1. Introduction
Achromatopsia (ACHM) is an inherited retinal disorder affecting retinal cones, the type of photoreceptors that mediate high acuity daylight vision. Cone outer segments, the specialized compartments of these photoreceptors, contain all proteins needed for light detection and conversion into chemical and electrical signals. Mutations in genes encoding key proteins of this cascade result in total colour blindness, also referred to as achromatopsia. Approximately 80 percent of ACHM patients carry mutations in one of the genes
CNGA3 or
CNGB3 [
1], which encode the two subunits of the cyclic nucleotide-gated (CNG) channel in cone photoreceptors [
2]. The cone CNG channel is part of the visual transduction cascade located in the cone outer segment and is the effector of cyclic guanosine monophosphate (cGMP), the key second messenger of this signaling cascade, which translates light signals into electrical and Ca
2+ signals [
2]. Four additional disease genes exist, among which
GNAT2,
PDE6C, and
PDE6H also encode proteins involved in the cone visual transduction cascade [
1]. The sixth known disease gene is
ATF6, encoding an endoplasmic reticulum (ER)-localized transmembrane transcription factor that can activate the unfolded protein response (UPR) and plays a role in ER homeostasis [
3,
4].
Like many other inherited disorders, ACHM manifests already in childhood with clinical symptoms that include lack of colour discrimination, poor visual acuity, extreme light sensitivity (photophobia), and involuntary eye movements (nystagmus) [
5]. Given the lack of cone photoreceptor function from beginning, there is no real progression of the clinical symptoms over time. However, animal experiments and morphological data from affected patients suggested a progressive degeneration and loss of cones over time [
6,
