Thioredoxin-Interacting Protein (TXNIP) with Focus on Brain and Neurodegenerative Diseases



Abstract
:
1. Introduction
2. Biochemistry of TXNIP: Overview
2.1. TXNIP and Thioredoxin System
2.2. TXNIP Interacting Proteins
3. Regulation of TXNIP Expression
3.1. Cell Types
3.1.1. Peripheral Cells
3.1.2. Neurons
3.1.3. Microglia
3.1.4. Astrocytes
3.1.5. Endothelial Cells
3.1.6. Retinal Pigment Epithelial Cells
3.1.7. Brain Cancer Cells
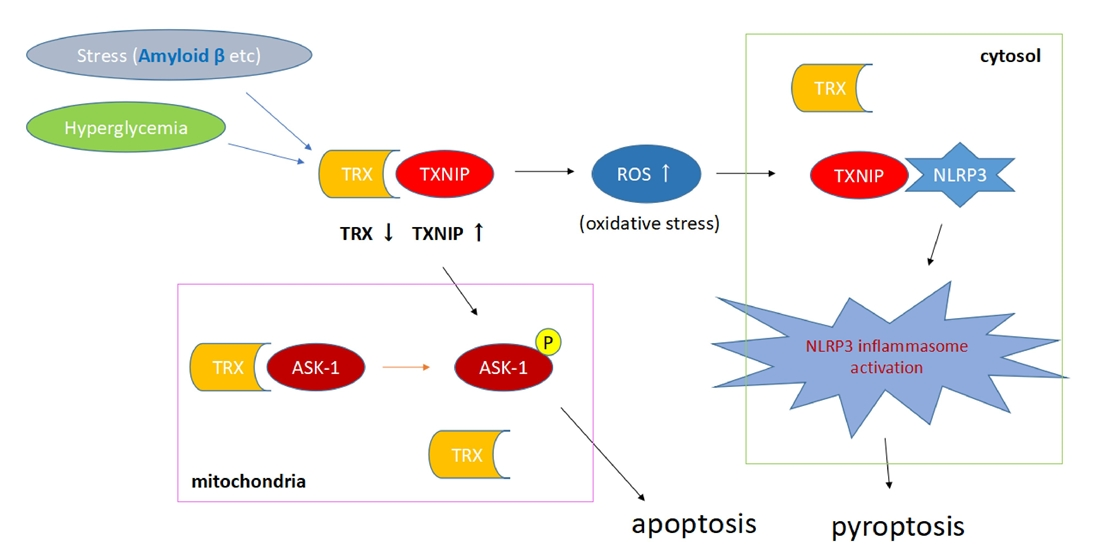
4. TXNIP, Hyperglycemia, and Oxidative Stress—NLRP3 Inflammasome Complex
5. TXNIP Expression in the Brain
5.1. TXNIP in Acute Neurological Conditions
5.2. TXNIP in Neurodegenerative Diseases (Animal and Cellular Models)
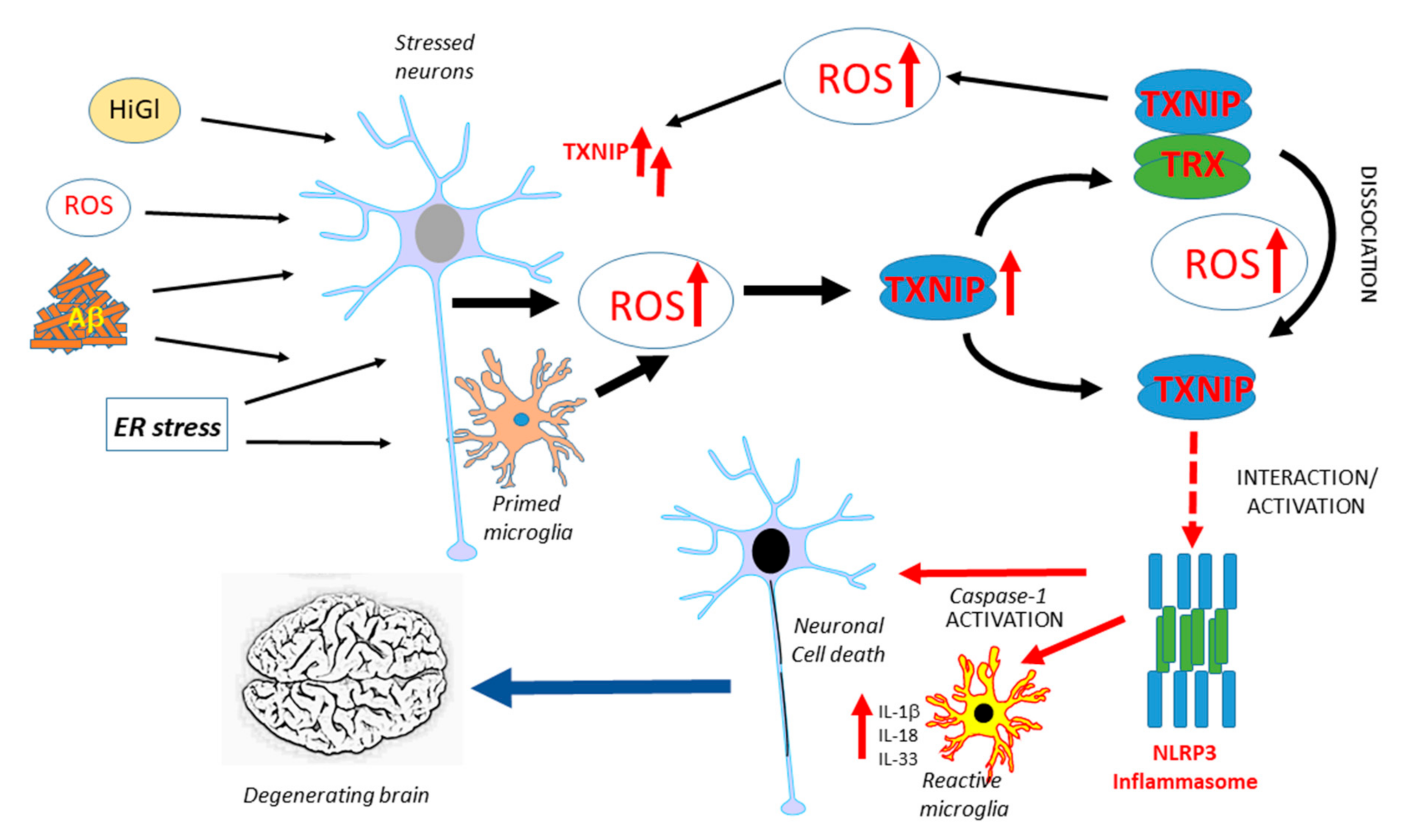
5.3. Linking Diabetes, TXNIP, and Alzheimer’s Disease: Hypothesis
6. Physiological Consequences of Loss of TXNIP
6.1. Experimental Animals
6.2. Human Subjects
7. Modulating TXNIP Expression
8. Summary
Funding
Conflicts of Interest
References
- Ramli, N.Z.; Yahaya, M.F.; Tooyama, I.; Damanhuri, H.A. A Mechanistic Evaluation of Antioxidant Nutraceuticals on Their Potential against Age-Associated Neurodegenerative Diseases. Antioxidants 2020, 9, 1019. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef] [PubMed]
- Pradeepkiran, J.A.; Reddy, P.H. Defective mitophagy in Alzheimer’s disease. Ageing Res. Rev. 2020, 64, 101191. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Jo, M.G.; Kim, S.Y.; Chung, C.G.; Lee, S.B. Dietary Antioxidants and the Mitochondrial Quality Control: Their Potential Roles in Parkinson’s Disease Treatment. Antioxidants 2020, 9, 1056. [Google Scholar] [CrossRef] [PubMed]
- López-Contreras, A.K.; Martínez-Ruiz, M.G.; Olvera-Montaño, C.; Robles-Rivera, R.R.; Arévalo-Simental, D.E.; Castellanos-González, J.A.; Hernández-Chávez, A.; Huerta-Olvera, S.G.; Cardona-Muñoz, E.G.; Rodríguez-Carrizalez, A.D. Importance of the Use of Oxidative Stress Biomarkers and Inflammatory Profile in Aqueous and Vitreous Humor in Diabetic Retinopathy. Antioxidants 2020, 9, 891. [Google Scholar] [CrossRef]
- Tian, Y.; Su, Y.; Ye, Q.; Chen, L.; Yuan, F.; Wang, Z. Silencing of TXNIP Alleviated Oxidative Stress Injury by Regulating MAPK-Nrf2 Axis in Ischemic Stroke. Neurochem. Res. 2020, 45, 428–436. [Google Scholar] [CrossRef]
- Su, C.; Shi, A.; Cao, G.; Tao, T.; Chen, R.; Hu, Z.; Shen, Z.; Tao, H.; Cao, B.; Hu, D.; et al. Fenofibrate suppressed proliferation and migration of human neuroblastoma cells via oxidative stress dependent of TXNIP upregulation. Biochem. Biophys. Res. Commun. 2015, 460, 983–988. [Google Scholar] [CrossRef]
- Pereira, J.D.; Fraga, V.G.; Santos, A.L.M.; Carvalho, M.D.G.; Caramelli, P.; Gomes, K.B. Alzheimer’s disease and type 2 diabetes mellitus: A systematic review of proteomic studies. J. Neurochem. 2020, 1–24. [Google Scholar] [CrossRef]
- Saleem, U.; Sabir, S.; Niazi, S.G.; Nadeem, M.; Ahmad, B. Role of Oxidative Stress and Antioxidant Defense Biomarkers in Neurodegenerative Diseases. Crit. Rev. Eukaryot. Gene Expr. 2020, 30, 311–322. [Google Scholar] [CrossRef]
- Nishizawa, K.; Nishiyama, H.; Matsui, Y.; Kobayashi, T.; Saito, R.; Kotani, H.; Masutani, H.; Oishi, S.; Toda, Y.; Fujii, N.; et al. Thioredoxin-interacting protein suppresses bladder carcinogenesis. Carcinogenesis 2011, 32, 1459–1466. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Xie, R.; Xie, S.; Wu, Y.; Wang, W.; Li, X.; Xu, Y.; Liu, B.; Zhou, Y.; Wang, T.; et al. Thioredoxin interacting protein (TXNIP) acts as a tumor suppressor in human prostate cancer. Cell Biol. Int. 2020, 44, 2094–2106. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, S.F.; Miele, M.E.; Hatta, N.; Takata, M.; Paquette-Straub, C.; Freedman, L.P.; Welch, D.R. Melanoma metastasis suppression by chromosome 6: Evidence for a pathway regulated by CRSP3 and TXNIP. Cancer Res. 2003, 63, 432–440. [Google Scholar] [PubMed]
- Chen, J.; Saxena, G.; Mungrue, I.N.; Lusis, A.J.; Shalev, A. Thioredoxin-interacting protein: A critical link between glucose toxicity and beta-cell apoptosis. Diabetes 2008, 57, 938–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, C.-R.; Chan, W.P.A.; Nguyen, T.H.; Liu, S.; Procter, N.E.K.; Ngo, D.T.; Sverdlov, A.L.; Chirkov, Y.Y.; Horowitz, J.D. Thioredoxin-Interacting Protein: Pathophysiology and Emerging Pharmacotherapeutics in Cardiovascular Disease and Diabetes. Cardiovasc. Drugs Ther. 2014, 28, 347–360. [Google Scholar] [CrossRef]
- Du, C.; Wu, M.; Liu, H.; Ren, Y.; Du, Y.; Wu, H.; Wei, J.; Liu, C.; Yao, F.; Wang, H.; et al. Thioredoxin-interacting protein regulates lipid metabolism via Akt/mTOR pathway in diabetic kidney disease. Int. J. Biochem. Cell Biol. 2016, 79, 1–13. [Google Scholar] [CrossRef]
- Devi, T.S.; Hosoya, K.-I.; Terasaki, T.; Singh, L.P. Critical role of TXNIP in oxidative stress, DNA damage and retinal pericyte apoptosis under high glucose: Implications for diabetic retinopathy. Exp. Cell Res. 2013, 319, 1001–1012. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Yu, Y. The Function of Thioredoxin-Binding Protein-2 (TBP-2) in Different Diseases. Oxidative Med. Cell. Longev. 2018, 2018, 4582130. [Google Scholar] [CrossRef] [Green Version]
- Alhawiti, N.M.; Al Mahri, S.; Aziz, M.A.; Malik, S.S.; Mohammad, S. TXNIP in Metabolic Regulation: Physiological Role and Therapeutic Outlook. Curr. Drug Targets 2017, 18, 1095–1103. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, A.; Matsui, M.; Iwata, S.; Hirota, K.; Masutani, H.; Nakamura, H.; Takagi, Y.; Sono, H.; Gon, Y.; Yodoi, J. Identification of Thioredoxin-binding Protein-2/Vitamin D(3)Up-regulated Protein 1 as a Negative Regulator of Thioredoxin Function and Expression. J. Biol. Chem. 1999, 274, 21645–21650. [Google Scholar] [CrossRef] [Green Version]
