrossRef]
- Patterson, C. World Alzheimer Report 2018—The State of the Art of Dementia Research: New Frontiers; Alzheimer’s Disease International: London, UK, 2018; pp. 1–48. [Google Scholar]
- Martin, P.; Anders, W.; Maëlenn, G.; Ali, G.; Yu-Tzu, W.; Matthew, P. World Alzheimer Report 2015: The Global Impact of Dementia—An Analysis of Prevalence, Incidence, Cost and Trends; Alzheimer’s Disease International: London, UK, 2015; pp. 1–87. [Google Scholar]
- Ledig, C.; Schuh, A.; Guerrero, R.; Heckemann, R.A.; Rueckert, D. Structural Brain Imaging in Alzheimer’s Disease and Mild Cognitive Impairment: Biomarker Analysis and Shared Morphometry Database. Sci. Rep. 2018, 8, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alzheimer’s Association. 2019 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Mensah, G.A.; Wei, G.S.; Sorlie, P.D.; Fine, L.J.; Rosenberg, Y.; Kaufmann, P.G.; Mussolino, M.E.; Hsu, L.L.; Addou, E.; Engelgau, M.M.; et al. Decline in Cardiovascular Mortality: Possible Causes and Implications. Circ. Res. 2017, 120, 366–380. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2020. Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Lackland, D.T.; Roccella, E.J.; Deutsch, A.F.; Fornage, M.; George, M.G.; Howard, G.; Kissela, B.M.; Kittner, S.J.; Lichtman, J.H.; Lisabeth, L.D.; et al. Factors Influencing the Decline in Stroke Mortality a Statement from the American Heart Association/American Stroke Association. Stroke 2014, 45, 315–353. [Google Scholar] [CrossRef] [Green Version]
- WHO. Dementia. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 21 September 2020).
- Murphy, M.P.; LeVine, H., III. Alzheimer’s Disease and the β-Amyloid Peptide. J. Alzheimer’s Dis. 2010, 19, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, J.; Allsop, D. Amyloid Deposition as the Central Event in the Aetiology of Alzheimer’s Disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Espíndola, S.L.; Damianich, A.; Alvarez, R.J.; Sartor, M.; Belforte, J.E.; Ferrario, J.E.; Gallo, J.M.; Avale, M.E. Modulation of Tau Isoforms Imbalance Precludes Tau Pathology and Cognitive Decline in a Mouse Model of Tauopathy. Cell Rep. 2018, 23, 709–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosik, K.S.; Joachim, C.L.; Selkoe, D.J. Microtubule-Associated Protein Tau (τ) Is a Major Antigenic Component of Paired Helical Filaments in Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1986, 83, 4044–4048. [Google Scholar] [CrossRef] [Green Version]
- Houck, A.L.; Hernández, F.; Ávila, J. A Simple Model to Study Tau Pathology. J. Exp. Neurosci. 2016, 10, 31–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a Central Mechanism in Alzheimer’s Disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, A.D.; Fonken, L.K. Glial Cells Shape Pathology and Repair After Spinal Cord Injury. Neurotherapeutics 2018, 15, 554–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawikr, Y.; Yarla, N.S.; Peluso, I.; Kamal, M.A.; Aliev, G.; Bishayee, A. Neuroinflammation in Alzheimer’s Disease: The Preventive and Therapeutic Potential of Polyphenolic Nutraceuticals. Adv. Protein Chem. Struct. Biol. 2017, 108, 33–57. [Google Scholar] [PubMed]
- Egan, M.F.; Kost, J.; Tariot, P.N.; Aisen, P.S.; Cummings, J.L.; Vellas, B.; Sur, C.; Mukai, Y.; Voss, T.; Furtek, C.; et al. Randomized Trial of Verubecestat for Mild-to-Moderate Alzheimer’s Disease. N. Engl. J. Med. 2018, 378, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Malone, E. Lilly/AstraZeneca’s Lanabecestat Becomes Latest BACE Inhibitor Casualty. Available online: https://scrip.pharmaintelligence.informa.com/SC123243/LillyAstraZenecas-Lanabecestat-BecomesLatest-BACE-Inhibitor-Casualty (accessed on 12 June 2018).
- Hung, S.Y.; Fu, W.M. Drug Candidates in Clinical Trials for Alzheimer’s Disease. J. Biomed. Sci. 2017, 24, 1–12. [Google Scholar] [CrossRef]
- Update on Janssen’s BACE Inhibitor Program Regarding the Dominantly Inherited Alzheimer’s Network Trial (DIAN-TU). Available online: https://www.janssen.com/neuroscience/update-janssens-bace-inhibitor-program-regarding-DIAN-TU (accessed on 17 May 2018).
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A Phase 3 Trial of Semagacestat for Treatment of Alzheimer’s Disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef]
- Green, R.C.; Schneider, L.S.; Amato, D.A.; Beelen, A.P.; Wilcock, G.; Swabb, E.A.; Zavitz, K.H. Effect of Tarenflurbil on Cognitive Decline and Activities of Daily Living in Patients with Mild Alzheimer Disease: A Randomized Controlled Trial. JAMA 2009, 302, 2557–2564. [Google Scholar] [CrossRef] [Green Version]
- Coric, V.; Van Dyck, C.H.; Salloway, S.; Andreasen, N.; Brody, M.; Richter, R.W.; Soininen, H.; Thein, S.; Shiovitz, T.; Pilcher, G.; et al. Safety and Tolerability of the γ-Secretase Inhibitor Avagacestat in a Phase 2 Study of Mild to Moderate Alzheimer Disease. Arch. Neurol. 2012, 69, 1430–1440. [Google Scholar] [CrossRef] [Green Version]
- Khorassani, F.; Hilas, O. Bapineuzumab, an Investigational Agent for Alzheimer’s Disease. Pharm. Ther. 2013, 38, 89–91. [Google Scholar]
- Ostrowitzki, S.; Lasser, R.A.; Dorflinger, E.; Scheltens, P.; Barkhof, F.; Nikolcheva, T.; Ashford, E.; Retout, S.; Hofmann, C.; Delmar, P.; et al. A Phase III Randomized Trial of Gantenerumab in Prodromal Alzheimer’s Disease. Alzheimer’s Res. Ther. 2017, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lannfelt, L.; Blennow, K.; Zetterberg, H.; Batsman, S.; Ames, D.; Harrison, J.; Masters, C.L.; Targum, S.; Bush, A.I.; Murdoch, R.; et al. Safety, Efficacy, and Biomarker Findings of PBT2 in Targeting Aβ as a Modifying Therapy for Alzheimer’s Disease: A Phase IIa, Double-Blind, Randomised, Placebo-Controlled Trial. Lancet Neurol. 2008, 7, 779–786. [Google Scholar] [CrossRef]
- DeVos, S.L.; Miller, R.L.; Schoch, K.M.; Holmes, B.B.; Kebodeaux, C.S.; Wegener, A.J.; Chen, G.; Shen, T.; Tran, H.; Nichols, B.; et al. Tau Reduction Prevents Neuronal Loss and Reverses Pathological Tau Deposition and Seeding in Mice with Tauopathy. Sci. Transl. Med. 2017, 9, eaag0481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamat, P.K.; Rai, S.; Nath, C. Okadaic Acid Induced Neurotoxicity: An Emerging Tool to Study Alzheimer’s Disease Pathology. Neurotoxicology 2013, 37, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, Y.; Fu, Z.; Li, Y.; Feng, J.; Luo, J.; Zhang, Q.; Wang, Q.; Tian, Q. Melatonin Ameliorates Alzheimer-like Pathological Changes and Spatial Memory Retention Impairment Induced by Calyculin A. J. Psychopharmacol. 2011, 25, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Chohan, M.O.; Khatoon, S.; Iqbal, I.G.; Iqbal, K. Involvement of I2PP2A in the Abnormal Hyperphosphorylation of Tau and Its Reversal by Memantine. FEBS Lett. 2006, 580, 3973–3979. [Google Scholar] [CrossRef] [Green Version]
- Giguère, F.S.-C.; Essis, S.A.; Chagniel, L.; Germain, M.; Cyr, M.; Massicotte, G. The Sphingosine-1-Phosphate Receptor 1 Agonist SEW2871 Reduces Tau-Ser262 Phosphorylation in Rat Hippocampal Slices. Brain Res. 2017, 1658, 51–59. [Google Scholar] [CrossRef]
- Xiong, Y.; Jing, X.P.; Zhou, X.W.; Wang, X.L.; Yang, Y.; Sun, X.Y.; Qiu, M.; Cao, F.Y.; Lu, Y.M.; Liu, R.; et al. Zinc Induces Protein Phosphatase 2A Inactivation and Tau Hyperphosphorylation through Src Dependent PP2A (Tyrosine 307) Phosphorylation. Neurobiol. Aging 2013, 34, 745–756. [Google Scholar] [CrossRef]
- Lovestone, S.; Boada, M.; Dubois, B.; Hüll, M.; Rinne, J.O.; Huppertz, H.J.; Calero, M.; Andrés, M.V.; Gómez-Carrillo, B.; León, T.; et al. A Phase II Trial of Tideglusib in Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 45, 75–88. [Google Scholar] [CrossRef]
- Harald, H.; Michael, E.; Katharina, B.; Peter, A.; Anette, M.; Anna, B.; Lutz, F.; Johannes, S.; Peter, S.; Matthias, W.R.; et al. Lithium Trial in Alzheimer’s Disease: A Randomized, Single-Blind, Placebo-Controlled, Multicenter 10-Week Study. J. Clin. Psychiatry 2009, 70, 922–931. [Google Scholar]
- Ratan, B.; Yafeng, X.; Stefan, B.; Sven, H.; Mats, O.; Yvonne, N.; Ann-Cathrin, R.; Eva, J.; Per-Olof, M.; Thomas, B.; et al. Structural Insights and Biological Effects of Glycogen Synthase Kinase 3-Specific Inhibitor AR-A014418. J. Biol. Chem. 2003, 278, 45937–45945. [Google Scholar]
- Martinez, A.; Alonso, M.; Castro, A.; Pérez, C.; Moreno, F.J. First Non-ATP Competitive Glycogen Synthase Kinase 3 β (GSK-3β) Inhibitors: Thiadiazolidinones (TDZD) as Potential Drugs for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2002, 45, 1292–1299. [Google Scholar] [CrossRef] [PubMed]
- Le Corre, S.; Klafki, H.W.; Plesnila, N.; Hübinger, G.; Obermeier, A.; Sahagún, H.; Monse, B.; Seneci, P.; Lewis, J.; Eriksen, J.; et al. An Inhibitor of Tau Hyperphosphorylation Prevents Severe Motor Impairments in Tau Transgenic Mice. Proc. Natl. Acad. Sci. USA 2006, 103, 9673–9678. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, A.; Tyzio, R.; Zilberter, Y.; Ben-Ari, Y. (R)-Roscovitine, a Cyclin-Dependent Kinase Inhibitor, Enhances Tonic GABA Inhibition in Rat Hippocampus. Neuroscience 2008, 156, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Mettey, Y.; Gompel, M.; Thomas, V.; Garnier, M.; Leost, M.; Ceballos-Picot, I.; Noble, M.; Endicott, J.; Vierfond, J.M.; Meijer, L. Aloisines, a New Family of CDK/GSK-3 Inhibitors. SAR Study, Crystal Structure in Complex with CDK2, Enzyme Selectivity, and Cellular Effects. J. Med. Chem. 2003, 46, 222–236. [Google Scholar] [CrossRef] [PubMed]