7]. While the principal development and morphology of affected cone photoreceptors is initially similar to non-affected cones, the diseased cones start degenerating during young adulthood and are eventually lost by induction of various cell death mechanisms [
8].
Currently, there is no treatment available for ACHM, but several groups are working on the development of gene supplementation therapies for both
CNGA3- and
CNGB3-linked ACHM [
9]. One phase I/II clinical trial testing the effect of an adeno-associated virus (AAV)-based gene therapy vector in patients with confirmed
CNGA3-linked ACHM was already completed and recently reported promising safety and efficacy data [
10]. Three additional ACHM gene therapy clinical trials are ongoing and expected to report first data soon.
The concept of gene supplementation with AAV vectors is only applicable at early stages of the disease and providing a sufficient number of morphologically intact and, thus, rescuable cone photoreceptors is still present [
9]. Given that affected ACHM cones degenerate over time, there is only a certain time window of opportunity for gene supplementation therapies. Unfortunately, the disease mechanisms involved in cone degeneration are only partially understood and there is a high need to better characterise the pathobiology in affected cone photoreceptors.
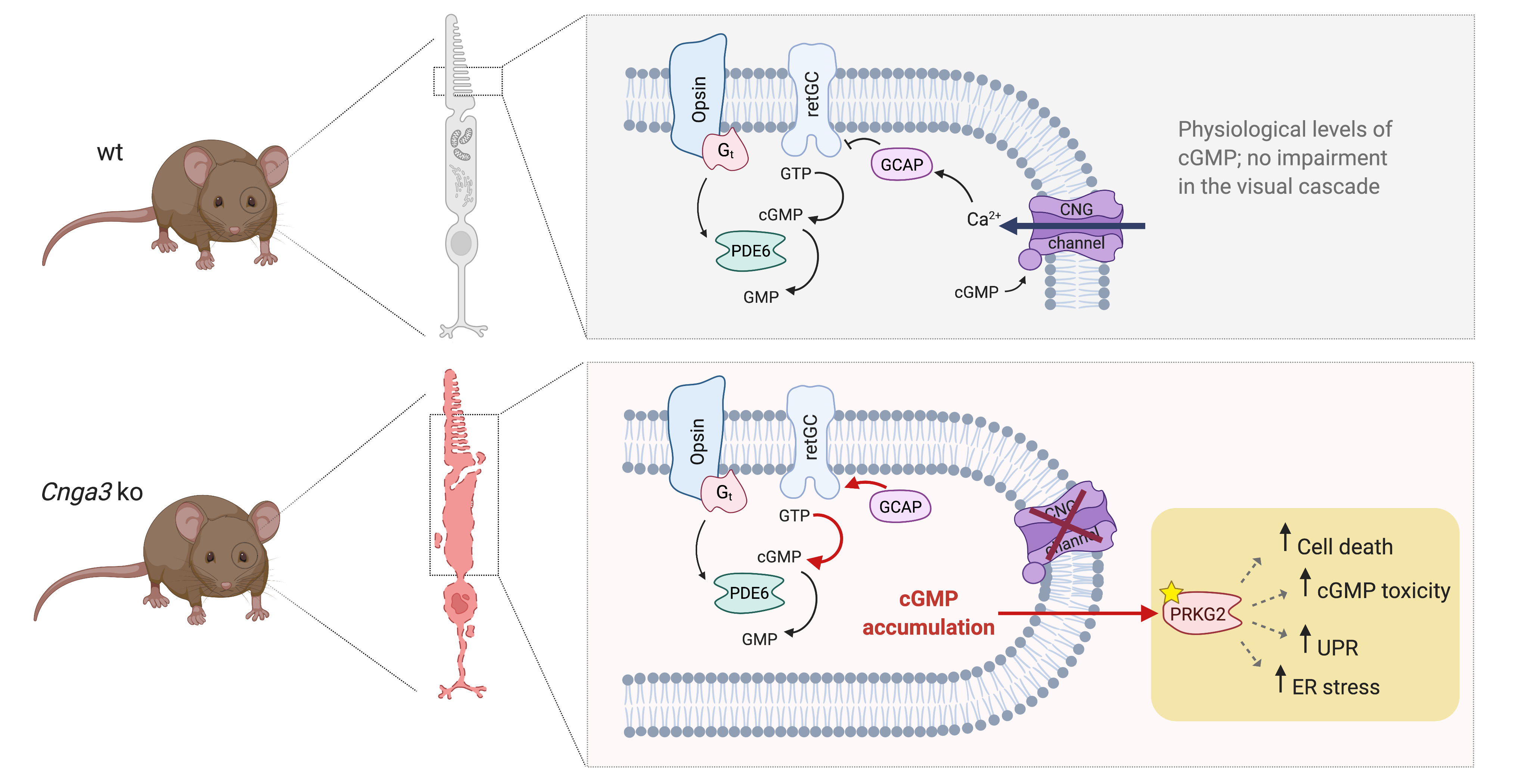
Here, we investigated the role of cGMP signalling on viability of cone photoreceptors in the Cnga3 knockout model of ACHM. We found that the membrane bound cGMP-dependent protein kinase 2 (Prkg2) is crucially involved in cGMP-mediated cytotoxicity in cones. Genetic depletion of Prkg2 resulted in a long-lasting preservation of cone photoreceptors. Mechanistically this neuroprotective effect of Prkg2 depletion on cones seems to involve inhibition of DNA damage, unfolded protein and ER-stress mechanisms. Our work highlights the cGMP kinase Prkg2 as a novel target for neuroprotective treatments aiming to preserve the morphology and structure of affected ACHM cone photoreceptors.
