Clinical Heterogeneity in Autosomal Recessive Bestrophinopathy with Biallelic Mutations in the BEST1 Gene

 , , , ,
, , , ,  ,
,
Abstract
:1. Introduction
2. Results
2.1. Patient History
2.2. Clinical Findings
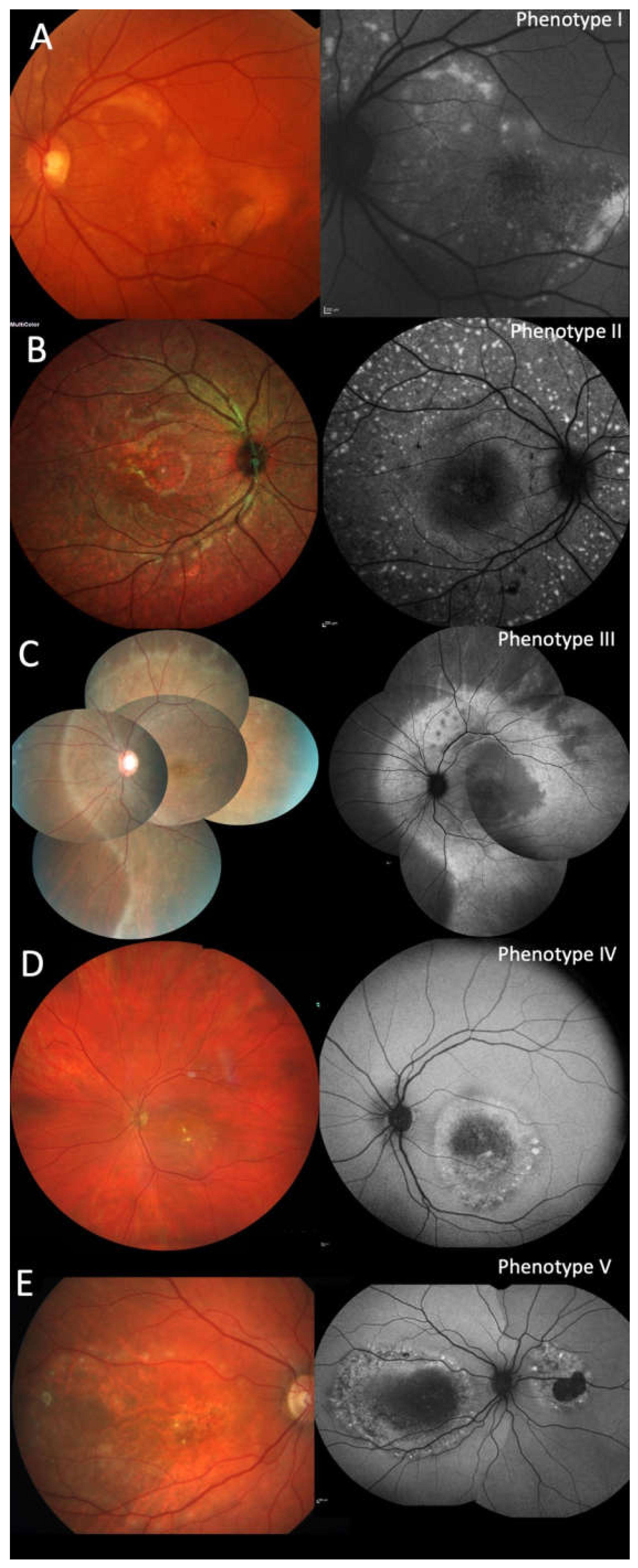
2.3. Clinical Phenotypes
2.4. Fundus Autofluorescence
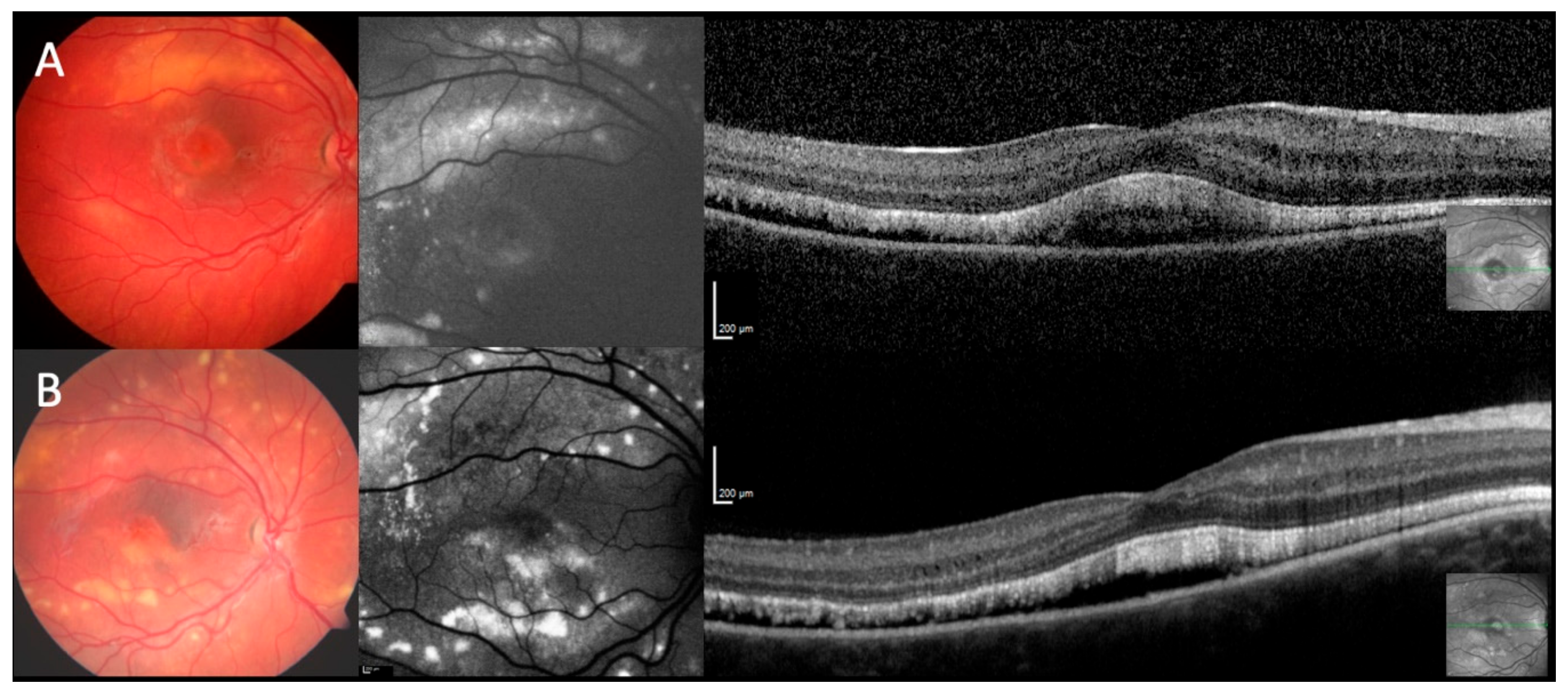
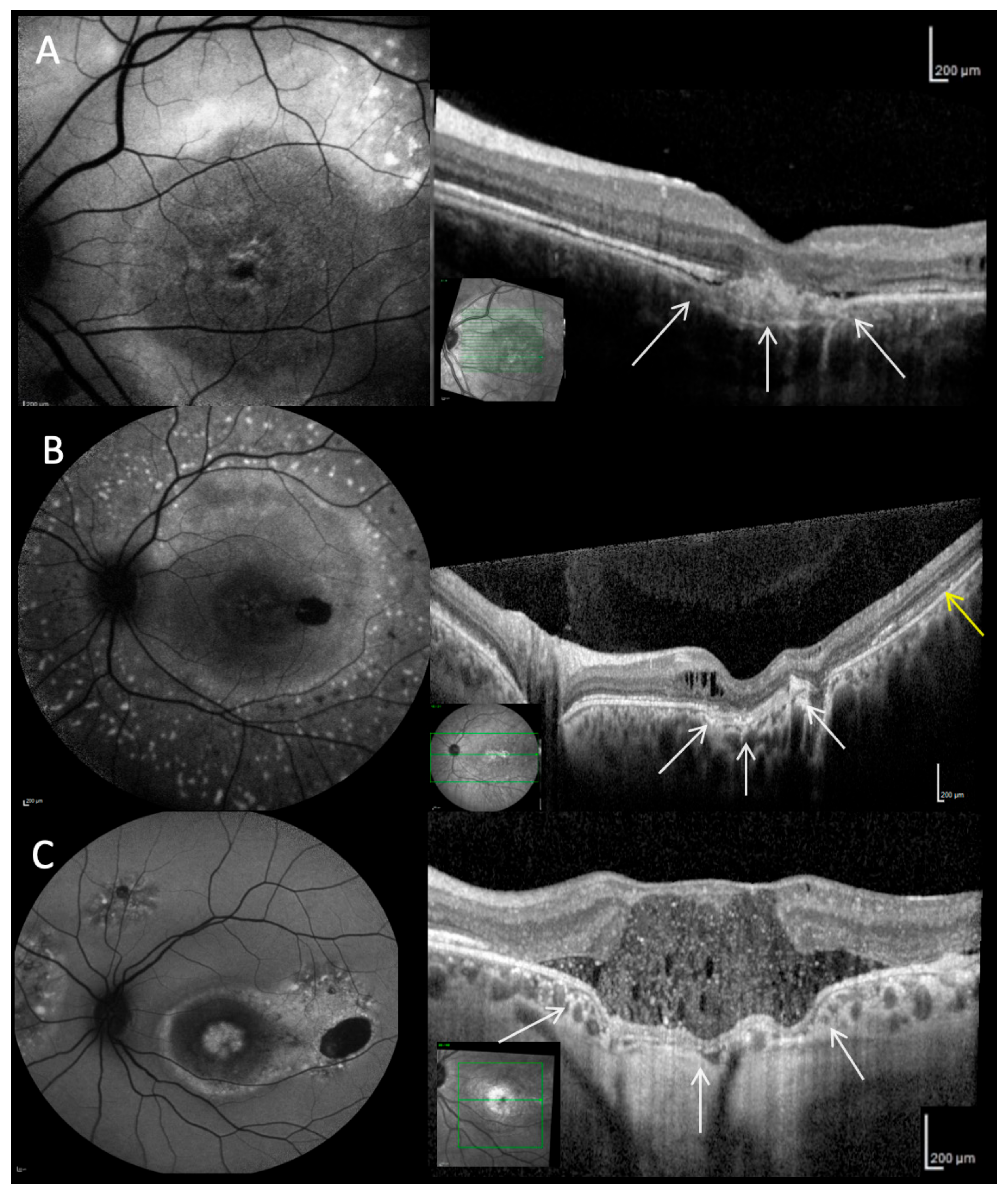
2.5. Optical Coherence Tomography
2.6. Fluorescein Angiography
2.7. Electrophysiology
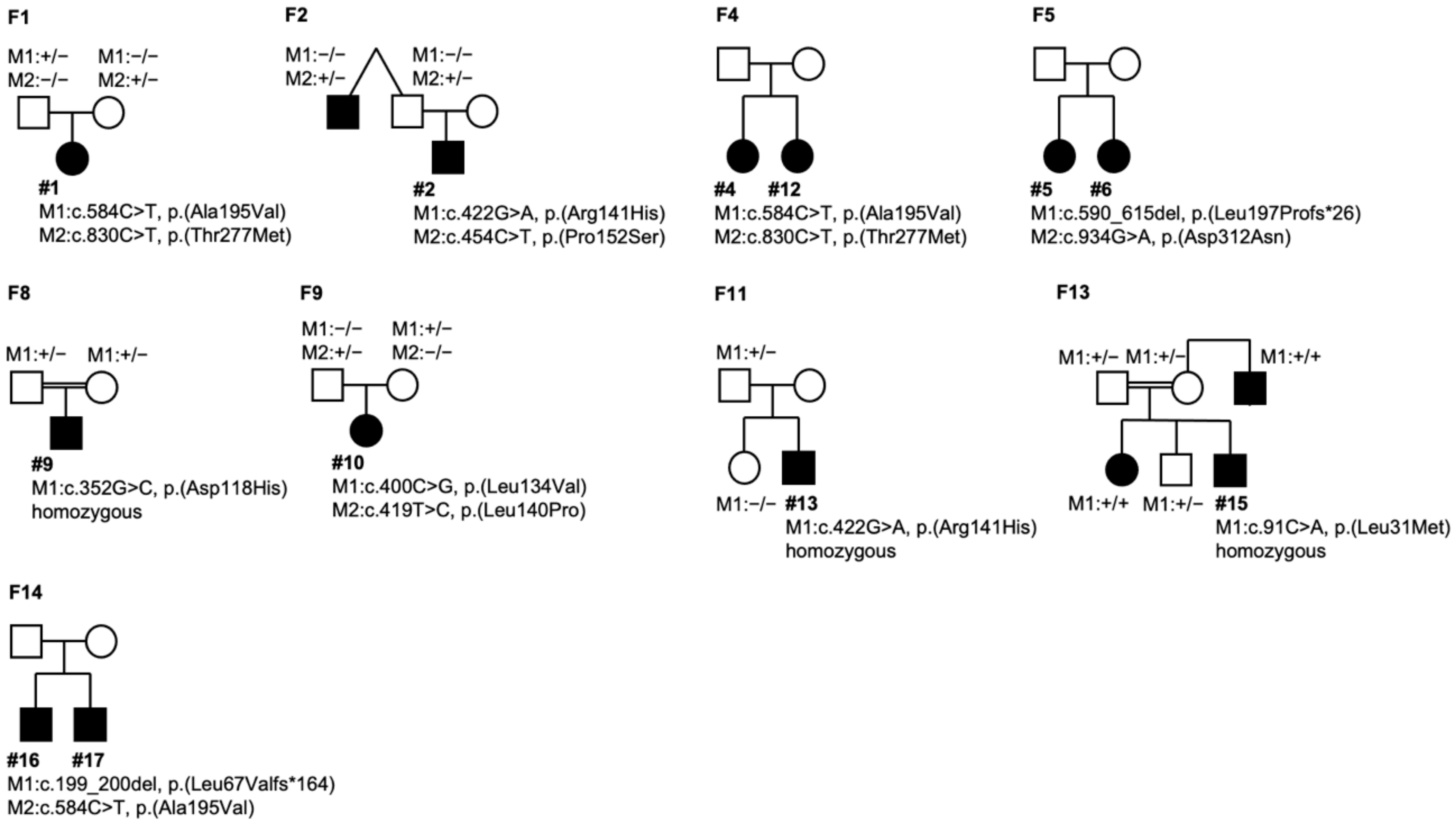
2.8. Molecular Genetics
3. Discussion
4. Materials and Methods
4.1. Ophthalmological Examination
4.2. Molecular Genetic Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADVIRC | Autosomal Dominant Vitreo-Retinochoroidopathy |
| ARB | Autosomal Recessive Bestrophinopathy |
| BCVA | Best-corrected visual acuity |
| BVMD | Best vitelliform macular dystrophy |
| CNV | Choroidal neovascularization |
| EOG | Electro-oculography |
| ERG | Full-field electroretinography |
| FAF | Fundus autofluorescence |
| mfERG | Multifocal electroretinography |
| MRCS | MRCS syndrome (microcornea, retinal dystrophy, cataract, posterior staphyloma) |
| OCT | Optical Coherence Tomography |
| PCR | Polymerase chain reaction |
| RPE | Retinal pigment epithelium |
References
- Petrukhin, K.; Koisti, M.J.; Bakall, B.; Li, W.; Xie, G.; Marknell, T.; Sandgren, O.; Forsman, K.; Holmgren, G.; Andreasson, S.; et al. Identification of the gene responsible for Best macular dystrophy. Nat. Genet. 1998, 19, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Yardley, J.; Leroy, B.P.; Hart-Holden, N.; Lafaut, B.A.; Loeys, B.; Messiaen, L.M.; Perveen, R.; Reddy, M.A.; Bhattacharya, S.S.; Traboulsi, E.; et al. Mutations of VMD2 splicing regulators cause nanophthalmos and autosomal dominant vitreoretinochoroidopathy (ADVIRC). Invest. Ophthalmol. Vis. Sci. 2004, 45, 3683–3689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaelides, M.; Urquhart, J.; Holder, G.E.; Restori, M.; Kayali, N.; Manson, F.D.C.; Black, G.C.M. Evidence of genetic heterogeneity in MRCS (microcornea, rod-cone dystrophy, cataract, and posterior staphyloma) syndrome. Am. J. Ophthalmol. 2006, 141, 418–420. [Google Scholar] [CrossRef] [PubMed]