- Min, S.W.; Chen, X.; Tracy, T.E.; Li, Y.; Zhou, Y.; Wang, C.; Shirakawa, K.; Minami, S.S.; Defensor, E.; Mok, S.A.; et al. Critical Role of Acetylation in Tau-Mediated Neurodegeneration and Cognitive Deficits. Nat. Med. 2015, 21, 1154–1162. [Google Scholar] [CrossRef] [Green Version]
- Sanders, O.; Rajagopal, L. Phosphodiesterase Inhibitors for Alzheimer’s Disease: A Systematic Review of Clinical Trials and Epidemiology with a Mechanistic Rationale. J. Alzheimer’s Dis. Rep. 2020, 4, 185–215. [Google Scholar] [CrossRef]
- Gauthier, S.; Feldman, H.H.; Schneider, L.S.; Wilcock, G.K.; Frisoni, G.B.; Hardlund, J.H.; Moebius, H.J.; Bentham, P.; Kook, K.A.; Wischik, D.J.; et al. Efficacy and Safety of Tau-Aggregation Inhibitor Therapy in Patients with Mild or Moderate Alzheimer’s Disease: A Randomised, Controlled, Double-Blind, Parallel-Arm, Phase 3 Trial. Lancet 2016, 388, 2873–2884. [Google Scholar] [CrossRef] [Green Version]
- Pradhan, K.; Das, G.; Kar, C.; Mukherjee, N.; Khan, J.; Mahata, T.; Barman, S.; Ghosh, S. Rhodamine-Based Metal Chelator: A Potent Inhibitor of Metal-Catalyzed Amyloid Toxicity. ACS Omega 2020, 5, 18958–18967. [Google Scholar] [CrossRef]
- Pickhardt, M.; Biernat, J.; Khlistunova, I.; Wang, Y.-P.; Gazova, Z.; Mandelkow, E.-M.; Mandelkow, E. N-Phenylamine Derivatives as Aggregation Inhibitors in Cell Models of Tauopathy. Curr. Alzheimer Res. 2007, 4, 397–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wischik, C.M.; Staff, R.T.; Wischik, D.J.; Bentham, P.; Murray, A.D.; Storey, J.M.D.; Kook, K.A.; Harrington, C.R. Tau Aggregation Inhibitor Therapy: An Exploratory Phase 2 Study in Mild or Moderate Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 44, 705–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Carroll, J.; Trojanowski, J.Q.; Yao, Y.; Iba, M.; Potuzak, J.S.; Hogan, A.M.L.; Xie, S.X.; Ballatore, C.; Smith, A.B.; et al. The Microtubule-Stabilizing Agent, Epothilone D, Reduces Axonal Dysfunction, Neurotoxicity, Cognitive Deficits, and Alzheimer-like Pathology in an Interventional Study with Aged Tau Transgenic Mice. J. Neurosci. 2012, 32, 3601–3611. [Google Scholar] [CrossRef] [PubMed]
- Quraishe, S.; Cowan, C.M.; Mudher, A. NAP (Davunetide) Rescues Neuronal Dysfunction in a Drosophila Model of Tauopathy. Mol. Psychiatry 2013, 18, 834–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, R.M.; Miller, Z.; Koestler, M.; Rojas, J.C.; Ljubenkov, P.A.; Rosen, H.J.; Rabinovici, G.D.; Fagan, A.M.; Cobigo, Y.; Brown, J.A.; et al. Reactions to Multiple Ascending Doses of the Microtubule Stabilizer TPI-287 in Patients with Alzheimer Disease, Progressive Supranuclear Palsy, and Corticobasal Syndrome: A Randomized Clinical Trial. JAMA Neurol. 2020, 77, 215–224. [Google Scholar] [CrossRef]
- Choi, M.; Kim, H.; Yang, E.J.; Kim, H.S. Inhibition of STAT3 Phosphorylation Attenuates Impairments in Learning and Memory in 5XFAD Mice, an Animal Model of Alzheimer’s Disease. J. Pharmacol. Sci. 2020, 143, 290–299. [Google Scholar] [CrossRef]
- Liu, X.W.; Ji, E.F.; He, P.; Xing, R.X.; Tian, B.X.; Li, X.D. Protective Effects of the P38 MAPK Inhibitor SB203580 on NMDA-Induced Injury in Primary Cerebral Cortical Neurons. Mol. Med. Rep. 2014, 10, 1942–1948. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Zhou, G.; Liu, H.; Zhang, B.; Li, J.; Cui, R.; Du, Y. Protective Effects of P38 MAPK Inhibitor SB202190 against Hippocampal Apoptosis and Spatial Learning and Memory Deficits in a Rat Model of Vascular Dementia. BioMed Res. Int. 2013, 2013, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Rehman, S.U.; Ahmad, A.; Yoon, G.H.; Khan, M.; Abid, M.N.; Kim, M.O. Inhibition of C-Jun N-Terminal Kinase Protects against Brain Damage and Improves Learning and Memory after Traumatic Brain Injury in Adult Mice. Cereb. Cortex 2018, 28, 2854–2872. [Google Scholar] [CrossRef]
- Gee, M.S.; Kim, S.W.; Kim, N.; Lee, S.J.; Oh, M.S.; Jin, H.K.; Bae, J.S.; Inn, K.S.; Kim, N.J.; Lee, J.K. A Novel and Selective P38 Mitogen-Activated Protein Kinase Inhibitor Attenuates LPS-Induced Neuroinflammation in BV2 Microglia and a Mouse Model. Neurochem. Res. 2018, 43, 2362–2371. [Google Scholar] [CrossRef]
- Rojanathammanee, L.; Floden, A.M.; Manocha, G.D.; Combs, C.K. Attenuation of Microglial Activation in a Mouse Model of Alzheimer’s Disease via NFAT Inhibition. J. Neuroinflamm. 2015, 12, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Massachusetts General Hospital. A Pilot Open Labeled Study of Tacrolimus in Alzheimer’s Disease. 2020; ClinicalTrials.gov Web Site. Available online: https://clinicaltrials.gov/ct2/show/NCT04263519 (accessed on 2 November 2020).
- Kim, J.; Eltorai, A.E.M.; Jiang, H.; Liao, F.; Verghese, P.B.; Kim, J.; Stewart, F.R.; Basak, J.M.; Holtzman, D.M. Anti-ApoE Immunotherapy Inhibits Amyloid Accumulation in a Transgenic Mouse Model of Aβ Amyloidosis. J. Exp. Med. 2012, 209, 2149–2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Q.; Danao, J.; Talreja, S.; Wen, P.; Yin, J.; Sun, N.; Li, C.M.; Chui, D.; Tran, D.; Koirala, S.; et al. TREM2-Activating Antibodies Abrogate the Negative Pleiotropic Effects of the Alzheimer’s Disease Variant Trem2R47H on Murine Myeloid Cell Function. J. Biol. Chem. 2018, 293, 12620–12633. [Google Scholar] [CrossRef] [Green Version]
- Olmos-Alonso, A.; Schetters, S.T.T.; Sri, S.; Askew, K.; Mancuso, R.; Vargas-Caballero, M.; Holscher, C.; Perry, V.H.; Gomez-Nicola, D. Pharmacological Targeting of CSF1R Inhibits Microglial Proliferation and Prevents the Progression of Alzheimer’s-like Pathology. Brain 2016, 139, 891–907. [Google Scholar] [CrossRef] [Green Version]
- Reichenbach, N.; Delekate, A.; Breithausen, B.; Keppler, K.; Poll, S.; Schulte, T.; Peter, J.; Plescher, M.; Hansen, J.N.; Blank, N.; et al. P2Y1 Receptor Blockade Normalizes Network Dysfunction and Cognition in an Alzheimer’s Disease Model. J. Exp. Med. 2018, 215, 1649–1663. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Arendash, G.W.; Dickson, A.; Mamcarz, M.B.; Lin, X.; Ethell, D.W. Aβ-Specific Th2 Cells Provide Cognitive and Pathological Benefits to Alzheimer’s Mice without Infiltrating the CNS Chuanhai. Neurobiol. Dis. 2009, 34, 63–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsh, S.E.; Abud, E.M.; Lakatos, A.; Karimzadeh, A.; Yeung, S.T.; Davtyan, H.; Fote, G.M.; Lau, L.; Weinger, J.G.; Lane, T.E.; et al. The Adaptive Immune System Restrains Alzheimer’s Disease Pathogenesis by Modulating Microglial Function. Proc. Natl. Acad. Sci. USA 2016, 113, E1316–E1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Fang, Y.; Lian, Y.; Chen, Y.; Wu, T.; Zheng, Y.; Zong, H.; Sun, L.; Zhang, R.; Wang, Z.; et al. Brain-Derived Neurotrophic Factor Ameliorates Learning Deficits in a Rat Model of Alzheimer’s Disease Induced by Aβ1-42. PLoS ONE 2015, 10, e0122415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capsoni, S.; Marinelli, S.; Ceci, M.; Vignone, D.; Amato, G.; Malerba, F.; Paoletti, F.; Meli, G.; Viegi, A.; Pavone, F.; et al. Intranasal “Painless” Human Nerve Growth Factors Slows Amyloid Neurodegeneration and Prevents Memory Deficits in App × PS1 Mice. PLoS ONE 2012, 7, e37555. [Google Scholar] [CrossRef]
- Rissman, R.A.; Siffert, J.; Aisen, P.S.; Team, A.S. Adeno-Associated Viral Vector (Serotype 2)—Nerve Growth Factor for Patients with Alzheimer Disease A Randomized Clinical Trial. JAMA Neurol. 2018, 75, 834–841. [Google Scholar]
- Devi, L.; Ohno, M. 7,8-Dihydroxyflavone, a Small-Molecule TrkB Agonist, Reverses Memory Deficits and BACE1 Elevation in a Mouse Model of Alzheimer’s Disease. Neuropsychopharmacology 2012, 37, 434–444. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.W.; Liu, X.; Chan, C.B.; France, S.A.; Sayeed, I.; Tang, W.; Lin, X.; Xiao, G.; Andero, R.; Chang, Q.; et al. Deoxygedunin, a Natural Product with Potent Neurotrophic Activity in Mice. PLoS ONE 2010, 5, e11528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massa, S.M.; Yang, T.; Xie, Y.; Shi, J.; Bilgen, M.; Joyce, J.N.; Nehama, D.; Rajadas, J.; Longo, F.M. Small Molecule BDNF Mimetics Activate TrkB Signaling and Prevent Neuronal Degeneration in Rodents. J. Clin. Investig. 2010, 120, 1774–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fletcher, J.M.; Morton, C.J.; Zwar, R.A.; Murray, S.S.; O’Leary, P.D.; Hughes, R.A. Design of a Conformationally Defined and Proteolytically Stable Circular Mimetic of Brain-Derived Neurotrophic Factor. J. Biol. Chem. 2008, 283, 33375–33383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrich, C.; Blum, R.; Gascón, S.; Masserdotti, G.; Tripathi, P.; Sánchez, R.; Tiedt, S.; Schroeder, T.; Götz, M.; Berninger, B. Directing Astroglia from the Cerebral Cortex into Subtype Specific Functional Neurons. PLoS Biol. 2010, 8, e1000373. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.H.; Li, W.; Zheng, J.J.; Xu, Y.G.; He, Q.; Chen, G. Differential Neuronal Reprogramming Induced by NeuroD1 from Astrocytes in Grey Matter versus White Matter. Neural Regen. Res. 2020, 15, 342–351. [Google Scholar] [CrossRef]
- Zhang, L.; Yin, J.C.; Yeh, H.; Ma, N.X.; Lee, G.; Chen, X.A.; Wang, Y.; Lin, L.; Chen, L.; Jin, P.; et al. Small Molecules Efficiently Reprogram Human Astroglial Cells into Functional Neurons. Cell Stem Cell 2015, 17, 735–747. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.C.; Zhang, L.; Ma, N.X.; Wang, Y.; Lee, G.; Hou, X.Y.; Lei, Z.F.; Zhang, F.Y.; Dong, F.P.; Wu, G.Y.; et al. Chemical Conversion of Human Fetal Astrocytes into Neurons through Modulation of Multiple Signaling Pathways. Stem Cell Rep. 2019, 12, 488–501. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Lim, I.J.; Park, S.W.; Kim, Y.B.; Ko, Y.; Kim, S.U. Human Neural Stem Cells Genetically Modified to Express Human Nerve Growth Factor (NGF) Gene Restore Cognition in the Mouse With Ibotenic Acid-Induced Cognitive Dysfunction. Cell Transplant. 2012, 21, 2487–2496. [Google Scholar] [CrossRef]