2. Results
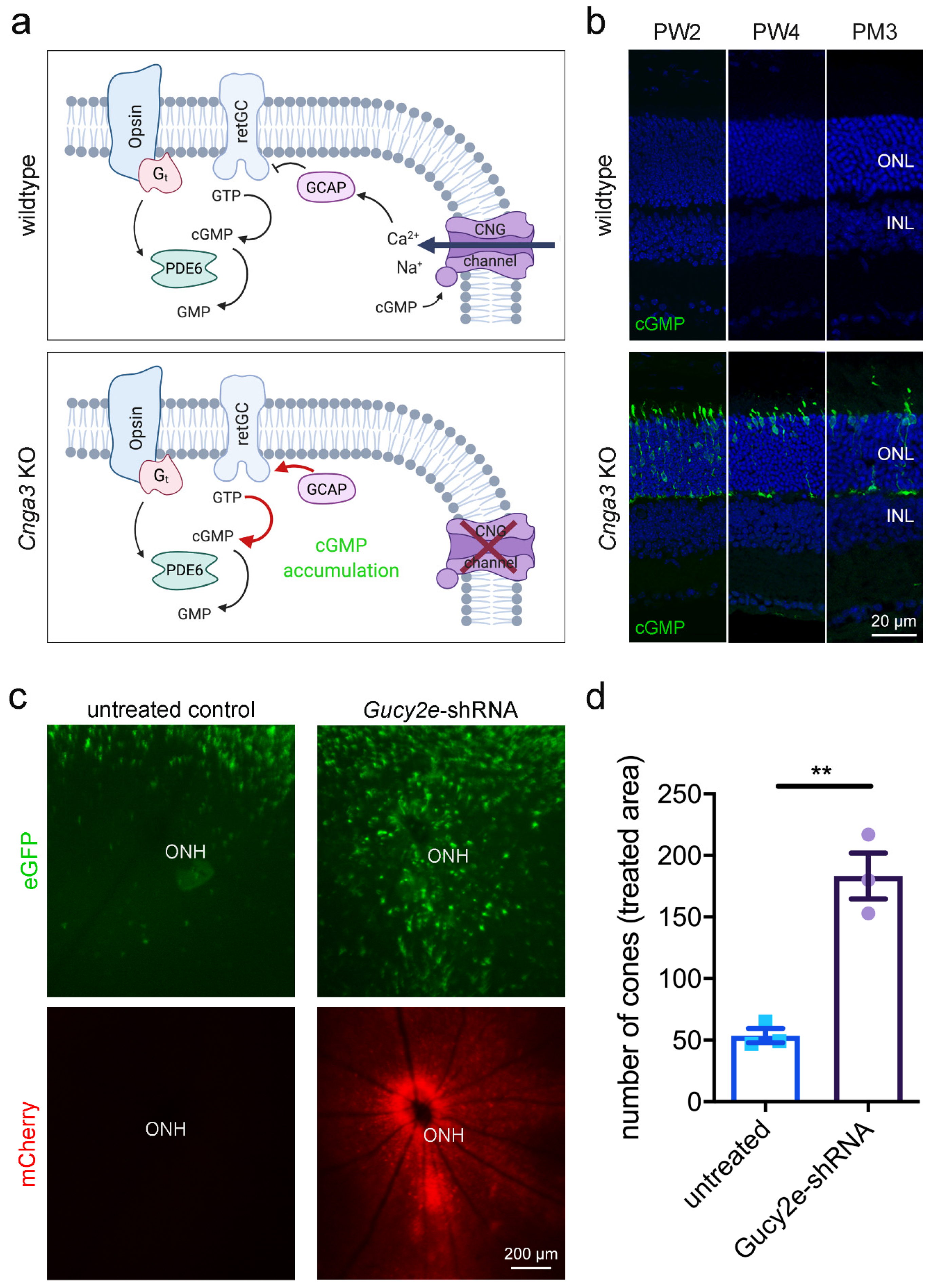
In cone outer segments, the second messenger cGMP is produced by the receptor guanylyl cyclase (retGC encoded by the
GUCY2E gene) and its levels are balanced by the cone phosphodiesterase (PDE6C;
Figure 1a). High levels of cGMP in the dark activate the cyclic nucleotide-gated (CNG) channel, which carries a mixed Na
+ and Ca
2+ inward current. Ca
2+-bound guanylyl cyclase activating protein (GCAP1) inhibits the retGC activity in a negative feedback mechanism. In cones missing the CNG channel, the lack in Ca
2+ influx would likely determine the constant activation of retGC by Gcap1 and the production of excessive amounts of cGMP (
Figure 1a). Indeed, immunolabeling of retinal cross-sections from
Cnga3 KO mice, which lack the cone CNG channel [
6], with a cGMP-specific antibody [
11] confirmed the accumulation of cGMP in cone photoreceptors (
Figure 1b) at eye opening and persisting at later timepoints. In parallel, the number of cGMP-positive cones decreased over time, reflecting the already described progressive cone degeneration in
Cnga3 KO mice [
6,
8]. Double labelling for cGMP and the cone marker glycogen phosphorylase confirmed that the cells showing high levels of cGMP are cone photoreceptors (
Supplementary Figure S1). This is also the case for those cGMP-positive cells localising in the lower part of the outer nuclear layer.