- Krämer, F.; White, K.; Pauleikhoff, D.; Gehrig, A.; Passmore, L.; Rivera, A.; Rudolph, G.; Kellner, U.; Andrassi, M.; Lorenz, B.; et al. Mutations in the VMD2 gene are associated with juvenile-onset vitelliform macular dystrophy (Best disease) and adult vitelliform macular dystrophy but not age-related macular degeneration. Eur. J. Hum. Genet. 2000, 8, 286–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schatz, P.; Klar, J.; Andréasson, S.; Ponjavic, V.; Dahl, N. Variant phenotype of Best vitelliform macular dystrophy associated with compound heterozygous mutations in VMD2. Ophthalmic Genet. 2006, 27, 51–56. [Google Scholar] [CrossRef]
- Burgess, R.; Millar, I.D.; Leroy, B.P.; Urquhart, J.E.; Fearon, I.M.; De Baere, E.; Brown, P.D.; Robson, A.G.; Wright, G.A.; Kestelyn, P.; et al. Biallelic mutation of BEST1 causes a distinct retinopathy in humans. Am. J. Hum. Genet. 2008, 82, 19–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquardt, A.; Stöhr, H.; Passmore, L.A.; Krämer, F.; Rivera, A.; Weber, B.H. Mutations in a novel gene, VMD2, encoding a protein of unknown properties cause juvenile-onset vitelliform macular dystrophy (Best’s disease). Hum. Mol. Genet. 1998, 7, 1517–1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Tsunenari, T.; Yau, K.-W.; Nathans, J. The vitelliform macular dystrophy protein defines a new family of chloride channels. Proc. Natl. Acad. Sci. USA 2002, 99, 4008–4013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milenkovic, A.; Brandl, C.; Milenkovic, V.M.; Jendryke, T.; Sirianant, L.; Wanitchakool, P.; Zimmermann, S.; Reiff, C.M.; Horling, F.; Schrewe, H.; et al. Bestrophin 1 is indispensable for volume regulation in human retinal pigment epithelium cells. Proc. Natl. Acad. Sci. USA 2015, 112, E2630–E2639. [Google Scholar] [CrossRef] [Green Version]
- Hartzell, H.C.; Qu, Z.; Yu, K.; Xiao, Q.; Chien, L.-T. Molecular physiology of bestrophins: Multifunctional membrane proteins linked to best disease and other retinopathies. Physiol. Rev. 2008, 88, 639–672. [Google Scholar] [CrossRef]
- Marmorstein, A.D.; Kinnick, T.R. Focus on molecules: Bestrophin (best-1). Exp. Eye Res. 2007, 85, 423–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nachtigal, A.-L.; Milenkovic, A.; Brandl, C.; Schulz, H.L.; Duerr, L.M.J.; Lang, G.E.; Reiff, C.; Herrmann, P.; Kellner, U.; Weber, B.H.F. Mutation-Dependent Pathomechanisms Determine the Phenotype in the Bestrophinopathies. Int. J. Mol. Sci. 2020, 21, 1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milenkovic, A.; Milenkovic, V.M.; Wetzel, C.H.; Weber, B.H.F. BEST1 protein stability and degradation pathways differ between autosomal dominant Best disease and autosomal recessive bestrophinopathy accounting for the distinct retinal phenotypes. Hum. Mol. Genet. 