- Wu, C.C.; Lien, C.C.; Hou, W.H.; Chiang, P.M.; Tsai, K.J. Gain of BDNF Function in Engrafted Neural Stem Cells Promotes the Therapeutic Potential for Alzheimer’s Disease. Sci. Rep. 2016, 6, 1–16. [Google Scholar] [CrossRef]
- Li, T.; Yu, Y.; Cai, H. Effects of Brain-Derived Neurotrophic Factor-Pretreated Neuron Stem Cell Transplantation on Alzheimer’s Disease Model Mice. Int. J. Clin. Exp. Med. 2015, 8, 21947–21955. [Google Scholar]
- Alfred, A. Human Cortical Neural Stem Cells Expressing Insulin-Like Growth Factor-I: A Novel Cellular Therapy for Alzheimer’s Disease. Stem Cells Transl. Med. 2016, 5, 379–391. [Google Scholar]
- Garcia, K.O.; Ornellas, F.L.M.; Martin, P.K.M.; Patti, C.L.; Mello, L.E.; Frussa-filho, R.; Han, S.W.; Longo, B.M. Therapeutic Effects of the Transplantation of VEGF Overexpressing Bone Marrow Mesenchymal Stem Cells in the Hippocampus of Murine Model of Alzheimer’ s Disease. Front. Aging Neurosci. 2014, 6, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klinge, P.M.; Harmening, K.; Miller, M.C.; Heile, A.; Wallrapp, C.; Geigle, P.; Brinker, T. Encapsulated Native and Glucagon-like Peptide-1 Transfected Human Mesenchymal Stem Cells in a Transgenic Mouse Model of Alzheimer’s Disease. Neurosci. Lett. 2011, 497, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Blurton-Jones, M.; Spencer, B.; Michael, S.; Castello, N.A.; Agazaryan, A.A.; Davis, J.L.; Müller, F.; Loring, J.F.; Masliah, E.; LaFerla, F.M. Neural Stem Cells Genetically-Modified to Express Neprilysin Reduce Pathology in Alzheimer Transgenic Models. Stem Cell Res. Ther. 2014, 5, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Fu, Z.; Meng, L.; He, M.; Zhang, Z. The Early Events That Initiate β-Amyloid Aggregation in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 359. [Google Scholar] [CrossRef] [PubMed]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular Amyloid-β in Alzheimer’s Disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef]
- Zhang, C.; Tanzi, R.E. Natural Modulators of Amyloid-Beta Precursor Protein Processing. Curr. Alzheimer Res. 2012, in press. [Google Scholar]
- O’Brein, R.J.; Wong, P.C. Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar]
- Chen, G.-F.; Xu, T.-H.; Yan, Y.; Zhou, Y.-R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid Beta: Structure, Biology and Structure-Based Therapeutic Development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Citron, M.; Diehl, T.S.; Gordon, G.; Biere, A.L.; Seubert, P.; Selkoe, D.J. Evidence That the 42- and 40-Amino Acid Forms of Amyloid β Protein Are Generated from the β-Amyloid Precursor Protein by Different Protease Activities. Proc. Natl. Acad. Sci. USA 1996, 93, 13170–13175. [Google Scholar] [CrossRef] [Green Version]
- Querfurth, H.W.; LaFerla, F.M. Mechanisms of Disease Alzheimer’s Disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; McInnes, J.; Wierda, K.; Holt, M.; Herrmann, A.G.; Jackson, R.J.; Wang, Y.C.; Swerts, J.; Beyens, J.; Miskiewicz, K.; et al. Tau Association with Synaptic Vesicles Causes Presynaptic Dysfunction. Nat. Commun. 2017, 8, 15295. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Yepes, J.; Burns, M.; Anandhan, A.; Khalimonchuk, O.; del Razo, L.M.; Quintanilla-Vega, B.; Pappa, A.; Panayiotidis, M.I.; Franco, R. Oxidative Stress, Redox Signaling, and Autophagy: Cell Death versus Survival. Antioxid. Redox Signal. 2014, 21, 66–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattson, M.P. Pathways towards and Away from Alzheimer’s Disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef] [Green Version]
- Haroon, E.; Miller, A.H.; Sanacora, G. Inflammation, Glutamate, and Glia: A Trio of Trouble in Mood Disorders. Neuropsychopharmacology 2017, 42, 193–215. [Google Scholar] [CrossRef]
- Evans, L.D.; Wassmer, T.; Fraser, G.; Smith, J.; Perkinton, M.; Billinton, A.; Livesey, F.J. Extracellular Monomeric and Aggregated Tau Efficiently Enter Human Neurons through Overlapping but Distinct Pathways. Cell Rep. 2018, 22, 3612–3624. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Dickson, D.W.; Trojanowski, J.Q.; Lee, V.M.Y. The Levels of Soluble versus Insoluble Brain Aβ Distinguish Alzheimer’s Disease from Normal and Pathologic Aging. Exp. Neurol. 1999, 158, 328–337. [Google Scholar] [CrossRef]
- Weggen, S.; Beher, D. Molecular Consequences of Amyloid Precursor Protein and Presenilin Mutations Causing Autosomal-Dominant Alzheimer’s Disease. Alzheimer’s Res. Ther. 2012, 4, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, A.; Matsumura, S.; Dezawa, M.; Tada, M.; Yanazawa, M.; Ito, A.; Akioka, M.; Kikuchi, S.; Sato, M.; Noda, M.; et al. Isolation and Characterization of Patient-Derived, Toxic, High Mass Amyloid β-Protein (Aβ) Assembly from Alzheimer Disease Brains. J. Biol. Chem. 2009, 284, 32895–32905. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.Y.; Maruff, P.; Pietrzak, R.H.; Ellis, K.A.; Darby, D.; Ames, D.; Harrington, K.; Martins, R.N.; Masters, C.L.; Szoeke, C.; et al. Aβ and Cognitive Change: Examining the Preclinical and Prodromal Stages of Alzheimer’s Disease. Alzheimer’s Dement. 2014, 10, 743–751.e1. [Google Scholar] [CrossRef]
- Klementieva, O.; Willén, K.; Martinsson, I.; Israelsson, B.; Engdahl, A.; Cladera, J.; Uvdal, P.; Gouras, G.K. Pre-Plaque Conformational Changes in Alzheimer’s Disease-Linked Aβ and APP. Nat. Commun. 2017, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Cao, L.; Liu, L.; Zhang, C.; Kalionis, B.; Tai, X.; Li, Y.; Xia, S. Aβ1-42 Oligomer-Induced Leakage in an in Vitro Blood-Brain Barrier Model Is Associated with up-Regulation of RAGE and Metalloproteinases, and down-Regulation of Tight Junction Scaffold Proteins. J. Neurochem. 2015, 134, 382–393. [Google Scholar] [CrossRef] [PubMed]
- Elvis, C.; Hector, R.; Susan, M.B.; Manue, A.R.; Aida, G.; Syed, Z.; Syed, F.A.; Sumit, S. Amyloid Beta 25–35 Induces Blood-Brain Barrier Disruption in Vitro. Metab. Brain Dis. 2019, 34, 1365–1374. [Google Scholar]
- Querol-Vilaseca, M.; Colom-Cadena, M.; Pegueroles, J.; Nuñez-Llaves, R.; Luque-Cabecerans, J.; Muñoz-Llahuna, L.; Andilla, J.; Belbin, O.; Spires-Jones, T.L.; Gelpi, E.; et al. Nanoscale Structure of Amyloid-β Plaques in Alzheimer’s Disease. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Pickett, E.K.; Koffie, R.M.; Wegmann, S.; Henstridge, C.M.; Herrmann, A.G.; Colom-Cadena, M.; Lleo, A.; Kay, K.R.; Vaught, M.; Soberman, R.; et al. Non-Fibrillar Oligomeric Amyloid-β within Synapses. J. Alzheimer’s Dis. 2016, 53, 787–800. [Google Scholar] [CrossRef] [Green Version]
- Pickett, E.K.; Herrmann, A.G.; McQueen, J.; Abt, K.; Dando, O.; Tulloch, J.; Jain, P.; Dunnett, S.; Sohrabi, S.; Fjeldstad, M.P.; et al. Amyloid Beta and Tau Cooperate to Cause Reversible Behavioral and Transcriptional Deficits in a Model of Alzheimer’s Disease. Cell Rep. 2019, 29, 3592–3604.e5. [Google Scholar] [CrossRef] [Green Version]
- Hur, J.Y.; Frost, G.R.; Wu, X.; Crump, C.; Pan, S.J.; Wong, E.; Barros, M.; Li, T.; Nie, P.; Zhai, Y.; et al. The Innate Immunity Protein IFITM3 Modulates γ-Secretase in Alzheimer’s Disease. Nature 2020, 586, 735–740. [Google Scholar] [CrossRef]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef] [Green Version]
- Tai, L.M.; Bilousova, T.; Jungbauer, L.; Roeske, S.K.; Youmans, K.L.; Yu, C.; Poon, W.W.; Cornwell, L.B.; Miller, C.A.; Vinters, H.V.; et al. Levels of Soluble Apolipoprotein E/Amyloid-β (Aβ) Complex Are Reduced and Oligomeric Aβ Increased with APOE4 and Alzheimer Disease in a Transgenic Mouse Model and Human Samples. J. Biol. Chem. 2013, 288, 5914–5926. [Google Scholar] [CrossRef] [Green Version]
- Alzheimer’s Association. 2017 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2017, 13, 325–373. [Google Scholar] [CrossRef]
- Holland, D.; Desikan, R.S.; Dale, A.M.; McEvoy, L.K. Rates of Decline in Alzheimer Disease Decrease with Age. PLoS ONE 2012, 7, e42325. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.-L.; Wang, N.; Sun, F.-R.; Cao, X.-P.; Zhang, W.; Yu, J.-T. Tau in Neurodegenerative Disease. Ann. Transl. Med. 2018, 6, 175. [Google Scholar] [CrossRef]
- Alonso, A.D.C.; Zaidi, T.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Hyperphosphorylation Induces Self-Assembly of τ into Tangles of Paired Helical Filaments/Straight Filaments. Proc. Natl. Acad. Sci. USA 2001, 98, 6923–6928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, S.J.; de Ture, M.A.; McBride, M.; Dickson, D.W.; Petrucelli, L. Three Repeat Isoforms of Tau Inhibit Assembly of Four Repeat Tau Filaments. PLoS ONE 2010, 5, e10810. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Martinez-Vicente, M.; Krüger, U.; Kaushik, S.; Wong, E.; Mandelkow, E.M.; Cuervo, A.M.; Mandelkow, E. Tau Fragmentation, Aggregation and Clearance: The Dual Role of Lysosomal Processing. Hum. Mol. Genet. 2009, 18, 4153–4170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Lu, F.; Wang, J.Z.; Gong, C.X. Concurrent Alterations of O-GlcNAcylation and Phosphorylation of Tau in Mouse Brains during Fasting. Eur. J. Neurosci. 2006, 23, 2078–2086. [Google Scholar] [CrossRef]