- Junn, E.; Han, S.H.; Im, J.Y.; Yang, Y.; Cho, E.W.; Um, H.D.; Kim, D.K.; Lee, K.W.; Han, P.L.; Rhee, S.G.; et al. Vitamin D3 Up-Regulated Protein 1 Mediates Oxidative Stress Via Suppressing the Thioredoxin Function. J. Immunol. 2000, 164, 6287–6295. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, H.; Maehira, F.; Oshiro, M.; Asato, T.; Yanagawa, Y.; Takei, H.; Nakashima, Y. A Possible Interaction of Thioredoxin with VDUP1 in HeLa Cells Detected in a Yeast Two-Hybrid System. Biochem. Biophys. Res. Commun. 2000, 271, 796–800. [Google Scholar] [CrossRef] [PubMed]
- Farrell, M.R.; Rogers, L.K.; Liu, Y.; Welty, S.E.; Tipple, T.E. Thioredoxin-interacting protein inhibits hypoxia-inducible factor transcriptional activity. Free Radic. Biol. Med. 2010, 49, 1361–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroder, K.; Zhou, R.; Tschopp, J. The NLRP3 Inflammasome: A Sensor for Metabolic Danger? Science 2010, 327, 296–300. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Gao, J.; Wang, X.; Wen, W.; Yang, H.; Tian, Y.; Liu, N.; Wang, Z.; Liu, H.; Zhang, Y.; et al. A novel indication of thioredoxin-interacting protein as a tumor suppressor gene in malignant glioma. Oncol. Lett. 2017, 14, 2053–2058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, K.Y.; Roy, J.; Fung, M.L.; Heng, B.C.; Zhang, C.; Lim, L.W. Relationships between Mitochondrial Dysfunction and Neurotransmission Failure in Alzheimer’s Disease. Aging Dis. 2020, 11, 1291–1316. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ismael, S.; Nasoohi, S.; Sakata, K.; Liao, F.-F.; McDonald, M.P.; Ishrat, T. Thioredoxin-Interacting Protein (TXNIP) Associated NLRP3 Inflammasome Activation in Human Alzheimer’s Disease Brain. J. Alzheimers Dis. 2019, 68, 255–265. [Google Scholar] [CrossRef]
- Gąsecka, A.; Siwik, D.; Gajewska, M.; Jaguszewski, M.J.; Mazurek, T.; Filipiak, K.J.; Postuła, M.; Eyileten, C. Early Biomarkers of Neurodegenerative and Neurovascular Disorders in Diabetes. J. Clin. Med. 2020, 9, 2807. [Google Scholar] [CrossRef]
- Vionnet, N.; Hani, E.H.; Dupont, S.; Gallina, S.; Francke, S.; Dotte, S.; De Matos, F.; Durand, E.; Leprêtre, F.; Lecoeur, C.; et al. Genomewide Search for Type 2 Diabetes–Susceptibility Genes in French Whites: Evidence for a Novel Susceptibility Locus for Early-Onset Diabetes on Chromosome 3q27-qter and Independent Replication of a Type 2–Diabetes Locus on Chromosome 1q21–q24. Am. J. Hum. Genet. 2000, 67, 1470–1480. [Google Scholar] [CrossRef]
- Abu El Maaty, M.A.; Almouhanna, F.; Wölfl, S. Expression of TXNIP in Cancer Cells and Regulation by 1,25(OH)2D3: Is It Really the Vitamin D3 Upregulated Protein? Int. J. Mol. Sci. 2018, 19, 796. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, D.L.; Kotanides, H.; Le, T.; Chavkin, D.; Bohlen, P.; Witte, L. Cloning, genetic characterization, and chromosomal mapping of the mouse VDUP1 gene. Gene 2001, 269, 103–112. [Google Scholar] [CrossRef]
- Patwari, P.; Higgins, L.J.; Chutkow, W.A.; Yoshioka, J.; Lee, R.T. The interaction of thioredoxin with Txnip. Evidence for formation of a mixed disulfide by disulfide exchange. J. Biol. Chem. 2006, 281, 21884–21891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, G.; Chen, J.; Shalev, A. Intracellular Shuttling and Mitochondrial Function of Thioredoxin-interacting Protein. J. Biol. Chem. 2010, 285, 3997–4005. [Google Scholar] [CrossRef] [Green Version]
- Luthman, M.; Holmgren, A. Rat liver thioredoxin and thioredoxin reductase: Purification and characterization. Biochemistry 1982, 21, 6628–6633. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, A.; Fagerstedt, M. The in vivo distribution of oxidized and reduced thioredoxin in Escherichia coli. J. Biol. Chem. 1982, 257, 6926–6930. [Google Scholar]
- Berstermann, A.; Vogt, K.; Follmann, H. Plant Seeds Contain Several Thioredoxins of Regular Size. Eur. J. Biochem. 1983, 131, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Chen, X.; Sun, J.; Li, Q.; Lian, X.; Li, S.; Wang, Y.; Tian, L. Inhibition of nuclear thioredoxin aggregation attenuates PM(2.5)-induced NF-κB activation and pro-inflammatory responses. Free Radic. Biol. Med. 2019, 130, 206–214. [Google Scholar] [CrossRef]
- Obikane, H.; Abiko, Y.; Ueno, H.; Kusumi, Y.; Esumi, M.; Mitsumata, M. Effect of endothelial cell proliferation on atherogenesis: A role of p21(Sdi/Cip/Waf1) in monocyte adhesion to endothelial cells. Atherosclerosis 2010, 212, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Bodenstein-Lang, J.; Buch, A.; Follmann, H. Animal and plant mitochondria contain specific thioredoxins. FEBS Lett. 1989, 258, 22–26. [Google Scholar] [CrossRef] [Green Version]
- Bodenstein, J.; Follmann, H. Characterization of Two Thioredoxins in Pig Heart Including a New Mitochondrial Protein. Zeitschrift für Naturforschung C 1991, 46, 270–279. [Google Scholar] [CrossRef]
- Rozell, B.; Hansson, H.A.; Luthman, M.; Holmgren, A. Immunohistochemical localization of thioredoxin and thioredoxin reductase in adult rats. Eur. J. Cell Biol. 1985, 38, 79–86. [Google Scholar] [PubMed]
- Lippoldt, A.; Padilla, C.A.; Gerst, H.; Andbjer, B.; Richter, E.; Holmgren, A.; Fuxe, K. Localization of thioredoxin in the rat brain and functional implications. J. Neurosci. 1995, 15, 6747–6756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rybnikova, E.; Damdimopoulos, A.E.; Gustafsson, J.Å.; Spyrou, G.; Pelto-Huikko, M. Expression of novel antioxidant thioredoxin-2 in the rat brain. Eur. J. Neurosci. 2000, 12, 1669–1678. [Google Scholar] [CrossRef] [PubMed]
- Mitsui, A.; Hamuro, J.; Nakamura, H.; Kondo, N.; Hirabayashi, Y.; Ishizaki-Koizumi, S.; Hirakawa, T.; Inoue, T.; Yodoi, J. Overexpression of Human Thioredoxin in Transgenic Mice Controls Oxidative Stress and Life Span. Antioxid. Redox Signal. 2002, 4, 693–696. [Google Scholar] [CrossRef] [PubMed]