2018, 27, 1630–1641. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, A.; Ueno, S.; Hayashi, T.; Katagiri, S.; Kominami, T.; Ito, Y.; Gekka, T.; Masuda, Y.; Tsuneoka, H.; Shinoda, K.; et al. Clinical and Genetic Findings of Autosomal Recessive Bestrophinopathy in Japanese Cohort. Am. J. Ophthalmol. 2016, 168, 86–94. [Google Scholar] [CrossRef]
- Luo, J.; Lin, M.; Guo, X.; Xiao, X.; Li, J.; Hu, H.; Xiao, H.; Xu, X.; Zhong, Y.; Long, S.; et al. Novel BEST1 mutations and special clinical characteristics of autosomal recessive bestrophinopathy in Chinese patients. Acta Ophthalmol. 2019, 97, 247–259. [Google Scholar] [CrossRef]
- Shah, M.; Broadgate, S.; Shanks, M.; Clouston, P.; Yu, J.; MacLaren, R.E.; Németh, A.H.; Halford, S.; Downes, S.M. Association of Clinical and Genetic Heterogeneity with BEST1 Sequence Variations. JAMA Ophthalmol. 2020, 138, 544–551. [Google Scholar] [CrossRef]
- Jaffal, L.; Joumaa, W.H.; Assi, A.; Helou, C.; Condroyer, C.; El Dor, M.; Cherfan, G.; Zeitz, C.; Audo, I.; Zibara, K.; et al. Novel Missense Mutations in BEST1 Are Associated with Bestrophinopathies in Lebanese Patients. Genes 2019, 10, 151. [Google Scholar] [CrossRef] [Green Version]
- Casalino, G.; Khan, K.N.; Armengol, M.; Wright, G.; Pontikos, N.; Georgiou, M.; Webster, A.R.; Robson, A.G.; Grewal, P.S.; Michaelidis, M. Autosomal recessive bestrophinopathy: Clinical features, natural history and genetic findings in preparation for clinical trials. Ophthalmology 2020. online ahead of print. [Google Scholar] [CrossRef]
- Milenkovic, A.; Brandl, C.; Nachtigal, A.-L.; Kellner, U.; Weber, B.H.F. Mutation-Dependent Mechanisms and Their Impact on Targeted Therapeutic Strategies with Reference to Bestrophin 1 and the Bestrophinopathies. Klin. Monbl. Augenheilkd. 2020, 237, 259–266. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Tian, L.; Sun, T.; Xu, K.; Zhang, X.; Peng, X.; Li, Y. Screening of BEST1 Gene in a Chinese Cohort with Best Vitelliform Macular Dystrophy or Autosomal Recessive Bestrophinopathy. Invest. Ophthalmol. Vis. Sci. 2017, 58, 3366–3375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, F.-J.; Qi, Y.-H.; Hu, F.-Y.; Wang, D.-D.; Xu, P.; Guo, J.-L.; Li, J.-K.; Zhang, Y.-J.; Li, W.; Chen, F.; et al. Mutation spectrum of the bestrophin-1 gene in a large Chinese cohort with bestrophinopathy. Br. J. Ophthalmol. 2020, 104, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Zatreanu, L.; Freund, K.B.; Leong, B.C.S.; Yu, H.G.; Teke, M.Y.; Yzer, S.; Sadda, S.R.; Sarraf, D. Serous macular detachment in best disease: A masquerade syndrome. Retina 2020, 40, 1456–1470. [Google Scholar] [CrossRef] [PubMed]