To investigate the effect of acute downregulation of retGC, we generated an adeno-associated virus (AAV) vector encoding a shRNA that targets the endogenous
Gucy2e mRNA and we delivered it via subretinal injection into the eyes of 2-week-old
Cnga3 KO mice expressing an eGFP reporter under the control of the cone-specific human red/green opsin (RG) promoter [
12]. This treatment resulted in preservation of a higher number of cones in areas of
Gucy2e downregulation (
Figure 1c,d). This is in line with a previous report showing that constitutive genetic inactivation of
Gucy2e in
Cnga3 KO mice delayed degeneration and preserved biochemical markers of cone photoreceptors [
13].
The key effector of cGMP in cone photoreceptors is the CNG channel [
2]. Since the CNG channel is absent from
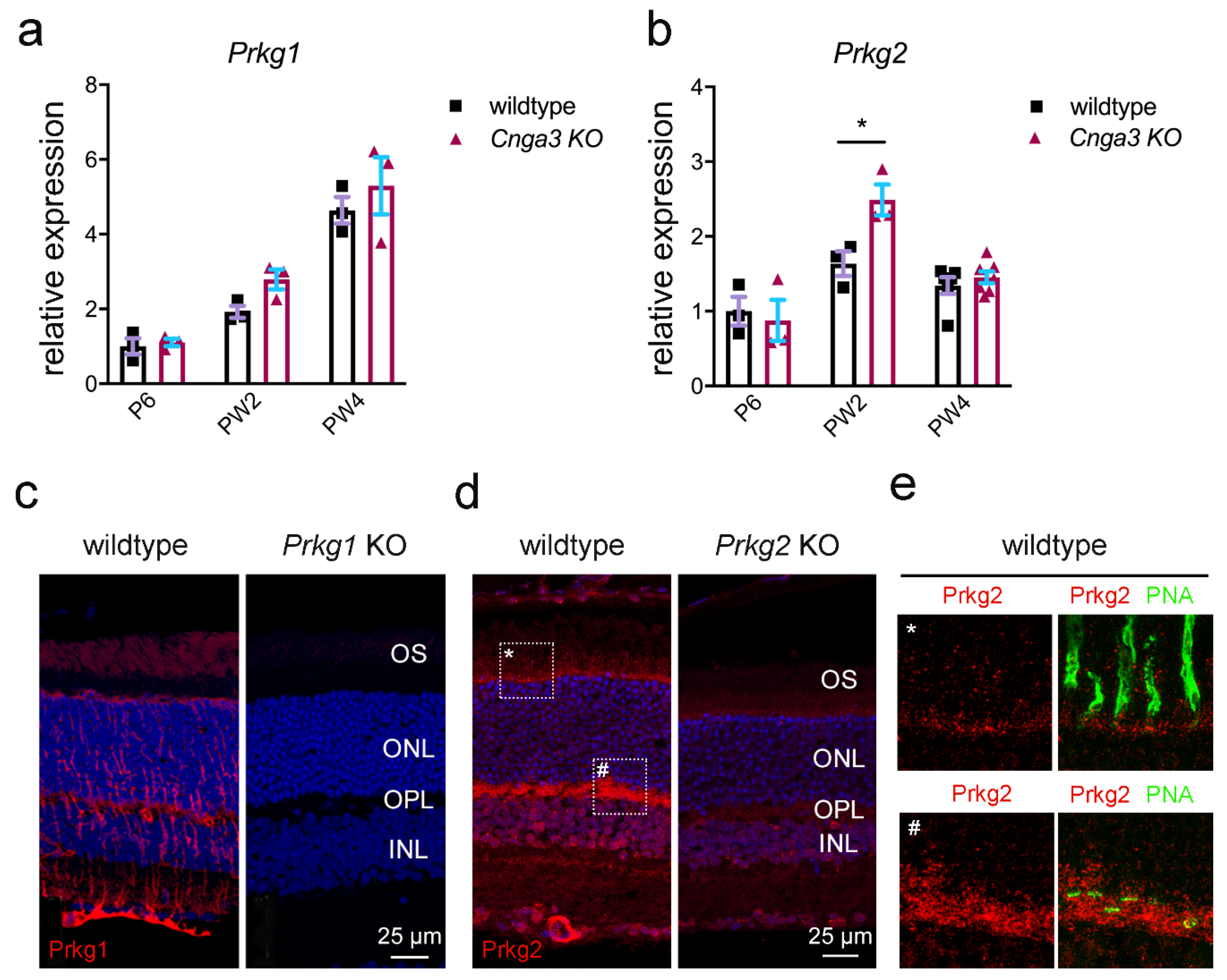
Cnga3 KO cones, we explored the expression of the cGMP-dependent kinases, namely Prkg1 and Prkg2. As shown in
Figure 2, both kinases are expressed in the mouse retina. When analysing the transcript levels in
Cnga3 KO retina at different postnatal time points, we observed a transient upregulation of
Prkg2 transcript on week 2 (
Figure 2b), whereas