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of Tau Misfolding from the Outside to the inside of a Cell. J. Biol. Chem. 2009, 284, 12845–12852. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Tseng, I.C.; Heyser, C.J.; Rockenstein, E.; Mante, M.; Adame, A.; Zheng, Q.; Huang, T.; Wang, X.; Arslan, P.E.; et al. Appoptosin-Mediated Caspase Cleavage of Tau Contributes to Progressive Supranuclear Palsy Pathogenesis. Neuron 2015, 87, 963–975. [Google Scholar] [CrossRef] [Green Version]
- Bardai, F.H.; Wang, L.; Mutreja, Y.; Yenjerla, M.; Gamblin, T.C.; Feany, M.B. A Conserved Cytoskeletal Signaling Cascade Mediates Neurotoxicity of FTDP-17 Tau Mutations in Vivo. J. Neurosci. 2018, 38, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; del Tredici, K.; et al. Correlation of Alzheimer Disease Neuropathologic Changes with Cognitive Status: A Review of the Literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef]
- David, A.B.; Julie, A.S.; Robert, S.W.; Julia, L.B.; Steven, E.A. Neurofibrillary Tangles Mediate the Association of Amyloid Load with Clinical Alzheimer Disease and Level of Cognitive Function. Arch. Neurol. 2004, 61, 378–384. [Google Scholar]
- Braak, H.; Braak, E. Neuropathological Stageing of Alzheimer-Related Changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Malpetti, M.; Kievit, R.A.; Passamonti, L.; Jones, P.S.; Tsvetanov, K.A.; Rittman, T.; Mak, E.; Nicastro, N.; Bevan-Jones, W.R.; Su, L.; et al. Microglial Activation and Tau Burden Predict Cognitive Decline in Alzheimer’s Disease. Brain 2020, 143, 1588–1602. [Google Scholar] [CrossRef]
- Simoes, S.; Neufeld, J.L.; Triana-Baltzer, G.; Moughadam, S.; Chen, E.I.; Kothiya, M.; Qureshi, Y.H.; Patel, V.; Honig, L.S.; Kolb, H.; et al. Tau and Other Protein Found in Alzheimer’s Disease Spinal Fluid Are Linked to Retromer-mediated Endosomal Traffic in Mice and Humans. Sci. Trans. Med. 2020, 12, eaba6334. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Cha, M.; Lee, B.H. Neuroprotective Effect of Antioxidants in the Brain. Int. J. Mol. Sci. 2020, 21, 7152. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Guo, C.; Kong, J. Oxidative Stress in Neurodegenerative Diseases. Neural Regen. Res. 2012, 7, 376–385. [Google Scholar] [PubMed]
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of Oxidative Stress in Alzheimer’s Disease (Review). Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef] [Green Version]
- Sonnen, J.A.; Breitner, J.C.; Lovell, M.A.; Markesbery, W.R.; Quinn, J.F.; Montine, T.J. Free Radical-Mediated Damage to Brain in Alzheimer’s Disease and Its Transgenic Mouse Models. Free Radic. Biol. Med. 2008, 45, 219–230. [Google Scholar] [CrossRef] [Green Version]
- Markesbery, W.R. Oxidative Stress Hypothesis in Alzheimer’s Disease. Free Radic. Biol. Med. 1997, 23, 134–147. [Google Scholar] [CrossRef]
- Chun, H.; Im, H.; Kang, Y.J.; Kim, Y.; Shin, J.H.; Won, W.; Lim, J.; Ju, Y.; Park, Y.M.; Kim, S.; et al. Severe Reactive Astrocytes Precipitate Pathological Hallmarks of Alzheimer’s Disease via H2O2− Production. Nat. Neurosci. 2020, 23, 1555–1566. [Google Scholar] [CrossRef]
- Mastroeni, D.; Khdour, O.M.; Delvaux, E.; Nolz, J.; Olsen, G.; Berchtold, N.; Cotman, C.; Hecht, S.M.; Coleman, P.D. Nuclear but Not Mitochondrial-Encoded Oxidative Phosphorylation Genes Are Altered in Aging, Mild Cognitive Impairment, and Alzheimer’s Disease. Alzheimer’s Dement. 2017, 13, 510–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldeiras, I.; Santana, I.; Proença, M.T.; Garrucho, M.H.; Pascoal, R.; Rodrigues, A.; Duro, D.; Oliveira, C.R. Peripheral Oxidative Damage in Mild Cognitive Impairment and Mild Alzheimer’s Disease. J. Alzheimer’s Dis. 2008, 15, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Youssef, P.; Chami, B.; Lim, J.; Middleton, T.; Sutherland, G.T.; Witting, P.K. Evidence Supporting Oxidative Stress in a Moderately Affected Area of the Brain in Alzheimer’s Disease. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Craddock, T.J.A.; Tuszynski, J.A.; Chopra, D.; Casey, N.; Goldstein, L.E.; Hameroff, S.R.; Tanzi, R.E. The Zinc Dyshomeostasis Hypothesis of Alzheimer’s Disease. PLoS ONE 2012, 7, e33552. [Google Scholar] [CrossRef]
- Darshpreet, K.; Vivek, S.; Rahul, D. Activation of Microglia and Astrocytes: A Roadway to Neuroinflammation and Alzheimer’s Disease. Inflammopharmacology 2019, 27, 663–677. [Google Scholar]
- Galloway, D.A.; Phillips, A.E.M.; Owen, D.R.J.; Moore, C.S. Phagocytosis in the Brain: Homeostasis and Disease. Front. Immunol. 2019, 10, 790. [Google Scholar] [CrossRef] [Green Version]
- Weldon, D.T.; Rogers, S.D.; Ghilardi, J.R.; Finke, M.P.; Cleary, J.P.; O’Hare, E.; Esler, W.P.; Maggio, J.E.; Mantyh, P.W. Fibrillar β-Amyloid Induces Microglial Phagocytosis, Expression of Inducible Nitric Oxide Synthase, and Loss of a Select Population of Neurons in the Rat CNS in Vivo. J. Neurosci. 1998, 18, 2161–2173. [Google Scholar] [CrossRef] [Green Version]
- Amor, S.; Puentes, F.; Baker, D.; Van Der Valk, P. Inflammation in Neurodegenerative Diseases. Immunology 2010, 129, 154–169. [Google Scholar] [CrossRef]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s Disease. J. Clin. Invest. 2017, 127, 3240–3249. [Google Scholar] [CrossRef]
- Koenigsknecht, J.; Landreth, G. Microglial Phagocytosis of Fibrillar β-Amyloid through a Β1 Integrin-Dependent Mechanism. J. Neurosci. 2004, 24, 9838–9846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheppard, O.; Coleman, M.P.; Durrant, C.S. Lipopolysaccharide-Induced Neuroinflammation Induces Presynaptic Disruption through a Direct Action on Brain Tissue Involving Microglia-Derived Interleukin 1 Beta. J. Neuroinflamm. 2019, 16, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, J.G.; Mrak, R.E.; Griffin, W.S.T. Glial-Neuronal Interactions in Alzheimer Disease: Progressive Association of IL−1α+ Microglia and S100β+ Astrocytes with Neurofibrillary Tangle Stages. J. Neuropathol. Exp. Neurol. 1997, 56, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of Pro-Inflammatory Cytokines Released from Microglia in Alzheimer’s Disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar]
- Brosseron, F.; Krauthausen, M.; Kummer, M.; Heneka, M.T. Body Fluid Cytokine Levels in Mild Cognitive Impairment and Alzheimer’s Disease: A Comparative Overview. Mol. Neurobiol. 2014, 50, 534–544. [Google Scholar] [CrossRef] [Green Version]
- Wood, H. Alzheimer Disease: Twin Peaks of Microglial Activation Observed in Alzheimer Disease. Nat. Rev. Neurol. 2017, 13, 129. [Google Scholar]
- Sanchez, J.R.; Marsh, S.; McIntyre, L.; Davtyan, H.; Walsh, C.; Blurton-Jones, M. Cytotoxic T Cells Infiltrate the Brain and Interact with Microglia to Reduce Alzheimer’s Disease Pathogenesis. J. Immunol. 2020, 204, 64.4. [Google Scholar]
- Fisher, Y.; Nemirovsky, A.; Baron, R.; Monsonego, A. T Cells Specifically Targeted to Amyloid Plaques Enhance Plaque Clearance in a Mouse Model of Alzheimer’s Disease. PLoS ONE 2010, 5, e10830. [Google Scholar] [CrossRef] [Green Version]
- Togo, T.; Akiyama, H.; Iseki, E.; Kondo, H.; Ikeda, K.; Kato, M.; Oda, T.; Tsuchiya, K.; Kosaka, K. Occurrence of T Cells in the Brain of Alzheimer’s Disease and Other Neurological Diseases. J. Neuroimmunol. 2002, 124, 83–92. [Google Scholar] [CrossRef]
- Bulati, M.; Martorana, A.; Gervasi, F.; Camarda, C.; Azzarello, D.M.; Monastero, R.; Caruso, C.; Colonna-Romano, G. Double Negative (IgG+IgD−CD27−) B Cells Are Increased in a Cohort of Moderate-Severe Alzheimer’s Disease Patients and Show a pro-Inflammatory Trafficking Receptor Phenotype. J. Alzheimer’s Dis. 2015, 44, 1241–1251. [Google Scholar] [CrossRef]
- Söllvander, S.; Ekholm-Pettersson, F.; Brundin, R.M.; Westman, G.; Kilander, L.; Paulie, S.; Lannfelt, L.; Sehlin, D. Increased Number of Plasma B Cells Producing Autoantibodies against Aβ 42 Protofibrils in Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 48, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Dionisio-Santos, D.A.; Olschowka, J.A.; O’Banion, M.K. Exploiting Microglial and Peripheral Immune Cell Crosstalk to Treat Alzheimer’s Disease. J. Neuroinflamm. 2019, 16, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panza, F.; Lozupone, M.; Solfrizzi, V.; Sardone, R.; Piccininni, C.; Dibello, V.; Stallone, R.; Giannelli, G.; Bellomo, A.; Greco, A.; et al. BACE Inhibitors in Clinical Development for the Treatment of Alzheimer’s Disease. Expert Rev. Neurother. 2018, 18, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.E.; Stamford, A.W.; Chen, X.; Cox, K.; Cumming, J.N.; Dockendorf, M.F.; Egan, M.; Ereshefsky, L.; Hodgson, R.A.; Hyde, L.A.; et al. The BACE1 Inhibitor Verubecestat (MK-8931) Reduces CNS b-Amyloid in Animal Models and in Alzheimer’s Disease Patients. Sci. Transl. Med. 2016, 8, 363ra150. [Google Scholar] [CrossRef] [PubMed]
- Cebers, G.; Lejeune, T.; Attalla, B.; Soderberg, M.; Alexander, R.C.; Haeberlein, S.B.; Kugler, A.R.; Ingersoll, E.W.; Platz, S.; Scott, C.W. Reversible and Species-Specific Depigmentation Effects of AZD3293, a BACE Inhibitor for the Treatment of Alzheimer’s Disease, Are Related to BACE2 Inhibition and Confined to Epidermis and Hair. J. Prev. Alzheimer’s Dis. 2016, 3, 202–218. [Google Scholar]
- US National Library of Medicine. A 24-Month Study to Evaluate the Efficacy and Safety of Elenbecestat (E2609) in Subjects with Early Alzheimer’s Disease (MissionAD1). 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT03036280 (accessed on 28 February 2020).