- Boon, C.J.F.; van den Born, L.I.; Visser, L.; Keunen, J.E.E.; Bergen, A.A.B.; Booij, J.C.; Riemslag, F.C.; Florijn, R.J.; van Schooneveld, M.J. Autosomal recessive bestrophinopathy: Differential diagnosis and treatment options. Ophthalmology 2013, 120, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Boon, C.J.F.; Klevering, B.J.; Leroy, B.P.; Hoyng, C.B.; Keunen, J.E.E.; den Hollander, A.I. The spectrum of ocular phenotypes caused by mutations in the BEST1 gene. Prog. Retin. Eye Res. 2009, 28, 187–205. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Poornachandra, B.; Verma, A.; Mehta, R.A.; Phalke, S.; Battu, R.; Ramprasad, V.L.; Peterson, A.S.; Ghosh, A.; Seshagiri, S. Next generation sequencing identifies novel disease-associated BEST1 mutations in Bestrophinopathy patients. Sci. Rep. 2018, 8, 10176. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.; Yang, G.; Wang, J.; Chen, Y. Screening for BEST1 gene mutations in Chinese patients with bestrophinopathy. Mol. Vis. 2014, 20, 1594–1604. [Google Scholar]
- Davidson, A.E.; Millar, I.D.; Urquhart, J.E.; Burgess-Mullan, R.; Shweikh, Y.; Parry, N.; O’Sullivan, J.; Maher, G.J.; McKibbin, M.; Downes, S.M.; et al. Missense mutations in a retinal pigment epithelium protein, bestrophin-1, cause retinitis pigmentosa. Am. J. Hum. Genet. 2009, 85, 581–592. [Google Scholar] [CrossRef] [Green Version]
- Chibani, Z.; Abid, I.Z.; Molbaek, A.; Söderkvist, P.; Feki, J.; Hmani-Aifa, M. Novel BEST1 gene mutations associated with two different forms of macular dystrophy in Tunisian families. Clin. Exp. Ophthalmol. 2019, 47, 1063–1073. [Google Scholar] [CrossRef]
- Hirawat, R.S.; Nagesha, C.K.; Divakar, M.M. Autosomal recessive bestrophinopathy with macular hole. Indian J. Ophthalmol. 2020, 68, 1173–1175. [Google Scholar] [CrossRef]
- Cascavilla, M.L.; Querques, G.; Stenirri, S.; Battaglia Parodi, M.; Querques, L.; Bandello, F. Unilateral vitelliform phenotype in autosomal recessive bestrophinopathy. Ophthalmic Res. 2012, 48, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Chatra, K. Fibrotic pillar leads to focal choroidal excavation in Best vitelliform dystrophy. Graefes Arch. Clin. Exp. Ophthalmol. 2018, 256, 2083–2087. [Google Scholar] [CrossRef] [PubMed]