- Takagi, Y.; Mitsui, A.; Nishiyama, A.; Nozaki, K.; Sono, H.; Gon, Y.; Hashimoto, N.; Yodoi, J. Overexpression of thioredoxin in transgenic mice attenuates focal ischemic brain damage. Proc. Natl. Acad. Sci. USA 1999, 96, 4131–4136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattori, I.; Takagi, Y.; Nakamura, H.; Nozaki, K.; Bai, J.; Kondo, N.; Sugino, T.; Nishimura, M.; Hashimoto, N.; Yodoi, J. Intravenous Administration of Thioredoxin Decreases Brain Damage Following Transient Focal Cerebral Ischemia in Mice. Antioxid. Redox Signal. 2004, 6, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Xie, C.; Gabbita, S.P.; Markesbery, W.R. Decreased thioredoxin and increased thioredoxin reductase levels in alzheimer’s disease brain. Free Radic. Biol. Med. 2000, 28, 418–427. [Google Scholar] [CrossRef]
- Arodin, L.; Lamparter, H.; Karlsson, H.; Nennesmo, I.; Björnstedt, M.; Schröder, J.; Fernandes, A.P. Alteration of Thioredoxin and Glutaredoxin in the Progression of Alzheimer’s Disease. J. Alzheimers Dis. 2014, 39, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Nishinaka, Y.; Masutani, H.; Oka, S.-I.; Matsuo, Y.; Yamaguchi, Y.; Nishio, K.; Ishii, Y.; Yodoi, J. Importin α1 (Rch1) Mediates Nuclear Translocation of Thioredoxin-binding Protein-2/Vitamin D(3)-up-regulated Protein 1. J. Biol. Chem. 2004, 279, 37559–37565. [Google Scholar] [CrossRef] [Green Version]
- Suh, H.-W.; Yun, S.; Song, H.; Jung, H.; Park, Y.-J.; Kim, T.-D.; Yoon, S.R.; Choi, I. TXNIP interacts with hEcd to increase p53 stability and activity. Biochem. Biophys. Res. Commun. 2013, 438, 264–269. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Kim, K.-Y.; Shin, S.M.; Kim, J.K.; Paik, S.G.; Yang, Y.; Choi, I. Heat shock factor regulates VDUP1 gene expression. Biochem. Biophys. Res. Commun. 2004, 315, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Schulze, P.C.; Liu, H.; Choe, E.; Yoshioka, J.; Shalev, A.; Bloch, K.D.; Lee, R.T. Nitric Oxide–Dependent Suppression of Thioredoxin-Interacting Protein Expression Enhances Thioredoxin Activity. Arter. Thromb. Vasc. Biol. 2006, 26, 2666–2672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.-X.; Goh, S.-R.; Dai, R.-P.; Luo, Y. Adenosine-Containing Molecules Amplify Glucose Signaling and Enhance Txnip Expression. Mol. Endocrinol. 2009, 23, 932–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, M.K.C.; Wu, J.; Chang, E.; Wang, B.; Katzenberg-Clark, R.; Ishii-Watabe, A.; Cooke, J.P. A Central Role for Nicotinic Cholinergic Regulation of Growth Factor–Induced Endothelial Cell Migration. Arter. Thromb. Vasc. Biol. 2007, 27, 106–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masaki, S.; Masutani, H.; Yoshihara, E.; Yodoi, J. Deficiency of Thioredoxin Binding Protein-2 (TBP-2) Enhances TGF-β Signaling and Promotes Epithelial to Mesenchymal Transition. PLoS ONE 2012, 7, e39900. [Google Scholar] [CrossRef] [PubMed]
- DeRoo, B.J.; Hewitt, S.C.; Peddada, S.D.; Korach, K.S. Estradiol Regulates the Thioredoxin Antioxidant System in the Mouse Uterus. Endocrinology 2004, 145, 5485–5492. [Google Scholar] [CrossRef] [Green Version]
- Saitoh, T.; Tanaka, S.; Koike, T. Rapid induction and Ca(2+) influx-mediated suppression of vitamin D3 up-regulated protein 1 (VDUP1) mRNA in cerebellar granule neurons undergoing apoptosis. J. Neurochem. 2001, 78, 1267–1276. [Google Scholar] [CrossRef] [Green Version]
- Sbai, O.; Devi, T.S.; Melone, M.A.B.; Féron, F.; Khrestchatisky, M.; Singh, L.P.; Perrone, L. RAGE-TXNIP axis is required for S100B-promoted Schwann cell migration, fibronectin expression and cytokine secretion. J. Cell Sci. 2010, 123, 4332–4339. [Google Scholar] [CrossRef] [Green Version]
- Parikh, H.; Carlsson, E.; Chutkow, W.A.; Johansson, L.E.; Storgaard, H.; Poulsen, P.; Saxena, R.; Ladd, C.; Schulze, P.C.; Mazzini, M.J.; et al. TXNIP Regulates Peripheral Glucose Metabolism in Humans. PLoS Med. 2007, 4, e158. [Google Scholar] [CrossRef] [Green Version]
- Perrone, L.; Devi, T.S.; Hosoya, K.-I.; Terasaki, T.; Singh, L.P. Thioredoxin interacting protein (TXNIP) induces inflammation through chromatin modification in retinal capillary endothelial cells under diabetic conditions. J. Cell. Physiol. 2009, 221, 262–272. [Google Scholar] [CrossRef]
- Chen, J.; Fontes, G.; Saxena, G.; Poitout, V.; Shalev, A. Lack of TXNIP protects against mitochondria-mediated apoptosis but not against fatty acid-induced ER stress-mediated beta-cell death. Diabetes 2010, 59, 440–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandala, A.; Das, N.; Bhattacharjee, S.; Mukherjee, B.; Mukhopadhyay, S.; Roy, S.S. Thioredoxin interacting protein mediates lipid-induced impairment of glucose uptake in skeletal muscle. Biochem. Biophys. Res. Commun. 2016, 479, 933–939. [Google Scholar] [CrossRef]
- Martel, M.-A.; Soriano, F.X.; Baxter, P.; Rickman, C.; Duncan, R.R.; Wyllie, D.J.A.; Hardingham, G.E. Inhibiting pro-death NMDA receptor signaling dependent on the NR2 PDZ ligand may not affect synaptic function or synaptic NMDA receptor signaling to gene expression. Channels 2009, 3, 12–15. [Google Scholar] [CrossRef] [Green Version]
- Lerner, A.G.; Upton, J.-P.; Praveen, P.V.K.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M.; et al. IRE1α Induces Thioredoxin-Interacting Protein to Activate the NLRP3 Inflammasome and Promote Programmed Cell Death under Irremediable ER Stress. Cell Metab. 2012, 16, 250–264. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.-X.; Luo, Y. Tandem ChoRE and CCAAT Motifs and Associated Factors Regulate Txnip Expression in Response to Glucose or Adenosine-Containing Molecules. PLoS ONE 2009, 4, e8397. [Google Scholar] [CrossRef] [PubMed]