- Janssen Research and Development. An Efficacy and Safety Study of Atabecestat in Participants Who Are Asymptomatic at Risk for Developing Alzheimer’s Dementia. Available online: https://clinicaltrials.gov/ct2/show/NCT02569398 (accessed on 20 February 2020).
- Novartis Pharmaceuticals; Banner Alzheimer’s Institute. A Study of CNP520 Versus Placebo in Participants at Risk for the Onset of Clinical Symptoms of Alzheimer’s Disease. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT03131453 (accessed on 19 August 2020).
- Vladimir, C.; Stephen, S.; Christopher, H.V.D.; Bruno, D.; Niels, A.; Mark, B.; Craig, C.; Hilkka, S.; Stephen, T.; Thomas, S.; et al. Targeting Prodromal Alzheimer Disease with Avagacestat: A Randomized Clinical Trial. JAMA Neurol. 2015, 72, 1324–1333. [Google Scholar]
- Tanokashira, D.; Mamada, N.; Yamamoto, F.; Taniguchi, K.; Tamaoka, A.; Lakshmana, M.K.; Araki, W. The Neurotoxicity of Amyloid β-Protein Oligomers Is Reversible in a Primary Neuron Model. Mol. Brain 2017, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Montoliu-Gaya, L.; Villegas, S. Aβ-Immunotherapeutic Strategies: A Wide Range of Approaches for Alzheimer’s Disease Treatment. Expert Rev. Mol. Med. 2016, 18, e13. [Google Scholar] [CrossRef]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The Antibody Aducanumab Reduces Aβ Plaques in Alzheimer’s Disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Loeffler, D.A. Intravenous Immunoglobulin and Alzheimer’s Disease: What Now? J. Neuroinflamm. 2013, 10, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Price, K.A.; Crouch, P.J.; White, A.R. Therapeutic Treatment of Alzheimer’s Disease Using Metal Complexing Agents. Recent Pat. CNS Drug Discov. 2007, 2, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, C.W.; Bush, A.I.; Mackinnon, A.; Macfarlane, S.; Mastwyk, M.; MacGregor, L.; Kiers, L.; Cherny, R.; Li, Q.X.; Tammer, A.; et al. Metal-Protein Attenuation with Iodochlorhydroxyquin (Clioquinol) Targeting Aβ Amyloid Deposition and Toxicity in Alzheimer Disease: A Pilot Phase 2 Clinical Trial. Arch. Neurol. 2003, 60, 1685–1691. [Google Scholar] [CrossRef]
- Arbiser, J.L.; Kraeft, S.K.; Van Leeuwen, R.; Hurwitz, S.J.; Selig, M.; Dickersin, G.R.; Flint, A.; Byers, H.R.; Chen, L.B. Clioquinol-Zinc Chelate: A Candidate Causative Agent of Subacute Myelo-Optic Neuropathy. Mol. Med. 1998, 4, 665–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makin, S. The Amyloid Hypothesis on Trial. Nature 2018, 559, S4–S7. [Google Scholar] [CrossRef] [Green Version]
- Lansdall, C.J. An Effective Treatment for Alzheimer’s Disease Must Consider Both Amyloid and Tau. Biosci. Horiz. 2014, 7, hzu002. [Google Scholar] [CrossRef] [Green Version]
- Panza, F.; Lozupone, M.; Logroscino, G.; Imbimbo, B.P. A Critical Appraisal of Amyloid-β-Targeting Therapies for Alzheimer Disease. Nat. Rev. Neurol. 2019, 15, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Piette, F.; Belmin, J.; Vincent, H.; Schmidt, N.; Pariel, S.; Verny, M.; Marquis, C.; Mely, J.; Hugonot-Diener, L.; Kinet, J.P.; et al. Masitinib as an Adjunct Therapy for Mild-to-Moderate Alzheimer’s Disease: A Randomised, Placebo-Controlled Phase 2 Trial. Alzheimer’s Res. Ther. 2011, 3, 16. [Google Scholar] [CrossRef]
- Congdon, E.E.; Sigurdsson, E.M. Tau-Targeting Therapies for Alzheimer Disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef]
- Augustinack, J.C.; Sanders, J.L.; Tsai, L.H.; Hyman, B.T. Colocalization and Fluorescence Resonance Energy Transfer between Cdk5 and AT8 Suggests a Close Association in Pre-Neurofibrillary Tangles and Neurofibrillary Tangles. J. Neuropathol. Exp. Neurol. 2002, 61, 557–564. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, A.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Regulation of Phosphorylation of Tau by Cyclin-Dependent Kinase 5 and Glycogen Synthase Kinase-3 at Substrate Level. FEBS Lett. 2006, 580, 5925–5933. [Google Scholar] [CrossRef] [Green Version]
- Caccamo, A.; Oddo, S.; Tran, L.X.; LaFerla, F.M. Lithium Reduces Tau Phosphorylation but Not Aβ or Working Memory Deficits in a Transgenic Model with Both Plaques and Tangles. Am. J. Pathol. 2007, 170, 1669–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.K.; Kim, N.J. Recent Advances in the Inhibition of P38 MAPK as a Potential Strategy for the Treatment of Alzheimer’s Disease. Molecules 2017, 22, 1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadikar, H.; Torres, I.; Aiello, G.; Kurup, M.; Yang, Z.; Lin, F.; Kobeissy, F.; Yost, R.; Wang, K. Screening of Tau Protein Kinase Inhibitors in a Tauopathy-Relevant Cell-Based Model of Tau Hyperphosphorylation and Oligomerization. PLoS ONE 2020, 15, e0224952. [Google Scholar] [CrossRef] [PubMed]
- Sontag, J.M.; Sontag, E. Protein Phosphatase 2A Dysfunction in Alzheimer’s Disease. Front. Mol. Neurosci. 2014, 7, 16. [Google Scholar] [CrossRef]
- Millward, T.A.; Zolnierowicz, S.; Hemmings, B.A. Regulation of Protein Kinase Cascades by Protein Phosphatase 2A. Trends Biochem. Sci. 1999, 24, 186–191. [Google Scholar] [CrossRef]
- Gong, C.-X.; Shaikh, S.; Wang, J.-Z.; Zaidi, T.; Grundke-Iqbal, I.; Iqbal, K. Phosphatase Activity Toward Abnormally Phosphorylated τ: Decrease in Alzheimer Disease Brain. J. Neurochem. 1995, 65, 732–738. [Google Scholar] [CrossRef]
- Braithwaite, S.P.; Stock, J.B.; Lombroso, P.J.; Nairn, A.C. Protein Phosphatases and Alzheimer’s Disease. Prog. Mol. Biol. Transl. Sci. 2012, 106, 343–379. [Google Scholar]
- Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Contributions of Protein Phosphatases PP1, PP2A, PP2B and PP5 to the Regulation of Tau Phosphorylation. Eur. J. Neurosci. 2005, 22, 1942–1950. [Google Scholar] [CrossRef]
- McKenzie-Nickson, S.; Chan, J.; Perez, K.; Hung, L.W.; Cheng, L.; Sedjahtera, A.; Gunawan, L.; Adlard, P.A.; Hayne, D.J.; McInnes, L.E.; et al. Modulating Protein Phosphatase 2A Rescues Disease Phenotype in Neurodegenerative Tauopathies. ACS Chem. Neurosci. 2018, 9, 2731–2740. [Google Scholar] [CrossRef]
- Morita, K.; He, S.; Nowak, R.P.; Wang, J.; Zimmerman, M.W.; Fu, C.; Durbin, A.D.; Martel, M.W.; Prutsch, N.; Gray, N.S.; et al. Allosteric Activators of Protein Phosphatase 2A Display Broad Antitumor Activity Mediated by Dephosphorylation of MYBL2. Cell 2020, 181, 702–715.e20. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F.; Gong, C.-X.; Grundke-Iqbal, I. Tau in Alzheimer Disease and Related Tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef] [Green Version]
- Khanna, M.R.; Kovalevich, J.; Lee, V.M.Y.; Trojanowski, J.Q.; Brunden, K.R. Therapeutic Strategies for the Treatment of Tauopathies: Hopes and Challenges. Alzheimer’s Dement. 2016, 12, 1051–1065. [Google Scholar] [CrossRef] [Green Version]
- Bulic, B.; Pickhardt, M.; Schmidt, B.; Mandelkow, E.M.; Waldmann, H.; Mandelkow, E. Development of Tau Aggregation Inhibitors for Alzheimer’s Disease. Angew. Chem. 2009, 48, 1740–1752. [Google Scholar] [CrossRef]
- Akoury, E.; Pickhardt, M.; Gajda, M.; Biernat, J.; Mandelkow, E.; Zweckstetter, M. Mechanistic Basis of Phenothiazine-Driven Inhibition of Tau Aggregation. Angew. Chem. 2013, 52, 3511–3515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jadhav, S.; Avila, J.; Schöll, M.; Kovacs, G.G.; Kövari, E.; Skrabana, R.; Evans, L.D.; Kontsekova, E.; Malawska, B.; de Silva, R.; et al. A Walk through Tau Therapeutic Strategies. Acta Neuropathol. Commun. 2019, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Kebede, M.T.; Kemeh, M.M.; Islam, S.; Lee, B.; Bleck, S.D.; Wurfl, L.A.; Lazo, N.D. Inhibition of the Self-Assembly of Aβ and of Tau by Polyphenols: Mechanistic Studies. Molecules 2019, 24, 2316. [Google Scholar] [CrossRef] [Green Version]
- van Bebber, F.; Paquet, D.; Hruscha, A.; Schmid, B.; Haass, C. Methylene Blue Fails to Inhibit Tau and Polyglutamine Protein Dependent Toxicity in Zebrafish. Neurobiol. Dis. 2010, 39, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, C.; Praça, C.; Ferreira, R.; Santos, T.; Ferreira, L.; Bernardino, L. Nanoparticle-Mediated Brain Drug Delivery: Overcoming Blood-Brain Barrier to Treat Neurodegenerative Diseases. J. Control. Release 2016, 235, 34–47. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, S.; Suzuki, N.; Masuda, M.; Hisanaga, S.I.; Iwatsubo, T.; Goedert, M.; Hasegawa, M. Inhibition of Heparin-Induced Tau Filament Formation by Phenothiazines, Polyphenols, and Porphyrins. J. Biol. Chem. 2004, 280, 7614–7623. [Google Scholar] [CrossRef] [Green Version]
- Perez-Nievas, B.G.; Serrano-Pozo, A. Deciphering the Astrocyte Reaction in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 114. [Google Scholar] [CrossRef] [Green Version]
- Sriram, K.; Benkovic, S.A.; Hebert, M.A.; Miller, D.B.; O’Callaghan, J.P. Induction of Gp130-Related Cytokines and Activation of JAK2/STAT3 Pathway in Astrocytes Precedes up-Regulation of Glial Fibrillary Acidic Protein in the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Model of Neurodegeneration: Key Signaling Pathway for Ast. J. Biol. Chem. 2004, 279, 19936–19947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; He, G.; Hao, Y.; Chen, C.; Li, M.; Wang, Y.; Zhang, G.; Yu, Z. The Role of the JAK2-STAT3 Pathway in pro-Inflammatory Responses of EMF-Stimulated N9 Microglial Cells. J. Neuroinflamm. 2010, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceyzériat, K.; Ben Haim, L.; Denizot, A.; Pommier, D.; Matos, M.; Guillemaud, O.; Palomares, M.A.; Abjean, L.; Petit, F.; Gipchtein, P.; et al. Modulation of Astrocyte Reactivity Improves Functional Deficits in Mouse Models of Alzheimer’s Disease. Acta Neuropathol. Commun. 2018, 6, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichenbach, N.; Delekate, A.; Plescher, M.; Schmitt, F.; Krauss, S.; Blank, N.; Halle, A.; Petzold, G.C. Inhibition of Stat3-mediated Astrogliosis Ameliorates Pathology in an Alzheimer’s Disease Model. EMBO Mol. Med. 2019, 11, 1–16. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent Advances in the Mechanisms of NLRP3 Inflammasome Activation and Its Inhibitors. Cell Death Dis. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 Is Activated in Alzheimer’s Disease and Contributes to Pathology in APP/PS1 Mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Dugan, L.L.; Ali, S.S.; Shekhtman, G.; Roberts, A.J.; Lucero, J.; Quick, K.L.; Behrens, M.M. IL-6 Mediated Degeneration of Forebrain GABAergic Interneurons and Cognitive Impairment in Aged Mice through Activation of Neuronal NADPH Oxidase. PLoS ONE 2009, 4, e5518. [Google Scholar] [CrossRef]