- Birtel, J.; Gliem, M.; Herrmann, P.; MacLaren, R.E.; Bolz, H.J.; Charbel Issa, P. Peripapillary Sparing in Autosomal Recessive Bestrophinopathy. Ophthalmol. Retina 2020, 4, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Battaglia Parodi, M.; Casalino, G.; Iacono, P.; Introini, U.; Adamyan, T.; Bandello, F. The expanding clinical spectrum of choroidal excavation in macular dystrophies. Retina 2018, 38, 2030–2034. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Kumar, V.; Azad, S.; Bhayana, A.A.; Surve, A.; Kumar, S.; Agarwal, P.; Chawla, R.; Venkatesh, P. Focal choroidal excavation: Review of literature. Br. J. Ophthalmol. 2020. online ahead of print. [Google Scholar] [CrossRef]
- Framme, C.; Wolf, S.; Wolf-Schnurrbusch, U. Small dense particles in the retina observable by spectral-domain optical coherence tomography in age-related macular degeneration. Invest. Ophthalmol. Vis. Sci. 2010, 51, 5965–5969. [Google Scholar] [CrossRef] [Green Version]
- Framme, C.; Schweizer, P.; Imesch, M.; Wolf, S.; Wolf-Schnurrbusch, U. Behavior of SD-OCT-detected hyperreflective foci in the retina of anti-VEGF-treated patients with diabetic macular edema. Invest. Ophthalmol. Vis. Sci. 2012, 53, 5814–5818. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Taylor, R.L.; Baines, R.A.; Swanton, L.; Freeman, S.; Corneo, B.; Patel, A.; Marmorstein, A.; Knudsen, T.; Black, G.C.; et al. Small Molecules Restore Bestrophin 1 Expression and Function of Both Dominant and Recessive Bestrophinopathies in Patient-Derived Retinal Pigment Epithelium. Invest. Ophthalmol. Vis. Sci. 2020, 61, 28. [Google Scholar] [CrossRef]
- Renner, A.B.; Tillack, H.; Kraus, H.; Kohl, S.; Wissinger, B.; Mohr, N.; Weber, B.H.F.; Kellner, U.; Foerster, M.H. Morphology and functional characteristics in adult vitelliform macular dystrophy. Retina 2004, 24, 929–939. [Google Scholar] [CrossRef] [Green Version]
- Renner, A.B.; Walter, A.; Fiebig, B.S.; Jägle, H. Gyrate atrophy: Clinical and genetic findings in a female without arginine-restricted diet during her first 39 years of life and report of a new OAT gene mutation. Doc. Ophthalmol. 2012, 125, 81–89. [Google Scholar] [CrossRef]