- Heman-Ackah, S.M.; Manzano, R.; Hoozemans, J.J.M.; Scheper, W.; Flynn, R.; Haerty, W.; Cowley, S.A.; Bassett, A.R.; Wood, M.J.A. Alpha-synuclein induces the unfolded protein response in Parkinson’s disease SNCA triplication iPSC-derived neurons. Hum. Mol. Genet. 2017, 26, 4441–4450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poirier, Y.; Grimm, A.; Schmitt, K.; Eckert, A. Link between the unfolded protein response and dysregulation of mitochondrial bioenergetics in Alzheimer’s disease. Cell. Mol. Life Sci. 2019, 76, 1419–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; He, F.; Hu, L.; Hai, L.; Huang, M.; Xu, Z.; Zhang, J.; Zhou, Z.; Liu, F.; Dai, Y.-S. Transcription Factor Ets1 Regulates Expression of Thioredoxin-Interacting Protein and Inhibits Insulin Secretion in Pancreatic β-Cells. PLoS ONE 2014, 9, e99049. [Google Scholar] [CrossRef]
- Ling, C. Epigenetic regulation of insulin action and secretion – role in the pathogenesis of type 2 diabetes. J. Intern. Med. 2020, 288, 158–167. [Google Scholar] [CrossRef]
- Wondafrash, D.Z.; Nire’A, A.T.; Tafere, G.G.; Desta, D.M.; Asmerom, D.; Zewdie, K.A. Thioredoxin-Interacting Protein as a Novel Potential Therapeutic Target in Diabetes Mellitus and Its Underlying Complications. Diabetes Metab. Syndr. Obesity Targets Ther. 2020, 13, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Billiet, L.; Furman, C.; Larigauderie, G.; Copin, C.; Page, S.; Fruchart, J.; Brand, K.; Rouis, M. Enhanced VDUP-1 gene expression by PPARgamma agonist induces apoptosis in human macrophage. J. Cell. Physiol. 2008, 214, 183–191. [Google Scholar] [CrossRef]
- Billiet, L.; Furman, C.; Cuaz-Pérolin, C.; Paumelle, R.; Raymondjean, M.; Simmet, T.; Rouis, M. Thioredoxin-1 and its natural inhibitor, vitamin D3 up-regulated protein 1, are differentially regulated by PPARalpha in human macrophages. J. Mol. Biol. 2008, 384, 564–576. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, F.; Takata, M.; Kamitori, K.; Nonaka, M.; Dong, Y.; Sui, L.; Tokuda, M. Rare sugar D-allose induces specific up-regulation of TXNIP and subsequent G1 cell cycle arrest in hepatocellular carcinoma cells by stabilization of p27kip1. Int. J. Oncol. 2008, 32, 377–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, F.; Kamitori, K.; Sanada, K.; Horii, M.; Dong, Y.; Sui, L.; Tokuda, M. Rare sugar d-allose enhances anti-tumor effect of 5-fluorouracil on the human hepatocellular carcinoma cell line HuH-7. J. Biosci. Bioeng. 2008, 106, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Hoshikawa, H.; Mori, T.; Mori, N. In vitro and in vivo effects of D-allose: Up-regulation of thioredoxin-interacting protein in head and neck cancer cells. Ann. Otol. Rhinol. Laryngol. 2010, 119, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Saito, M.; Tsukamoto, I.; Yamaguchi, F.; Sui, L.; Kamitori, K.; Dong, Y.; Uehara, E.; Konishi, R.; Janjua, N.; et al. Analysis of the inhibitory mechanism of d-allose on MOLT-4F leukemia cell proliferation. J. Biosci. Bioeng. 2009, 107, 562–568. [Google Scholar] [CrossRef]
- Stoltzman, C.A.; Kaadige, M.R.; Peterson, C.W.; Ayer, D.E. MondoA senses non-glucose sugars: Regulation of thioredoxin-interacting protein (TXNIP) and the hexose transport curb. J. Biol. Chem. 2011, 286, 38027–38034. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.-J.; Yoon, S.-J.; Suh, H.-W.; Kim, D.O.; Park, J.-R.; Jung, H.; Kim, T.-D.; Yoon, S.R.; Min, J.-K.; Na, H.-J.; et al. TXNIP Deficiency Exacerbates Endotoxic Shock via the Induction of Excessive Nitric Oxide Synthesis. PLoS Pathog. 2013, 9, e1003646. [Google Scholar] [CrossRef]
- Kanari, Y.; Sato, Y.; Aoyama, S.; Muta, T. Thioredoxin-Interacting Protein Gene Expression via MondoA Is Rapidly and Transiently Suppressed during Inflammatory Responses. PLoS ONE 2013, 8, e59026. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Joe, Y.; Jeong, S.O.; Zheng, M.; Back, S.H.; Park, S.W.; Ryter, S.W.; Chung, H.T. Endoplasmic reticulum stress is sufficient for the induction of IL-1β production via activation of the NF-κB and inflammasome pathways. Innate Immun. 2014, 20, 799–815. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Li, J.; Li, S.; Li, Y.; Wang, X.; Liu, B.; Fu, Q.; Ma, S. Curcumin attenuates glutamate neurotoxicity in the hippocampus by suppression of ER stress-associated TXNIP/NLRP3 inflammasome activation in a manner dependent on AMPK. Toxicol. Appl. Pharmacol. 2015, 286, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Mendsaikhan, A.; Takeuchi, S.; Walker, D.G.; Tooyama, I. Differences in Gene Expression Profiles and Phenotypes of Differentiated SH-SY5Y Neurons Stably Overexpressing Mitochondrial Ferritin. Front. Mol. Neurosci. 2018, 11, 470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.; Zuo, D.; Yu, L.; Zhang, L.; Tang, J.; Cui, C.; Bao, L.; Zan, K.; Zhang, Z.; Yang, X.; et al. ROS/TXNIP pathway contributes to thrombin induced NLRP3 inflammasome activation and cell apoptosis in microglia. Biochem. Biophys. Res. Commun. 2017, 485, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Zhang, L. Resveratrol Suppresses Aβ-Induced Microglial Activation Through the TXNIP/TRX/NLRP3 Signaling Pathway. DNA Cell Biol. 2019, 38, 874–879. [Google Scholar] [CrossRef] [PubMed]
- Bharti, V.; Tan, H.; Zhou, H.; Wang, J.-F. Txnip mediates glucocorticoid-activated NLRP3 inflammatory signaling in mouse microglia. Neurochem. Int. 2019, 131, 104564. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, I.N.; Hafez, S.S.; Fairaq, A.; Ergul, A.; Imig, J.D.; El-Remessy, A.B. Thioredoxin-interacting protein is required for endothelial NLRP3 inflammasome activation and cell death in a rat model of high-fat diet. Diabetologia 2014, 57, 413–423. [Google Scholar] [CrossRef]