- Valerio, A.; Boroni, F.; Benarese, M.; Sarnico, I.; Ghisi, V.; Bresciani, L.G.; Ferrario, M.; Borsani, G.; Spano, P.; Pizzi, M. NF-ΚB Pathway: A Target for Preventing β-Amyloid (Aβ)-Induced Neuronal Damage and Aβ42 Production. Eur. J. Neurosci. 2006, 23, 1711–1720. [Google Scholar] [CrossRef]
- Bales, K.R.; Du, Y.; Dodel, R.C.; Yan, G.M.; Hamilton-Byrd, E.; Paul, S.M. The NF-ΚB/Rel Family of Proteins Mediates A β-Induced Neurotoxicity and Glial Activation. Mol. Brain Res. 1998, 57, 63–72. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-ΚB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Shen, B.; Lin, A. Novel Strategies for Inhibition of the P38 MAPK Pathway. Trends Pharmacol. Sci. 2007, 28, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Coulthard, L.R.; White, D.E.; Jones, D.L.; McDermott, M.F.; Burchill, S.A. P38MAPK: Stress Responses from Molecular Mechanisms to Therapeutics. Trends Mol. Med. 2009, 15, 369–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kheiri, G.; Dolatshahi, M.; Rahmani, F.; Rezaei, N. Role of P38/MAPKs in Alzheimer’s Disease: Implications for Amyloid Beta Toxicity Targeted Therapy. Rev. Neurosci. 2018, 30, 9–30. [Google Scholar] [CrossRef] [PubMed]
- Gee, M.S.; Son, S.H.; Jeon, S.H.; Do, J.; Kim, N.; Ju, Y.J.; Lee, S.J.; Chung, E.K.; Inn, K.S.; Kim, N.J.; et al. A Selective P38α/β MAPK Inhibitor Alleviates Neuropathology and Cognitive Impairment, and Modulates Microglia Function in 5XFAD Mouse. Alzheimer’s Res. Ther. 2020, 12, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Y.; Du, Y.; Zhang, Y.; Huang, Z.; Fu, M.; Li, J.; Pang, Y.; Lei, P.; Wang, Y.T.; Song, W.; et al. MKP-1 Reduces Aβ Generation and Alleviates Cognitive Impairments in Alzheimer’s Disease Models. Signal Transduct. Target. Ther. 2019, 4, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sompol, P.; Norris, C.M. Ca2+, Astrocyte Activation and Calcineurin/NFAT Signaling in Age-Related Neurodegenerative Diseases. Front. Aging Neurosci. 2018, 10, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagamoto-Combs, K.; Combs, C.K. Microglial Phenotype Is Regulated by Activity of the Transcription Factor, NFAT (Nuclear Factor of Activated T Cells). J. Neurosci. 2010, 30, 9641–9646. [Google Scholar] [CrossRef] [Green Version]
- Furman, J.L.; Sama, D.M.; Gant, J.C.; Beckett, T.L.; Murphy, M.P.; Bachstetter, A.D.; van Eldik, L.J.; Norris, C.M. Targeting Astrocytes Ameliorates Neurologic Changes in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2012, 32, 16129–16140. [Google Scholar] [CrossRef] [Green Version]
- Taglialatela, G.; Rastellini, C.; Cicalese, L. Reduced Incidence of Dementia in Solid Organ Transplant Patients Treated with Calcineurin Inhibitors. J. Alzheimer’s Dis. 2015, 47, 329–333. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Wu, X.; Li, X.; Jiang, L.L.; Gui, X.; Liu, Y.; Sun, Y.; Zhu, B.; Piña-Crespo, J.C.; Zhang, M.; et al. TREM2 Is a Receptor for β-Amyloid That Mediates Microglial Function. Neuron 2018, 97, 1023–1031.e7. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Tan, L.; Zhu, X.C.; Zhou, J.S.; Cao, L.; Tan, M.S.; Wang, H.F.; Chen, Q.; Zhang, Y.D.; Yu, J.T. Silencing of TREM2 Exacerbates Tau Pathology, Neurodegenerative Changes, and Spatial Learning Deficits in P301S Tau Transgenic Mice. Neurobiol. Aging 2015, 36, 3176–3186. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Zhang, Y.D.; Chen, Q.; Gao, Q.; Zhu, X.C.; Zhou, J.S.; Shi, J.Q.; Lu, H.; Tan, L.; Yu, J.T. TREM2 Modifies Microglial Phenotype and Provides Neuroprotection in P301S Tau Transgenic Mice. Neuropharmacology 2016, 105, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Bisht, K.; Sharma, K.P.; Lecours, C.; Sánchez, M.G.; El Hajj, H.; Milior, G.; Olmos-Alonso, A.; Gómez-Nicola, D.; Luheshi, G.; Vallières, L.; et al. Dark Microglia: A New Phenotype Predominantly Associated with Pathological States. Glia 2016, 64, 826–839. [Google Scholar] [CrossRef] [Green Version]
- Fitz, N.F.; Wolfe, C.M.; Playso, B.E.; Biedrzycki, R.J.; Lu, Y.; Nam, K.N.; Lefterov, I.; Koldamova, R. Trem2 Deficiency Differentially Affects Phenotype and Transcriptome of Human APOE3 and APOE4 Mice. Mol. Neurodegener. 2020, 15, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Yeh, F.L.; Hansen, D.V.; Sheng, M. TREM2, Microglia, and Neurodegenerative Diseases. Trends Mol. Med. 2017, 23, 512–533. [Google Scholar] [CrossRef] [PubMed]
- Korvatska, O.; Leverenz, J.B.; Jayadev, S.; McMillan, P.; Kurtz, I.; Guo, X.; Rumbaugh, M.; Matsushita, M.; Girirajan, S.; Dorschner, M.O.; et al. R47H Variant of TREM2 Associated With Alzheimer Disease in a Large Late-Onset Family. JAMA Neurol. 2015, 72, 920. [Google Scholar] [CrossRef] [Green Version]
- Ren, S.; Yao, W.; Tambini, M.D.; Yin, T.; Norris, K.A.; D’Adamio, L. Microglia TREM2R47H Alzheimer-Linked Variant Enhances Excitatory Transmission and Reduces LTP via Increased TNF-α Levels. eLife 2020, 9, 1–18. [Google Scholar] [CrossRef]
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.K.; Younkin, S.; et al. TREM2 Variants in Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Tan, L.; Zhu, X.C.; Zhang, Q.Q.; Cao, L.; Tan, M.S.; Gu, L.Z.; Wang, H.F.; Ding, Z.Z.; Zhang, Y.D.; et al. Upregulation of TREM2 Ameliorates Neuropathology and Rescues Spatial Cognitive Impairment in a Transgenic Mouse Model of Alzheimer’s Disease. Neuropsychopharmacology 2014, 39, 2949–2962. [Google Scholar] [CrossRef] [Green Version]
- Zhong, L.; Xu, Y.; Zhuo, R.; Wang, T.; Wang, K.; Huang, R.; Wang, D.; Gao, Y.; Zhu, Y.; Sheng, X.; et al. Soluble TREM2 Ameliorates Pathological Phenotypes by Modulating Microglial Functions in an Alzheimer’s Disease Model. Nat. Commun. 2019, 10, 1–16. [Google Scholar]
- Lee, C.Y.D.; Daggett, A.; Gu, X.; Jiang, L.; Langfelder, P.; Li, X.; Wang, N.; Zhao, Y.; Park, C.S.; Cooper, Y.; et al. Elevated TREM2 Gene Dosage Reprograms Microglia Responsivity and Ameliorates Pathological Phenotypes in Alzheimer’s Disease Models. Neuron 2018, 97, 1032–1048.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kielian, T. Toll-like Receptors in Central Nervous System Glial Inflammation and Homeostasis. J. Neurosci. Res. 2006, 83, 711–730. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Zhao, S.; Zhu, Y.; Fan, Z.; Wang, J.; Zhang, B.; Chen, Y. Microbiota-Gut-Brain Axis and Toll-like Receptors in Alzheimer’s Disease. Comput. Struct. Biotechnol. J. 2019, 17, 1309–1317. [Google Scholar] [CrossRef]
- Chen, K.; Iribarren, P.; Hu, J.; Chen, J.; Gong, W.; Cho, E.H.; Lockett, S.; Dunlop, N.M.; Ji, M.W. Activation of Toll-like Receptor 2 on Microglia Promotes Cell Uptake of Alzheimer Disease-Associated Amyloid β Peptide. J. Biol. Chem. 2006, 281, 3651–3659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, C.L.; Hennessy, E.; Rubio-Araiz, A.; Keogh, B.; McCormack, W.; McGuirk, P.; Reilly, M.; Lynch, M.A. Inhibiting TLR2 Activation Attenuates Amyloid Accumulation and Glial Activation in a Mouse Model of Alzheimer’s Disease. Brain Behav. Immun. 2016, 58, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Kazuki, T.; Hong-Duck, K.; Jing-Ji, J.; Adam, M.J.; Ling, L.; Ken-Ichiro, F. Role of Toll-like Receptor Signalling in Aβ Uptake and Clearance. Brain 2008, 129, 3006–3019. [Google Scholar]
- Hughes, C.; Choi, M.L.; Yi, J.H.; Kim, S.C.; Drews, A.; George-Hyslop, P.S.; Bryant, C.; Gandhi, S.; Cho, K.; Klenerman, D. Beta Amyloid Aggregates Induce Sensitised TLR4 Signalling Causing Long-Term Potentiation Deficit and Rat Neuronal Cell Death. Commun. Biol. 2020, 3, 1–7. [Google Scholar] [CrossRef]
- Deane, R.J. Is RAGE Still a Therapeutic Target for Alzheimers Disease? Future Med. Chem. 2012, 4, 915–925. [Google Scholar] [CrossRef] [Green Version]
- Fang, F.; Lue, L.-F.; Yan, S.; Xu, H.; Luddy, J.S.; Chen, D.; Walker, D.G.; Stern, D.M.; Yan, S.; Schmidt, A.M.; et al. RAGE-dependent Signaling in Microglia Contributes to Neuroinflammation, Aβ Accumulation, and Impaired Learning/Memory in a Mouse Model of Alzheimer’s Disease. FASEB J. 2010, 24, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Baek, H.; Ye, M.; Kang, G.; Lee, C.; Lee, G.; Choi, B.; Jung, J.; Kim, H.; Lee, S.; Kim, J.S.; et al. Neuroprotective Effects of CD4+ CD25+ Foxp3+ Regulatory T Cells in a 3xTg-AD Alzheimer’s Disease Model. Oncotarget 2016, 7, 69347–69357. [Google Scholar] [CrossRef] [Green Version]
- Dansokho, C.; Ahmed, D.A.; Aid, S.; Toly-Ndour, C.; Chaigneau, T.; Calle, V.; Cagnard, N.; Holzenberger, M.; Piaggio, E.; Aucouturier, P.; et al. Regulatory T Cells Delay Disease Progression in Alzheimer-like Pathology. Brain 2016, 139, 1237–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelhardt, B. T Cell Migration into the Central Nervous System during Health and Disease: Different Molecular Keys Allow Access to Different Central Nervous System Compartments. Clin. Exp. Neuroimmunol. 2010, 1, 79–93. [Google Scholar] [CrossRef]
- Fu, H.J.; Liu, B.; Frost, J.L.; Lemere, C.A. Amyloid-β Immunotherapy for Alzheimers Disease. CNS Neurol. Disord. Drug Targets 2010, 9, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Monsonego, A.; Imitola, J.; Zota, V.; Oida, T.; Weiner, H.L. Microglia-Mediated Nitric Oxide Cytotoxicity of T Cells Following Amyloid β Peptide Presentation to Th1 Cells. J. Immunol. 2004, 172, 717. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Noh, M.Y.; Kim, H.-J.; Oh, K.-W.; Park, J.; Lee, S.; Moon, Y.; Kim, Y.-E.; Bae, J.S.; Jin, H.K. A Therapeutic Strategy for Alzheimer’s Disease Focused on Immune-Inflammatory Modulation. Dement. Neurocogn. Disord. 2019, 18, 33–46. [Google Scholar] [CrossRef]
- NIH. Safety and Efficacy Study of ALZT-OP1 in Subjects with Evidence of Early Alzheimer’s Disease. 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT02547818 (accessed on 24 November 2020).