- Hood, D.C.; Bach, M.; Brigell, M.; Keating, D.; Kondo, M.; Lyons, J.S.; Marmor, M.F.; McCulloch, D.L.; Palmowski-Wolfe, A.M. ISCEV standard for clinical multifocal electroretinography (mfERG) (2011 edition). Doc. Ophthalmol. 2012, 124, 1–13. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, D.L.; Marmor, M.F.; Brigell, M.G.; Hamilton, R.; Holder, G.E.; Tzekov, R.; Bach, M. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc. Ophthalmol. 2015, 130, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Constable, P.A.; Bach, M.; Frishman, L.J.; Jeffrey, B.G.; Robson, A.G. International Society for Clinical Electrophysiology of Vision ISCEV Standard for clinical electro-oculography (2017 update). Doc. Ophthalmol. 2017, 134, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case ID | Family ID | Age (First Visit; y) | Sex | Family Members Affected | Age at Onset (y) | Follow-Up (y) | Symptoms | Right (OD) or Left (OS) Eye | Phenotype | BCVA (First Visit) | BCVA (Last Visit) | Refraction (D) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F1 | 3 | F | None | 3 | 2 | Photo-phobia | OD | I | 0.3 | 0.5 | +5.00/−1.25 |
| OS | I | 0.5 | 0.6 | +4.25/−1.00 | ||||||||
| 2 | F2 | 7 | M | Paternal uncle (BVMD) | 3 | 5 | blurred vision | OD | I | 0.7 | 0.4 | −1.00/−0.75 |
| OS | I | 0.1 | 0.25 | ±0.00/−0.75 | ||||||||
| 3 | F3 | 7 | M | None | 4 | 0 | blurred vision | OD | I | 0.3 | - | +3.50/−1.00 |
| OS | I | 1.0 | - | +2.50/±0.00 | ||||||||
| 4 | F4 | 7 | F | Sister (#12) | 6 | 21 | blurred vision | OD | I | 1.0 | 1.0 | +0.25/−1.50 |
| OS | I | 1.0 | 1.0 | +0.75/−1.75 | ||||||||
| 5 | F5 | 8 | F | Sister (#6) | 8 | 2 | none | OD | I | 1.0 | 1.0 | +3.75/−1.25 |
| OS | I | 1.0 | 1.0 | +3.75/−1.25 | ||||||||
| 6 | F5 | 9 | F | Sister (#5) | 9 | 2 | none | OD | V | 1.0 | 1.2 | +1.00/−0.50 |
| OS | V | 1.0 | 0.8 | +0.75/−0.75 | ||||||||
| 7 | F6 | 10 | F | None | 10 | 0 | blurred vision | OD | I | 0.2 | - | +3.75/−0.50 |
| OS | I | 0.4 | - | +4.00/−1.00 | ||||||||
| 8 | F7 | 20 | F | None | 12 | 4 | blurred vision | OD | II | 0.4 | 0.4 | +2.50/−0.50 |
| OS | II | 0.25 | 0.16 | +3.00/−0.25 | ||||||||
| 9 | F8 | 21 | M | None | 4 | 5 | blurred vision | OD | II | 0.5 | 0.4 | +0.25/−2.00 |
| OS | II | 0.4 | 0.3 | −0.25/−1.00 | ||||||||
| 10 | F9 | 25 | F | None | 17 | 7 | blurred vision, night blindness | OD | I | 0.5 | 0.2 | +1.00/−0.75 |
| OS | I | 0.6 | 0.2 | +1.00/±0.00 | ||||||||
| 11 | F10 | 25 | M | None | 6 | 26 | blurred vision | OD | IV | 0.8 | 0.125 | −3.25/−0.25 |
| OS | IV | 0.8 | 0.2 | −1.75/−1.00 | ||||||||
| 12 | F4 | 26 | F | Sister (#4) | 6 | 5 | none | OD | I | 1.0 | 1.0 | ±0 |
| OS | I | 1.0 | 1.0 | ±0 | ||||||||
| 13 | F11 | 28 | M | None | 6 | 5 | blurred vision | OD | II | 0.16 | 0.1 | −1.00/−1.00 |
| OS | II | 0.1 | 0.05 | −1.25/−0.75 | ||||||||