- Du, R.-H.; Wu, F.-F.; Lu, M.; Shu, X.-D.; Ding, J.-H.; Wu, G.; Hu, G. Uncoupling protein 2 modulation of the NLRP3 inflammasome in astrocytes and its implications in depression. Redox Biol. 2016, 9, 178–187. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Che, X.; Zhang, H.; Fan, P.; Tan, G.; Liu, L.; Jiang, D.; Zhao, J.; Xiang, X.; Liang, Y.; et al. Thioredoxin-interacting protein links endoplasmic reticulum stress to inflammatory brain injury and apoptosis after subarachnoid haemorrhage. J. Neuroinflammation 2017, 14, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Muraleedharan, R.; Gawali, M.V.; Tiwari, D.; Sukumaran, A.; Oatman, N.; Anderson, J.; Nardini, D.; Bhuiyan, M.A.N.; Tkáč, I.; Ward, A.L.; et al. AMPK-Regulated Astrocytic Lactate Shuttle Plays a Non-Cell-Autonomous Role in Neuronal Survival. Cell Rep. 2020, 32, 108092. [Google Scholar] [CrossRef] [PubMed]
- Dunn, L.L.; Buckle, A.M.; Cooke, J.P.; Ng, M.K.C. The Emerging Role of the Thioredoxin System in Angiogenesis. Arter. Thromb. Vasc. Biol. 2010, 30, 2089–2098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyandwi, J.B.; Ko, Y.S.; Jin, H.; Yun, S.P.; Park, S.W.; Kim, H.J. Rosmarinic acid inhibits oxLDL-induced inflammasome activation under high-glucose conditions through downregulating the p38-FOXO1-TXNIP pathway. Biochem. Pharmacol. 2020, 182, 114246. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, I.N.; Sheibani, N.; El-Remessy, A.B. Deletion of Thioredoxin-Interacting Protein (TXNIP) Abrogates High Fat Diet-Induced Retinal Leukostasis, Barrier Dysfunction and Microvascular Degeneration in a Mouse Obesity Model. Int. J. Mol. Sci. 2020, 21, 3983. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Kover, K.L.; Heruth, D.P.; Watkins, D.J.; Guo, Y.; Moore, W.V.; He, L.G.; Zang, M.; Clements, M.A.; Yan, Y. Thioredoxin-interacting protein promotes high-glucose-induced macrovascular endothelial dysfunction. Biochem. Biophys. Res. Commun. 2017, 493, 291–297. [Google Scholar] [CrossRef] [Green Version]
- Simó, R.; Villarroel, M.; Corraliza, L.; Hernández, C.; García-Ramírez, M. The Retinal Pigment Epithelium: Something More than a Constituent of the Blood-Retinal Barrier—Implications for the Pathogenesis of Diabetic Retinopathy. J. Biomed. Biotechnol. 2010, 2010, 190724. [Google Scholar] [CrossRef]
- Singh, L.P. Thioredoxin Interacting Protein (TXNIP) and Pathogenesis of Diabetic Retinopathy. J. Clin. Exp. Ophthalmol. 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Devi, T.S.; Yumnamcha, T.; Yao, F.; Somayajulu, M.; Kowluru, R.A.; Singh, L.P. TXNIP mediates high glucose-induced mitophagic flux and lysosome enlargement in human retinal pigment epithelial cells. Biol. Open 2019, 8, bio038521. [Google Scholar] [CrossRef] [Green Version]
- Cho, M.J.; Yoon, S.-J.; Kim, W.; Park, J.; Lee, J.; Park, J.-G.; Cho, Y.-L.; Kim, J.H.; Jang, H.; Park, Y.-J.; et al. Oxidative stress-mediated TXNIP loss causes RPE dysfunction. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Yao, A.; Storr, S.J.; Al-Hadyan, K.; Rahman, R.; Smith, S.; Grundy, R.; Paine, S.; Martin, S.G. Thioredoxin System Protein Expression Is Associated with Poor Clinical Outcome in Adult and Paediatric Gliomas and Medulloblastomas. Mol. Neurobiol. 2020, 57, 2889–2901. [Google Scholar] [CrossRef]
- Morrison, J.A.; Pike, L.A.; Sams, S.B.; Sharma, V.; Zhou, Q.; Severson, J.J.; Tan, A.C.; Wood, W.M.; Haugen, B.R. Thioredoxin interacting protein (TXNIP) is a novel tumor suppressor in thyroid cancer. Mol. Cancer 2014, 13, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koka, S.; Xia, M.; Chen, Y.; Bhat, O.M.; Yuan, X.; Boini, K.M.; Li, P.-L. Endothelial NLRP3 inflammasome activation and arterial neointima formation associated with acid sphingomyelinase during hypercholesterolemia. Redox Biol. 2017, 13, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.-J. Pathogenesis of Chronic Hyperglycemia: From Reductive Stress to Oxidative Stress. J. Diabetes Res. 2014, 2014, 137919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.-W.; Wu, J.; Zhang, Z.; Datta, P.K.; Ibrahimi, I.M.; Taniguchi, S.; Sagara, J.; Fernandes-Alnemri, T.; Alnemri, E.S. Cryopyrin and pyrin activate caspase-1, but not NF-κB, via ASC oligomerization. Cell Death Differ. 2006, 13, 236–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, P.; He, F.-F.; Tang, H.; Lei, C.-T.; Chen, S.; Meng, X.-F.; Su, H.; Zhang, C. NADPH Oxidase-Induced NALP3 Inflammasome Activation Is Driven by Thioredoxin-Interacting Protein Which Contributes to Podocyte Injury in Hyperglycemia. J. Diabetes Res. 2015, 2015, 504761. [Google Scholar] [CrossRef]
- Li, H.; Willingham, S.B.; Ting, J.P.-Y.; Re, F. Cutting Edge: Inflammasome Activation by Alum and Alum’s Adjuvant Effect Are Mediated by NLRP3. J. Immunol. 2008, 181, 17–21. [Google Scholar] [CrossRef] [Green Version]
- Luo, B.; Huang, F.; Liu, Y.; Liang, Y.; Wei, Z.; Ke, H.; Zeng, Z.; Huang, W.; He, Y. NLRP3 Inflammasome as a Molecular Marker in Diabetic Cardiomyopathy. Front. Physiol. 2017, 8, 519. [Google Scholar] [CrossRef] [Green Version]
- Levendusky, M.C.; Basle, J.; Chang, S.; Mandalaywala, N.V.; Voigt, J.M.; Dearborn, R.E.J. Expression and regulation of vitamin D3 upregulated protein 1 (VDUP1) is conserved in mammalian and insect brain. J. Comp. Neurol. 2009, 517, 581–600. [Google Scholar] [CrossRef]
- Aon-Bertolino, M.L.; Romero, J.I.; Galeano, P.; Holubiec, M.; Badorrey, M.S.; Saraceno, G.E.; Hanschmann, E.-M.; Lillig, C.H.; Capani, F. Thioredoxin and glutaredoxin system proteins—Immunolocalization in the rat central nervous system. Biochim. Biophys. Acta Gen. Subj. 2011, 1810, 93–110. [Google Scholar] [CrossRef]
- Blouet, C.; Schwartz, G.J. Nutrient-Sensing Hypothalamic TXNIP Links Nutrient Excess to Energy Imbalance in Mice. J. Neurosci. 2011, 31, 6019–6027. [Google Scholar] [CrossRef] [Green Version]
- Blouet, C.; Liu, S.-M.; Jo, Y.-H.; Chua, S.; Schwartz, G.J. TXNIP in Agrp Neurons Regulates Adiposity, Energy Expenditure, and Central Leptin Sensitivity. J. Neurosci. 2012, 32, 9870–9877. [Google Scholar] [CrossRef] [PubMed]