- Van Eldik, L.J.; Carrillo, M.C.; Cole, P.E.; Feuerbach, D.; Greenberg, B.D.; Hendrix, J.A.; Kennedy, M.; Kozauer, N.; Margolin, R.A.; Molinuevo, J.L.; et al. The Roles of Inflammation and Immune Mechanisms in Alzheimers Disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2016, 2, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Allen, S.J.; Watson, J.J.; Dawbarn, D. The Neurotrophins and Their Role in Alzheimers Disease. Curr. Neuropharmacol. 2011, 9, 559–573. [Google Scholar] [CrossRef] [Green Version]
- Hampel, H.; Caraci, F.; Cuello, A.C.; Caruso, G.; Nisticò, R.; Corbo, M.; Baldacci, F.; Toschi, N.; Garaci, F.; Chiesa, P.A.; et al. A Path Toward Precision Medicine for Neuroinflammatory Mechanisms in Alzheimer’s Disease. Front. Immunol. 2020, 11, 456. [Google Scholar] [CrossRef]
- Shohayeb, B.; Diab, M.; Ahmed, M.; Ng, D.C.H. Factors That Influence Adult Neurogenesis as Potential Therapy. Transl. Neurodegener. 2018, 7, 1–19. [Google Scholar] [CrossRef]
- Weissmiller, A.M.; Wu, C. Current Advances in Using Neurotrophic Factors to Treat Neurodegenerative Disorders. Transl. Neurodegener. 2012, 1, 14. [Google Scholar] [CrossRef] [Green Version]
- Laske, C.; Stransky, E.; Leyhe, T.; Eschweiler, G.W.; Wittorf, A.; Richartz, E.; Bartels, M.; Buchkremer, G.; Schott, K. Stage-Dependent BDNF Serum Concentrations in Alzheimer’s Disease. J. Neural Transm. 2006, 113, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- Alicia, M.; Jordan, S.; Maria-Jose, M.; Reginald, M.H.; Janet, K.; Judi, M.W.; John, E.L. The Effect of Dietary Supplementation on Brain-Derived Neurotrophic Factor and Cognitive Functioning in Alzheimer’s Dementia. J. Clin. Transl. Res. 2018, 3, 337–343. [Google Scholar]
- Jönhagen, M.E.; Nordberg, A.; Amberla, K.; Bäckman, L.; Ebendal, T.; Meyerson, B.; Olson, L.; Seiger, Å.; Shigeta, M.; Theodorsson, E.; et al. Intracerebroventricular Infusion of Nerve Growth Factor in Three Patients with Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. 1998, 9, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Yaar, M.; Zhai, S.; Panova, I.; Fine, R.E.; Eisenhauer, P.B.; Blusztajn, J.K. A Cyclic Peptide That Binds P75NTR Protects Neurones from Beta Amyloid (1–40)-Induced Cell Death. Neuropathol. Appl. Neurobiol. 2007, 33, 533–543. [Google Scholar] [PubMed]
- Rafii, M.S.; Baumann, T.L.; Bakay, R.A.E.; Ostrove, J.M.; Siffert, J.; Fleisher, A.S.; Herzog, C.D.; Barba, D.; Pay, M.; Salmon, D.P.; et al. A Phase1 Study of Stereotactic Gene Delivery of AAV2-NGF for Alzheimer’s Disease. Alzheimer’s Dement. 2014, 10, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D. Peptide Mimetics of Neurotrophins and Their Receptors. Curr. Pharm. Des. 2011, 17, 2704–2718. [Google Scholar] [CrossRef]
- Liu, X.; Chan, C.; Jang, S.; Pradoldej, S.; Huang, J.; He, K.; Phun, L.H. A Synthetic 7,8-Dihydroxyflavone Derivative Promotes Neurogenesis and Exhibits Potent Antidepressant Effect. J. Med. Chem. 2010, 53, 8274–8286. [Google Scholar] [CrossRef] [Green Version]
- Augusto-Oliveira, M.; Arrifano, G.P.; Lopes-Araújo, A.; Santos-Sacramento, L.; Takeda, P.Y.; Anthony, D.C.; Malva, J.O.; Crespo-Lopez, M.E. What Do Microglia Really Do in Healthy Adult Brain? Cells 2019, 8, 1293. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, G.M.; Avalos, P.; Ma, A.A.; Alkaslasi, M.; Cho, N.; Wyss, L.; Vit, J.P.; Godoy, M.; Suezaki, P.; Shelest, O.; et al. Transplantation of Neural Progenitor Cells Expressing Glial Cell Line-Derived Neurotrophic Factor into the Motor Cortex as a Strategy to Treat Amyotrophic Lateral Sclerosis. Stem Cells 2018, 36, 1122–1131. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, T.; Adler, A.F.; Mattsson, B.; Hoban, D.B.; Nolbrant, S.; Wahlestedt, J.N.; Kirkeby, A.; Grealish, S.; Björklund, A.; Parmar, M. Target-Specific Forebrain Projections and Appropriate Synaptic Inputs of HESC-Derived Dopamine Neurons Grafted to the Midbrain of Parkinsonian Rats. J. Comp. Neurol. 2018, 526, 2133–2146. [Google Scholar] [CrossRef]
- Henriques, D.; Moreira, R.; Schwamborn, J.; de Almeida, L.P.; Mendonça, L.S. Successes and Hurdles in Stem Cells Application and Production for Brain Transplantation. Front. Neurosci. 2019, 13, 1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Parry, M.; Hou, X.Y.; Liu, M.H.; Wang, H.; Cain, R.; Pei, Z.F.; Chen, Y.C.; Guo, Z.Y.; Abhijeet, S.; et al. Gene Therapy Conversion of Striatal Astrocytes into GABAergic Neurons in Mouse Models of Huntington’s Disease. Nat. Commun. 2020, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Ricci, G.; Volpi, L.; Pasquali, L.; Petrozzi, L.; Siciliano, G. Astrocyte—Neuron Interactions in Neurological Disorders. J. Biol. Phys. 2009, 35, 317–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parpura, V.; Heneka, M.T.; Montana, V.; Oliet, S.H.R.; Schousboe, A.; Haydon, P.G.; Stout, R.F.; Spray, D.C.; Reichenbach, A.; Pannicke, T.; et al. Glial Cells in (Patho)Physiology. J. Neurochem. 2012, 121, 4–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.H.; Tanzi, R.E. Is Alzheimer’s Disease a Neurogenesis Disorder? Cell Stem Cell 2019, 25, 7–8. [Google Scholar] [CrossRef]
- Lee, J.K.; Jin, H.K.; Endo, S.; Schuchman, E.H.; Carter, J.E.; Bae, J.S. Intracerebral Transplantation of Bone Marrow-Derived Mesenchymal Stem Cells Reduces Amyloid-Beta Deposition and Rescues Memory Deficits in Alzheimer’s Disease Mice by Modulation of Immune Responses. Stem Cells 2010, 28, 329–343. [Google Scholar] [CrossRef]
- Ziyuan, G.; Lei, Z.; Zheng, W.; Yuchen, C.; Fan, W.; Gong, C. In Vivo Direct Reprogramming of Reactive Glial Cells into Functional Neurons after Brain Injury and in an Alzheimer’s Disease Model. Cell Stem Cell 2014, 14, 188–202. [Google Scholar]
- Chen, Y.C.; Ma, N.X.; Pei, Z.F.; Wu, Z.; Do-Monte, F.H.; Keefe, S.; Yellin, E.; Chen, M.S.; Yin, J.C.; Lee, G.; et al. A NeuroD1 AAV-Based Gene Therapy for Functional Brain Repair after Ischemic Injury through In Vivo Astrocyte-to-Neuron Conversion. Mol. Ther. 2020, 28, 217–234. [Google Scholar] [CrossRef] [Green Version]
- Han, F.; Bi, J.; Qiao, L.; Arancio, O. Stem Cell Therapy for Alzheimer’s Disease. Adv. Exp. Med. Biol. 2020, 1266, 39–55. [Google Scholar]
- Santamaria, G.; Brandi, E.; La Vitola, P.; Grandi, F.; Ferrara, G.; Pischiutta, F.; Vegliante, G.; Zanier, E.R.; Re, F.; Uccelli, A.; et al. Intranasal Delivery of Mesenchymal Stem Cell Secretome Repairs the Brain of Alzheimer’s Mice. Cell Death Differ. 2020, in press. [Google Scholar]
- Li, H.; Chen, G. In Vivo Reprogramming for CNS Repair: Regenerating Neurons from Endogenous Glial Cells. Neuron 2016, 91, 728–738. [Google Scholar] [CrossRef] [Green Version]
- Das, G.; Gupta, V.; Ghosh, S. Glial-Neuron Transformation by “Chemical Cocktail”. ACS Chem. Neurosci. 2019, 10, 42–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunn, J.S.; Sakowski, S.A.; Hur, J.; Feldman, E.L. Stem Cell Technology for Neurodegenerative Diseases. Ann. Neurol. 2011, 70, 353–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urrutia, D.N.; Caviedes, P.; Mardones, R. Comparative Study of the Neural Differentiation Capacity of Mesenchymal Stromal Cells from Different Tissue Sources: An Approach for Their Use in Neural Regeneration Therapies. PLoS ONE 2019, 14, e0213032. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, T.; Kibayashi, T.; Katayama, T.; Yamashita, Y.; Suzuki, S.; Kawamata, J.; Honmou, O.; Minami, M.; Shimohama, S. Mesenchymal Stem Cells Transmigrate across Brain Microvascular Endothelial Cell Monolayers through Transiently Formed Inter-Endothelial Gaps. Neurosci. Lett. 2011, 502, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chang, K.A.; Kim, J.A.; Park, H.G.; Ra, J.C.; Kim, H.S.; Suh, Y.H. The Preventive and Therapeutic Effects of Intravenous Human Adipose-Derived Stem Cells in Alzheimer’s Disease Mice. PLoS ONE 2012, 7, e45757. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y.; Lin, H.T.; Lee, C.C.; Tsai, K.J. Effects of Neural Stem Cell Transplantation in Alzheimer’s Disease Models. J. Biomed. Sci. 2020, 27, 29. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.I.; Zaben, M.; Gray, W.P. Stem Cells in the Adult Human Brain. Br. J. Neurosurg. 2011, 25, 28–37. [Google Scholar] [CrossRef]