| 14 | F12 | 30 | M | None | 26 | 1 | blurred vision | OD | II | 0.3 | 0.25 | +4.50/−3.25 |
| OS | II | 0.08 | 0.05 | +6.00/−3.00 | ||||||||
| 15 | F13 | 33 | M | Sister, maternal uncle, brother (U) | 10 | 2 | night blindness | OD | III | 0.05 | 0.08 | +0.75/±0.00 |
| OS | III | 0.02 | 0.1 | +2.25/−0.50 | ||||||||
| 16 | F14 | 46 | M | Brother (#17) | 43 | 0 | blurred vision | OD | V | 0.6 | - | +4.00/−1.25 |
| OS | I | 0.2 | - | +4.25/−0.75 | ||||||||
| 17 | F14 | 50 | M | Brother (#16) | 50 | 10 | blurred vision | OD | I | 0.5 | 0.3 | +3.25/−0.50 |
| OS | I | 0.5 | 0.4 | +4.00/−1.75 | ||||||||
| 18 | F15 | 50 | F | Brother (MD) | 6 | 5 | blurred vision | OD | V | 0.1 | 0.125 | +1.5/−0.50 |
| Case ID | Age | Sex | Phenotype Classification | Family | Variant (NM_004183.4) | Protein | Reference (PMID) | Classification 1 |
|---|---|---|---|---|---|---|---|---|
| 1 | 3 | F | I | father, heterozygous | c.584C>T | p.(Ala195Val) | 10798642 | 5 |
| mother, heterozygous | c.830C>T | p.(Thr277Met) | 25474345 | 4 | ||||
| 2 | 7 | M | I | mother n.a. | c.422G>A | p.(Arg141His) | 18179881 | 5 |
| father/uncle heterozygous | c.454C>T | p.(Pro152Ser) | novel | 4 | ||||
| 3 | 7 | M | I | parents n.a. | c.422G>A | p.(Arg141His) | 18179881 | 5 |
| c.584C>T | p.(Ala195Val) | 10798642 | 5 | |||||
| 4 | 7 | F | I | Sister #12 | c.400C>G | p.(Leu134Val) | 17287362 | 4 |
| parents n.a. | c.422G>A | p.(Arg141His) | 18179881 | 5 | ||||
| 5 | 8 | F | I | Sister #6 | c.590_615del | p.(Leu197Profs*26) | novel | 5 |
| parents n.a. | c.934G>A | p.(Asp312Asn) | 18179881 | 5 | ||||
| 6 | 9 | F | V | Sister #5 | c.590_615del | p.(Leu197Profs*26) | novel | 5 |
| parents n.a. | c.934G>A | p.(Asp312Asn) | 18179881 | 5 | ||||
| 7 | 10 | F | I | mother heterozygous | c.620T>A | p.(Leu207His) | novel | 4 |
| father heterozygous | c.620T>A | p.(Leu207His) | novel | 4 | ||||
| 8 | 20 | F | II | parents n.a. | c.287_298del | p.(Gln96_Asn99del) | novel | 4 |
| - | c.287_298del | p.(Gln96_Asn99del) | novel | 4 | ||||
| 9 | 21 | M | II | mother, heterozygous | c.352G>C | p.(Asp118His) | 32141364 | 4 |
| father, heterozygous | c.352G>C | p.(Asp118His) | 32141364 | 4 | ||||
| 10 | 25 | F | I | mother, heterozygous | c.400C>G | p.(Leu134Val) | 17287362 | 4 |
| father, heterozygous | c.419T>C | p.(Leu140Pro) | 10798642 | 4 | ||||
| 11 | 25 | M | IV | parents n.a. | c.584C>T | p.(Ala195Val) | 10798642 | 5 |
| - | c.584C>T | p.(Ala195Val) | 10798642 | 5 | ||||