- Hand, L.E.; Saer, B.R.C.; Hui, S.T.; Jinnah, H.A.; Steinlechner, S.; Loudon, A.S.I.; Bechtold, D.A. Induction of the Metabolic Regulator Txnip in Fasting-Induced and Natural Torpor. Endocrinology 2013, 154, 2081–2091. [Google Scholar] [CrossRef] [PubMed]
- Ishrat, T.; Mohamed, I.N.; Pillai, B.; Soliman, S.; Fouda, A.Y.; Ergul, A.; El-Remessy, A.B.; Fagan, S.C. Thioredoxin-Interacting Protein: A Novel Target for Neuroprotection in Experimental Thromboembolic Stroke in Mice. Mol. Neurobiol. 2015, 51, 766–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Li, R.; Wang, X.; Fu, Q.; Ma, S. Umbelliferone ameliorates cerebral ischemia–reperfusion injury via upregulating the PPAR gamma expression and suppressing TXNIP/NLRP3 inflammasome. Neurosci. Lett. 2015, 600, 182–187. [Google Scholar] [CrossRef]
- Gamdzyk, M.; Doycheva, D.M.; Malaguit, J.; Enkhjargal, B.; Tang, J.; Zhang, J.H. Role of PPAR-β/δ/miR-17/TXNIP pathway in neuronal apoptosis after neonatal hypoxic-ischemic injury in rats. Neuropharmacology 2018, 140, 150–161. [Google Scholar] [CrossRef]
- Gamdzyk, M.; Doycheva, D.M.; Kang, R.; Tang, H.; Travis, Z.D.; Tang, J.; Zhang, J.H. GW0742 activates miR-17-5p and inhibits TXNIP/NLRP3-mediated inflammation after hypoxic-ischaemic injury in rats and in PC12 cells. J. Cell. Mol. Med. 2020, 24, 12318–12330. [Google Scholar] [CrossRef]
- Ding, R.; Ou, W.; Chen, C.; Liu, Y.; Li, H.; Zhang, X.; Chai, H.; Ding, X.; Wang, Q. Endoplasmic reticulum stress and oxidative stress contribute to neuronal pyroptosis caused by cerebral venous sinus thrombosis in rats: Involvement of TXNIP/peroxynitrite-NLRP3 inflammasome activation. Neurochem. Int. 2020, 141, 104856. [Google Scholar] [CrossRef]
- Su, C.-J.; Feng, Y.; Liu, T.-T.; Liu, X.; Bao, J.-J.; Shi, A.-M.; Hu, D.-M.; Liu, T.; Yu, Y.-L. Thioredoxin-interacting protein induced α-synuclein accumulation via inhibition of autophagic flux: Implications for Parkinson’s disease. CNS Neurosci. Ther. 2017, 23, 717–723. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Bharti, V.; Zhou, H.; Hoi, V.; Tan, H.; Wu, Z.; Nagakannan, P.; Eftekharpour, E.; Wang, J.-F. Upregulation of Thioredoxin-Interacting Protein in Brain of Amyloid-β Protein Precursor/Presenilin 1 Transgenic Mice and Amyloid-β Treated Neuronal Cells. J. Alzheimers Dis. 2019, 72, 139–150. [Google Scholar] [CrossRef]
- Zhou, H.; Tan, H.; Letourneau, L.; Wang, J.-F. Increased thioredoxin-interacting protein in brain of mice exposed to chronic stress. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 88, 320–326. [Google Scholar] [CrossRef]
- Pan, Q.; Guo, K.; Xue, M.; Tu, Q. Estrogen protects neuroblastoma cell from amyloid-β 42 (Aβ42)-induced apoptosis via TXNIP/TRX axis and AMPK signaling. Neurochem. Int. 2020, 135, 104685. [Google Scholar] [CrossRef] [PubMed]
- Fertan, E.; Rodrigues, G.J.; Wheeler, R.V.; Goguen, D.; Wong, A.A.; James, H.; Stadnyk, A.; Brown, R.E.; Weaver, I.C.G. Cognitive Decline, Cerebral-Spleen Tryptophan Metabolism, Oxidative Stress, Cytokine Production, and Regulation of the Txnip Gene in a Triple Transgenic Mouse Model of Alzheimer Disease. Am. J. Pathol. 2019, 189, 1435–1450. [Google Scholar] [CrossRef] [PubMed]
- Melone, M.A.B.; Dato, C.; Paladino, S.; Coppola, C.; Trebini, C.; Giordana, M.T.; Perrone, L. Verapamil Inhibits Ser202/Thr205 Phosphorylation of Tau by Blocking TXNIP/ROS/p38 MAPK Pathway. Pharm. Res. 2018, 35, 44. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Lučiūnaitė, A.; McManus, R.M.; Jankunec, M.; Rácz, I.; Dansokho, C.; Dalgėdienė, I.; Schwartz, S.; Brosseron, F.; Heneka, M.T. Soluble Aβ oligomers and protofibrils induce NLRP3 inflammasome activation in microglia. J. Neurochem. 2019, e14945. [Google Scholar] [CrossRef] [Green Version]
- Beyer, M.M.S.; Lonnemann, N.; Remus, A.; Latz, E.; Heneka, M.; Korte, M. Enduring Changes in Neuronal Function upon Systemic Inflammation Are NLRP3 Inflammasome Dependent. J. Neurosci. 2020, 40, 5480–5494. [Google Scholar] [CrossRef]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef]
- Mahmoudian, E.; Khalilnezhad, A.; Gharagozli, K.; Amani, D. Thioredoxin-1, redox factor-1 and thioredoxin-interacting protein, mRNAs are differentially expressed in Multiple Sclerosis patients exposed and non-exposed to interferon and immunosuppressive treatments. Gene 2017, 634, 29–36. [Google Scholar] [CrossRef]
- Patwari, P.; Chutkow, W.A.; Cummings, K.; Verstraeten, V.L.R.M.; Lammerding, J.; Schreiter, E.R.; Lee, R.T. Thioredoxin-independent Regulation of Metabolism by the α-Arrestin Proteins. J. Biol. Chem. 2009, 284, 24996–25003. [Google Scholar] [CrossRef] [Green Version]
- Wu, N.; Zheng, B.; Shaywitz, A.; Dagon, Y.; Tower, C.; Bellinger, G.; Shen, C.-H.; Wen, J.; Asara, J.; McGraw, T.E.; et al. AMPK-Dependent Degradation of TXNIP upon Energy Stress Leads to Enhanced Glucose Uptake via GLUT1. Mol. Cell 2013, 49, 1167–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, D.F.; Kuhl, D.E.; Hawkins, R.A.; Phelps, M.E.; Cummings, J.L.; Tsai, S.Y. The Fluorodeoxyglucose 18F Scan in Alzheimer’s Disease and Multi-infarct Dementia. Arch. Neurol. 1983, 40, 711–714. [Google Scholar] [CrossRef] [PubMed]
- Koepsell, H. Glucose transporters in brain in health and disease. Pflügers Arch. Eur. J. Physiol. 2020, 472, 1299–1343. [Google Scholar] [CrossRef] [PubMed]
- De La Monte, S.M. The Full Spectrum of Alzheimer’s Disease Is Rooted in Metabolic Derangements That Drive Type 3 Diabetes. Adv. Exp. Med. Biol. 2019, 1128, 45–83. [Google Scholar] [CrossRef]
- Bendlin, B.B. Antidiabetic therapies and Alzheimer disease. Dialogues Clin. Neurosci. 2019, 21, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Malek-Ahmadi, M.; Beach, T.; Obradov, A.; Sue, L.; Belden, C.M.; Davis, K.; Walker, D.G.; Lue, L.-F.; Adem, A.; Sabbagh, M.N. Increased Alzheimer’s Disease Neuropathology is Associated with Type 2 Diabetes and ApoE ε4 Carrier Status. Curr. Alzheimer Res. 2013, 10, 654–659. [Google Scholar] [CrossRef]