- Ager, R.R.; Davis, J.L.; Agazaryan, A.; Benavente, F.; Poon, W.W.; LaFerla, F.M.; Blurton-Jones, M. Human Neural Stem Cells Improve Cognition and Promote Synaptic Growth in Two Complementary Transgenic Models of Alzheimer’s Disease and Neuronal Loss. Hippocampus 2015, 25, 813–826. [Google Scholar] [CrossRef] [Green Version]
- Nori, S.; Okada, Y.; Nishimura, S.; Sasaki, T.; Itakura, G.; Kobayashi, Y.; Renault-mihara, F.; Shimizu, A.; Koya, I.; Yoshida, R.; et al. Long-Term Safety Issues of IPSC-Based Cell Therapy in a Spinal Cord Injury Model: Oncogenic Transformation with Epithelial-Mesenchymal Transition. Stem Cell Rep. 2015, 4, 360–373. [Google Scholar] [CrossRef] [Green Version]
- Dlouhy, B.J.; Awe, O.; Rao, R.C.; Kirby, P.A.; Hitchon, P.W. Autograft-Derived Spinal Cord Mass Following Olfactory Mucosal Cell Transplantation in a Spinal Cord Injury Patient. J. Neurosurg. 2014, 21, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Herberts, C.A.; Kwa, M.S.G.; Hermsen, H.P.H. Risk Factors in the Development of Stem Cell Therapy. J. Transl. Med. 2011, 9, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, L.H.; Richard, M.R.; Burkhard, B. Immune Attack: The Role of Inflammation in Alzheimer Disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar]

{kind=link}
| Class of Drugs | Compound | Mechanisms | Status | Details * | [Ref] |
|---|---|---|---|---|---|
| 1. Targeting Aβ pathology | |||||
| Inhibition of Aβ cascade | Verubecestat | β-secretase inhibitor | Phase II/III, failed | 1958/55.3/71.8 | [18] |
| Lanabecestat | β-secretase inhibitor | Phase II/III, failed | 2218/53.1/71.3 | [19] | |
| Elenbecestat | β-secretase inhibitor | Phase III | Not recruiting yet | [20] | |
| Atabecestat | β-secretase inhibitor | Phase II/III, failed | 63/54.8/69.1 | [21] | |
| CNP520 | β-secretase inhibitor | Phase III, failed | 1145/NA/NA | [22] | |
| Tarenflurbil | γ -secretase inhibitor | Phase III, failed | 1649/50.9/74.6 | [23] | |
| Semagacestat | γ -secretase inhibitor | Phase II, failed | 1534/53.0/73.2 | [24] | |
| Passive immunotherapy | Bapineuzumab | Clearance of oligomeric Aβ | Phase II/III, failed | 2204/53.9/72.4 | [25] |
| Gantenerumab | Clearance of oligomeric Aβ | Phase II/III, failed | 797/NA/70.3 | [26] | |
| Inhibition of Aβ aggregation | PBT2 | Anti-Aβ aggregation | Phase II | 78/86.7/71.9 | [27] |
| 2. Targeting tauopathy | |||||
| Tau expression inhibitors | Antisense nucleotides | Reducing tau mRNA and protein | In vitro | PS19 mice with Tau/NA | [28] |
| Phosphatase modifiers | Okadaic acid | PP2A inhibitor | In vitro & in vivo | Sparague-Dawley rats/Male | [29] |
| Calyculin A | PP2A inhibitor | In vitro & in vivo | Sparague-Dawley rats/NA | [30] | |
| Memantine | PP2A inhibitor | In vitro | PC12 cells | [31] | |
| SEW2871 | PP2A activiator | In vivo | Sparague-Dawley rats/Male | [32] | |
| Zinc Chelators | PP2A activiator | In vivo | Sparague-Dawley rats/Male | [33] | |
| Tau kinase inhibitors | Tideglusib | GSK3-β inhibitor | Phase II | 306/55.5/71.5 | [34] |
| Lithium | GSK3-β inhibitor | Phase II | 71/NA/NA | [35] | |
| Amino-thiazole | GSK3-β inhibitor | In vitro | Sf21 cells | [36] | |
| Thiadiazolidinone | GSK3-β inhibitor | In vitro | Kinase assay | [37] | |
| Sirenade | GSK3-β inhibitor | In vivo | Wistar rats/Male | [38] | |
| R-roscovitine | CDK inhibitor | In vivo | Wistar rats/Both | [39] | |
| Aloisine | CDK inhibitor | In vitro | NT2 cells | [40] | |
| Inhibition of tau acetylation | Salsalate | Tau acetylation inhibitor | In vivo | PS19 mice/NA | [41] |
| Tau glycosylation | p-diesterase IV inhibitor | cAMP activator | Phase III | 336/56.8/74.0 | [42] |
| Tau aggregation inhibitors | LMTX | Dissolving tau filaments | Phase III | 885/62.0/70.6 | [43] |
| Rhodamine | Dissolving tau oligomer | In vitro & in vivo | Neurons from PC12 cells | [44] | |
| N-Phenylamines | Inhibition of tau aggregation | In vitro | N2a cells expressing tau | [45] | |
| RemberTM | Dissolving aggregates | Phase II | 321/54.0/73.8 | [46] | |
| Microtubule stabilizers | Epothilone D | Increasing axonal MT density | In vivo | PS19 mice/Male | [47] |
| NAP | Tubulin stabilizer | In vitro & in vivo | Motor neurons with Tau mutant | [48] | |
| TPI 287 | Tubulin stabilizer | Phase I | 26/54.0/63.0 | [49] | |
| 3. Modulation of neuroinflammation | |||||
| Regulation of innate immunity | Stattic | JAK2/STAT3 inhibitor | In vivo | 5XFAD mice | [50] |
| SB203580 | p38 MAPK inhibitor | In vitro | Cortical neuron | [51] | |
| SB202190 | p38 MAPK inhibitor | In vivo | Rats with vascular dementia/NA | [52] | |
| SP600125 | p38 MAPK inhibitor | In vivo | Mice with traumatic injury/NA | [53] | |
| NJK14047 | p38 MAPK inhibitor | In vitro & in vivo | BV2 cells & Mice with LPS/NA | [54] | |
| VIVIT peptide | Calcineurin/NFAT inhibitor | In vivo model | APP/PS1 mice/NA | [55] | |
| Tacrolimus | Calcineurin/NFAT inhibitor | Phase II | Not recruiting yet | [56] | |
| Anti-apoE | TREM2 activator | In vivo | APP/PS1 mice/Male | [57] | |
| Anti-TREM2 | TREM2 activator | In vivo | Mice with TREM2 mutant/NA | [58] | |
| GW2580 | CSF1R inhibitor | In vivo | APP, PSEN, APP/PS1 mice/NA | [59] | |
| MRS2179 | P2Y1R antagonists | In vivo | APP/PS1 mice/NA | [60] | |
| Regulation of adaptive immunity | Aβ-specific Th2 lymphocytes | Regulation of adaptive immunity | In vivo | APP/PS1 mice/NA | [61] |
| anti-Aβ IgG | Neutralization of fibrinogenesis | In vivo | 5XFAD mice/NA | [62] | |
| 4. Neuroregeneration Restoring Cognitive Impairment | |||||
| Supplement of neurotrophins | BDNF | Neurotrophic factors | In vivo | Rats with Aβ1–42/NA | [63] |
| hNGFp | Neurotrophic factors | In vivo | APP/PS1 mice/NA | [64] | |
| AAV2-NGF | Neurotrophic factors | Phase II | 49/43.0/68.0 | [65] | |
| Neurotrophin mimetics | 7,8-DHF | BDNF mimetic | In vivo | 5XFAD mice/NA | [66] |
| Doxygedunin | BDNF mimetic | In vivo | Mice with TrkB mutant/NA | [67] | |
| LM22A-4 | BDNF mimetic | In vitro & in vivo | Mice with TrkB mutant/Male | [68] | |
| Bicyclic BDNF loop mimetic | BDNF mimetic | In vitro | Primary sensory neurons | [69] | |
| Conversion of glial cells to neuronal cells | Neurogenin 2 | Astrocytes to glutamatergic neurons | In vitro | Postnatal cortical astroglia | [70] |
| Dlx2 | Astrocytes to GABAergic neurons | In vitro | Postnatal cortical astroglia | [71] | |
| Combination of 9 molecules | Astrocytes to neurons | In vitro & in vivo | Astrocytes | [72] | |
| Combination of 4 molecules | Astrocytes to neurons | In vitro & in vivo | Cortical astrocytes | [73] | |
| Transplanting stem cells into brains | NSCs encoding with NGF | NSCs to neurons and astrocytes | In vivo | Mice with dementia/NA | [74] |
| NSCs encoding with BDNF | NSCs to neurons | In vivo | APP mice/NA | [75] | |
| NSCs pretreated with BDNF | NSCs to cholinergic neurons | In vivo | APP/PS1 mice/NA | [76] | |
| NSCs expressing IGF-I | Engraftment of HK532-IGF-I cells | In vitro & in vivo | APP/PS1 mice/NA | [77] | |
| BM-MSCs expressing VEGF | Decrease in senile plaque | In vivo | APP/PS1 mice/NA | [78] | |
| MSCs expressing GLP-1 | Decrease in Aβ plaque deposition | In vivo | APP mice/Male | [79] | |
| NSCs expressing neprilysin | Decrease in Aβ pathology | In vivo | APP mice/NA | [80] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, Y.J.; Diep, Y.N.; Tran, M.; Cho, H. Therapeutic Targeting Strategies for Early- to Late-Staged Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 9591. https://doi.org/10.3390/ijms21249591
Kang YJ, Diep YN, Tran M, Cho H. Therapeutic Targeting Strategies for Early- to Late-Staged Alzheimer’s Disease. International Journal of Molecular Sciences. 2020; 21(24):9591. https://doi.org/10.3390/ijms21249591
Chicago/Turabian StyleKang, You Jung, Yen N. Diep, Minh Tran, and Hansang Cho. 2020. "Therapeutic Targeting Strategies for Early- to Late-Staged Alzheimer’s Disease" International Journal of Molecular Sciences 21, no. 24: 9591. https://doi.org/10.3390/ijms21249591
APA StyleKang, Y. J., Diep, Y. N., Tran, M., & Cho, H. (2020). Therapeutic Targeting Strategies for Early- to Late-Staged Alzheimer’s Disease. International Journal of Molecular Sciences, 21(24), 9591. https://doi.org/10.3390/ijms21249591