| 12 | 26 | F | I | Sister #4 | c.400C>G | p.(Leu134Val) | 17287362 | 4 |
| parents n.a. | c.422G>A | p.(Arg141His) | 18179881 | 5 | ||||
| 13 | 28 | M | II | mother n.a. | c.422G>A | p.(Arg141His) | 18179881 | 5 |
| father, heterozygous | c.422G>A | p.(Arg141His) | 18179881 | 5 | ||||
| 14 | 30 | M | II | parents n.a. | c.346_355dup | p.(Glu119Glyfs*116) | 29781975 | 5 |
| c.524del | p.(Ser175Thrfs*19) | novel | 5 | |||||
| 15 | 33 | M | III | Mother/father/sister/brother, heterozygous | c.91C>A | p.(Leu31Met) | 31254423 | 4 |
| uncle, homozygous | c.91C>A | p.(Leu31Met) | 31254423 | 4 | ||||
| 16 | 46 | M | V/I | Brother #17 | c.199_200del | p.(Leu67Valfs*164) | novel | 5 |
| parents n.a. | c.584C>T | p.(Ala195Val) | 10798642 | 5 | ||||
| 17 | 50 | M | I | Brother #16 | c.199_200del | p.(Leu67Valfs*164) | novel | 5 |
| parents n.a. | c.584C>T | p.(Ala195Val) | 10798642 | 5 | ||||
| 18 | 50 | F | V | parents n.a. | c.140G>A | p.(Arg47His) | 10854112 | 4 |
| c.422G>A | p.(Arg141His) | 18179881 | 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hufendiek, K.; Hufendiek, K.; Jägle, H.; Stöhr, H.; Book, M.; Spital, G.; Rustambayova, G.; Framme, C.; Weber, B.H.F.; Renner, A.B.; et al. Clinical Heterogeneity in Autosomal Recessive Bestrophinopathy with Biallelic Mutations in the BEST1 Gene. Int. J. Mol. Sci. 2020, 21, 9353. https://doi.org/10.3390/ijms21249353
Hufendiek K, Hufendiek K, Jägle H, Stöhr H, Book M, Spital G, Rustambayova G, Framme C, Weber BHF, Renner AB, et al. Clinical Heterogeneity in Autosomal Recessive Bestrophinopathy with Biallelic Mutations in the BEST1 Gene. International Journal of Molecular Sciences. 2020; 21(24):9353. https://doi.org/10.3390/ijms21249353
Chicago/Turabian StyleHufendiek, Karsten, Katerina Hufendiek, Herbert Jägle, Heidi Stöhr, Marius Book, Georg Spital, Günay Rustambayova, Carsten Framme, Bernhard H. F. Weber, Agnes B. Renner, and et al. 2020. "Clinical Heterogeneity in Autosomal Recessive Bestrophinopathy with Biallelic Mutations in the BEST1 Gene" International Journal of Molecular Sciences 21, no. 24: 9353. https://doi.org/10.3390/ijms21249353
APA StyleHufendiek, K., Hufendiek, K., Jägle, H., Stöhr, H., Book, M., Spital, G., Rustambayova, G., Framme, C., Weber, B. H. F., Renner, A. B., & Kellner, U. (2020). Clinical Heterogeneity in Autosomal Recessive Bestrophinopathy with Biallelic Mutations in the BEST1 Gene. International Journal of Molecular Sciences, 21(24), 9353. https://doi.org/10.3390/ijms21249353