- Cardoso, S.; Moreira, P.I. Antidiabetic drugs for Alzheimer’s and Parkinson’s diseases: Repurposing insulin, metformin, and thiazolidinediones. Int. Rev. Neurobiol. 2020, 155, 37–64. [Google Scholar] [CrossRef]
- Wu, C.-Y.; Ouk, M.; Wong, Y.Y.; Anita, N.Z.; Edwards, J.D.; Yang, P.; Shah, B.R.; Herrmann, N.; Lanctôt, K.L.; Kapral, M.K.; et al. Relationships between memory decline and the use of metformin or DPP4 inhibitors in people with type 2 diabetes with normal cognition or Alzheimer’s disease, and the role APOE carrier status. Alzheimers Dement. 2020. [Google Scholar] [CrossRef]
- Shi, Q.; Liu, S.; Fonseca, V.A.; Thethi, T.K.; Shi, L. Effect of metformin on neurodegenerative disease among elderly adult US veterans with type 2 diabetes mellitus. BMJ Open 2019, 9, e024954. [Google Scholar] [CrossRef] [Green Version]
- Tang, G.; Duan, F.; Li, W.; Wang, Y.; Zeng, C.; Hu, J.; Li, H.; Zhang, X.; Chen, Y.; Tan, H. Metformin inhibited Nod-like receptor protein 3 inflammasomes activation and suppressed diabetes-accelerated atherosclerosis in apoE(-/-) mice. Biomed. Pharmacother. 2019, 119, 109410. [Google Scholar] [CrossRef]
- Hou, X.; Song, J.; Li, X.-N.; Zhang, L.; Wang, X.; Chen, L.; Shen, Y.H. Metformin reduces intracellular reactive oxygen species levels by upregulating expression of the antioxidant thioredoxin via the AMPK-FOXO3 pathway. Biochem. Biophys. Res. Commun. 2010, 396, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Shaked, M.; Ketzinel-Gilad, M.; Cerasi, E.; Kaiser, N.; Leibowitz, G. AMP-Activated Protein Kinase (AMPK) Mediates Nutrient Regulation of Thioredoxin-Interacting Protein (TXNIP) in Pancreatic Beta-Cells. PLoS ONE 2011, 6, e28804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.; Cui, R.; Wang, C.; Feng, Y.; Li, Z.; Tong, Y.; Qu, K.; Liu, C.; Zhang, J. Metformin protects against intestinal ischemia-reperfusion injury and cell pyroptosis via TXNIP-NLRP3-GSDMD pathway. Redox Biol. 2020, 32, 101534. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, J.S.; Chatterjee, A.; Castellani, L.W.; Ross, D.A.; Ohmen, J.; Cavalcoli, J.; Wu, C.; Dains, K.M.; Catanese, J.; Chu, M.; et al. Positional cloning of the combined hyperlipidemia gene Hyplip1. Nat. Genet. 2002, 30, 110–116. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, K.L.; Margosian, M.R.; Sheth, S.S.; Lusis, A.J.; Parks, E.J. Increased lipogenesis and fatty acid reesterification contribute to hepatic triacylglycerol stores in hyperlipidemic Txnip-/- mice. J. Nutr. 2004, 134, 1475–1480. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.N.; Kang, H.-S.; Jeon, J.-H.; Kim, E.-M.; Yoon, S.-R.; Song, H.; Lyu, C.-Y.; Piao, Z.-H.; Kim, S.-U.; Han, Y.-H.; et al. VDUP1 Is Required for the Development of Natural Killer Cells. Immunity 2005, 22, 195–208. [Google Scholar] [CrossRef] [Green Version]
- Okuyama, H.; Yoshida, T.; Son, A.; Oka, S.-I.; Wang, D.; Nakayama, R.; Masutani, H.; Nakamura, H.; Nabeshima, Y.-I.; Yodoi, J. Thioredoxin Binding Protein 2 Modulates Natural Killer T Cell-Dependent Innate Immunity in the Liver: Possible Link to Lipid Metabolism. Antioxid. Redox Signal. 2009, 11, 2585–2593. [Google Scholar] [CrossRef]
- Son, A.; Nakamura, H.; Okuyama, H.; Oka, S.-I.; Yoshihara, E.; Liu, W.; Matsuo, Y.; Kondo, N.; Masutani, H.; Ishii, Y.; et al. Dendritic cells derived from TBP-2-deficient mice are defective in inducing T cell responses. Eur. J. Immunol. 2008, 38, 1358–1367. [Google Scholar] [CrossRef]
- Oka, S.-I.; Yoshihara, E.; Bizen-Abe, A.; Liu, W.; Watanabe, M.; Yodoi, J.; Masutani, H. Thioredoxin Binding Protein-2/Thioredoxin-Interacting Protein Is a Critical Regulator of Insulin Secretion and Peroxisome Proliferator-Activated Receptor Function. Endocrinology 2008, 150, 1225–1234. [Google Scholar] [CrossRef] [Green Version]
- Chutkow, W.A.; Birkenfeld, A.L.; Brown, J.D.; Lee, H.-Y.; Frederick, D.W.; Yoshioka, J.; Patwari, P.; Kursawe, R.; Cushman, S.W.; Plutzky, J.; et al. Deletion of the α-Arrestin Protein Txnip in Mice Promotes Adiposity and Adipogenesis while Preserving Insulin Sensitivity. Diabetes 2010, 59, 1424–1434. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Ren, Y.; Zhao, L.; Du, C.; Wang, Y.; Zhang, Y.; Li, Y.; Zhao, S.; Duan, H. Knockdown of thioredoxin interacting protein attenuates high glucose-induced apoptosis and activation of ASK1 in mouse mesangial cells. FEBS Lett. 2011, 585, 1789–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Y.; Shi, Y.; Wang, Y.; Li, Y.; Wu, S.; Li, H.; Zhang, Y.; Duan, H. p38 MAPK pathway is involved in high glucose-induced thioredoxin interacting protein induction in mouse mesangial cells. FEBS Lett. 2010, 584, 3480–3485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, A.; Xia, L.; Masson, E.A.Y.; Gui, C.; Momen, A.; Shikatani, E.A.; Husain, M.; Quaggin, S.; John, R.; Fantus, I.G. Thioredoxin-Interacting Protein Deficiency Protects against Diabetic Nephropathy. J. Am. Soc. Nephrol. 2015, 26, 2963–2977. [Google Scholar] [CrossRef] [PubMed]
- Elshaer, S.L.; Mohamed, I.N.; Coucha, M.; Altantawi, S.; Eldahshan, W.; Bartasi, M.L.; Shanab, A.Y.; Lorys, R.; El-Remessy, A.B. Deletion of TXNIP Mitigates High-Fat Diet-Impaired Angiogenesis and Prevents Inflammation in a Mouse Model of Critical Limb Ischemia. Antioxidants 2017, 6, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsu-Jiménez, Y.; Vázquez-Calvo, C.; Maffezzini, C.; Halldin, M.; Peng, X.; Freyer, C.; Wredenberg, A.; Giménez-Cassina, A.; Wedell, A.; Arnér, E.S.J. Absence of TXNIP in Humans Leads to Lactic Acidosis and Low Serum Methionine Linked to Deficient Respiration on Pyruvate. Diabetes 2019, 68, 709–723. [Google Scholar] [CrossRef] [Green Version]
- Kaya, B.; Erdi, F.; Kılınc, I.; Keskin, F.; Feyzıoglu, B.; Esen, H.; Karataş, Y.; Uyar, M.; Kalkan, E.; Kilinc, I.; et al. Alterations of the thioredoxin system during subarachnoid hemorrhage-induced cerebral vasospasm. Acta Neurochir. 2015, 157, 793–800. [Google Scholar] [CrossRef]
- Yamagishi, K.; Yamamoto, K.; Mochizuki, Y.; Nakano, T.; Yamada, S.; Tokiwa, H. Flexible ligand recognition of peroxisome proliferator-activated receptor-gamma (PPARgamma). Bioorg. Med. Chem. Lett. 2010, 20, 3344–3347. [Google Scholar] [CrossRef]
- Qi, W.; Chen, X.; Holian, J.; Tan, C.Y.R.; Kelly, D.J.; Pollock, C.A. Transcription Factors Krüppel-Like Factor 6 and Peroxisome Proliferator-Activated Receptor-γ Mediate High Glucose-Induced Thioredoxin-Interacting Protein. Am. J. Pathol. 2009, 175, 1858–1867. [Google Scholar] [CrossRef] [Green Version]
- Shaked, M.; Ketzinel-Gilad, M.; Ariav, Y.; Cerasi, E.; Kaiser, N.; Leibowitz, G. Insulin counteracts glucotoxic effects by suppressing thioredoxin-interacting protein production in INS-1E beta cells and in Psammomys obesus pancreatic islets. Diabetologia 2009, 52, 636–644. [Google Scholar] [CrossRef] [Green Version]
- Nagaraj, K.; Lapkina-Gendler, L.; Sarfstein, R.; Gurwitz, D.; Pasmanik-Chor, M.; Laron, Z.; Yakar, S.; Werner, H. Identification of thioredoxin-interacting protein (TXNIP) as a downstream target for IGF1 action. Proc. Natl. Acad. Sci. USA 2018, 115, 1045–1050. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Cha-Molstad, H.; Szabo, A.; Shalev, A. Diabetes induces and calcium channel blockers prevent cardiac expression of proapoptotic thioredoxin-interacting protein. Am. J. Physiol. Metab. 2009, 296, E1133–E1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha-Molstad, H.; Xu, G.; Chen, J.; Jing, G.; Young, M.E.; Chatham, J.C.; Shalev, A. Calcium channel blockers act through nuclear factor Y to control transcription of key cardiac genes. Mol. Pharmacol. 2012, 82, 541–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buyandelger, U.; Walker, D.G.; Taguchi, H.; Yanagisawa, D.; Tooyama, I. Novel fluorinated derivative of curcumin negatively regulates thioredoxin-interacting protein expression in retinal pigment epithelial and macrophage cells. Biochem. Biophys. Res. Commun. 2020, 532, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Yang, Z.; Sun, Z.; Zhang, W.; Chen, X.; Nie, S. Curcumin relieves paraquat-induced lung injury through inhibiting the thioredoxin interacting protein/NLR pyrin domain containing 3-mediated inflammatory pathway. Mol. Med. Rep. 2019, 20, 5032–5040. [Google Scholar] [CrossRef]
- Lu, X.; Wu, F.; Jiang, M.; Sun, X.; Tian, G. Curcumin ameliorates gestational diabetes in mice partly through activating AMPK. Pharm. Biol. 2019, 57, 250–254. [Google Scholar] [CrossRef] [Green Version]
- Hoshikawa, H.; Kamitori, K.; Indo, K.; Mori, T.; Kamata, M.; Takahashi, T.; Tokuda, M. Combined treatment with D-allose, docetaxel and radiation inhibits the tumor growth in an in vivo model of head and neck cancer. Oncol. Lett. 2018, 15, 3422–3428. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Finding | Reference |
|---|---|
| TXNIP induced in cerebellar granular neurons by changes in potassium led to cell death | [57] |
| Glutamate-induced cell death in vivo and in vitro via TXNIP induced expression | [81] |
| TXNIP expression downregulated in vitro in neuronal cells overexpressing mitochondrial ferritin | [82] |
| TXNIP induction by Aβ in microglia resulting in activation inhibited by antioxidant resveratrol | [84] |
| High-fat-diet fed rats had increased expression of TXNIP in astrocytes, retinal endothelial cells, and microglia | [88] |
| Increased TXNIP expression in a rat model of subarachnoid hemorrhage in neurons, astrocytes, and microglia correlated with cell death, which was reduced by inhibiting TXNIP expression | [156] |
| Increased TXNIP expression in a rat model of subarachnoid hemorrhage in neurons, astrocytes, and microglia correlated with cell death which was reduced by inhibiting TXNIP expression | [156] |
| Downregulation of TXNIP in RPE cell line by oxidative stress reduced cell proliferation | [99] |
| Decreased expression of TXNIP in glioma cells correlated with poor patient outcome | [100] |
| TXNIP was immunolocalized to neurons and microglia in normal drosophila and rat brain tissue | [108] |
| Increased neuronal pyroptosis in a rat model of cerebral thrombosis mediated by elevated TXNIP expression and inflammasome activation | [117] |
| TXNIP overexpression in substantia nigra of mice caused the loss of dopaminergic neurons | [118] |
| Increased expression of TXNIP mRNA, protein, and numbers of immunopositive cells in AD brain | [26] |
| Increased TXNIP expression in the brain of an AD mouse model but reduced expression in the spleen | [122] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsubaki, H.; Tooyama, I.; Walker, D.G. Thioredoxin-Interacting Protein (TXNIP) with Focus on Brain and Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 9357. https://doi.org/10.3390/ijms21249357
Tsubaki H, Tooyama I, Walker DG. Thioredoxin-Interacting Protein (TXNIP) with Focus on Brain and Neurodegenerative Diseases. International Journal of Molecular Sciences. 2020; 21(24):9357. https://doi.org/10.3390/ijms21249357
Chicago/Turabian StyleTsubaki, Haruka, Ikuo Tooyama, and Douglas Gordon Walker. 2020. "Thioredoxin-Interacting Protein (TXNIP) with Focus on Brain and Neurodegenerative Diseases" International Journal of Molecular Sciences 21, no. 24: 9357. https://doi.org/10.3390/ijms21249357
APA StyleTsubaki, H., Tooyama, I., & Walker, D. G. (2020). Thioredoxin-Interacting Protein (TXNIP) with Focus on Brain and Neurodegenerative Diseases. International Journal of Molecular Sciences, 21(24), 9357. https://doi.org/10.3390/ijms21249357