Prkg1 did not reveal any genotype-specific changes (
Figure 2a).
We then analysed the immunolocalization of the two kinases on retinal cross-sections. The Prkg1 signal was primarily found in Müller glia cells and retinal blood vessels and only a faint signal could be detected in the outer segment layer of photoreceptors (
Figure 2c). Importantly, this signal was absent in cross-sections from
Prkg1 knockout animals (
Figure 2c). Prkg2 showed a distinct expression pattern with strong labelling of retinal blood vessels, but also of the photoreceptor synaptic layer (outer plexiform layer, OPL) and weaker labelling of the photoreceptor inner segments (
Figure 2d,e for high magnification view). While the Prkg2 antibody signal was not completely absent in tissue from
Prkg2 KO mice, it was substantially reduced compared to the wildtype condition (
Figure 2d).
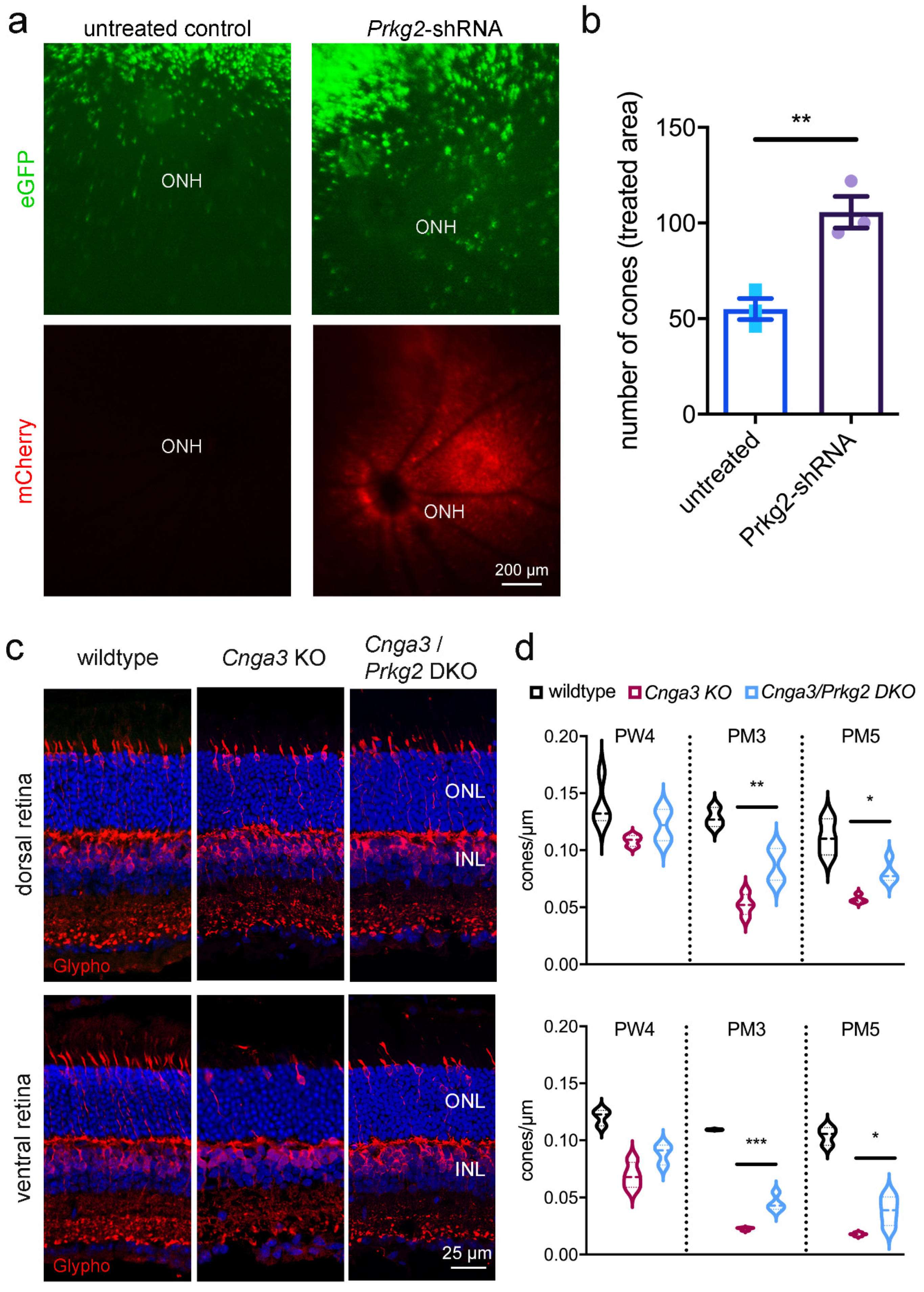
Given the exuberant accumulation of cGMP in Cnga3 KO cones and the transient upregulation of the potential effector Prkg2, we wondered whether this kinase plays any role for the survival of affected cones. We, therefore, generated an AAV vector encoding a shRNA that targets the Prkg2 mRNA and delivered it via subretinal injection into the eyes of 2-week-old RG-eGFP/Cnga3 KO mice.
This treatment led to significant preservation of the number of cones at 3 months of age (
Figure 3a,b), suggesting that the
Prkg2 knockdown has a neuroprotective effect on
Cnga3 KO cones. To corroborate this finding, we cross-bred
Cnga3 KO mice with mice lacking Prkg2 [
14] and analysed the survival of cone photoreceptors in the resulting
Cnga3/
Prkg2 double knockout (DKO) as well as in
Cnga3 KO and wildtype mice (
Figure 3c,d). To determine the cell density of cones, we immunolabeled retinal cross-sections from these mice for glycogen phosphorylase, which, in addition to bipolar cells, labels the entire cytoplasm of cones from the inner segment throughout the synapse and thus facilitated quantification [
15]. While at 4 weeks of age the cone density did not substantially differ among genotypes, the number of cones was significantly higher in
Cnga3/
Prkg2 DKO than in
Cnga3 KO mice at 3 and 5 months of age (
Figure 3d). This was evident in the dorsal retina, but also in the faster degenerating ventral part (
Figure 3d). Thus, constitutive knockout or acute knockdown of
Prkg2 protected Cnga3 KO cone photoreceptors from degeneration.
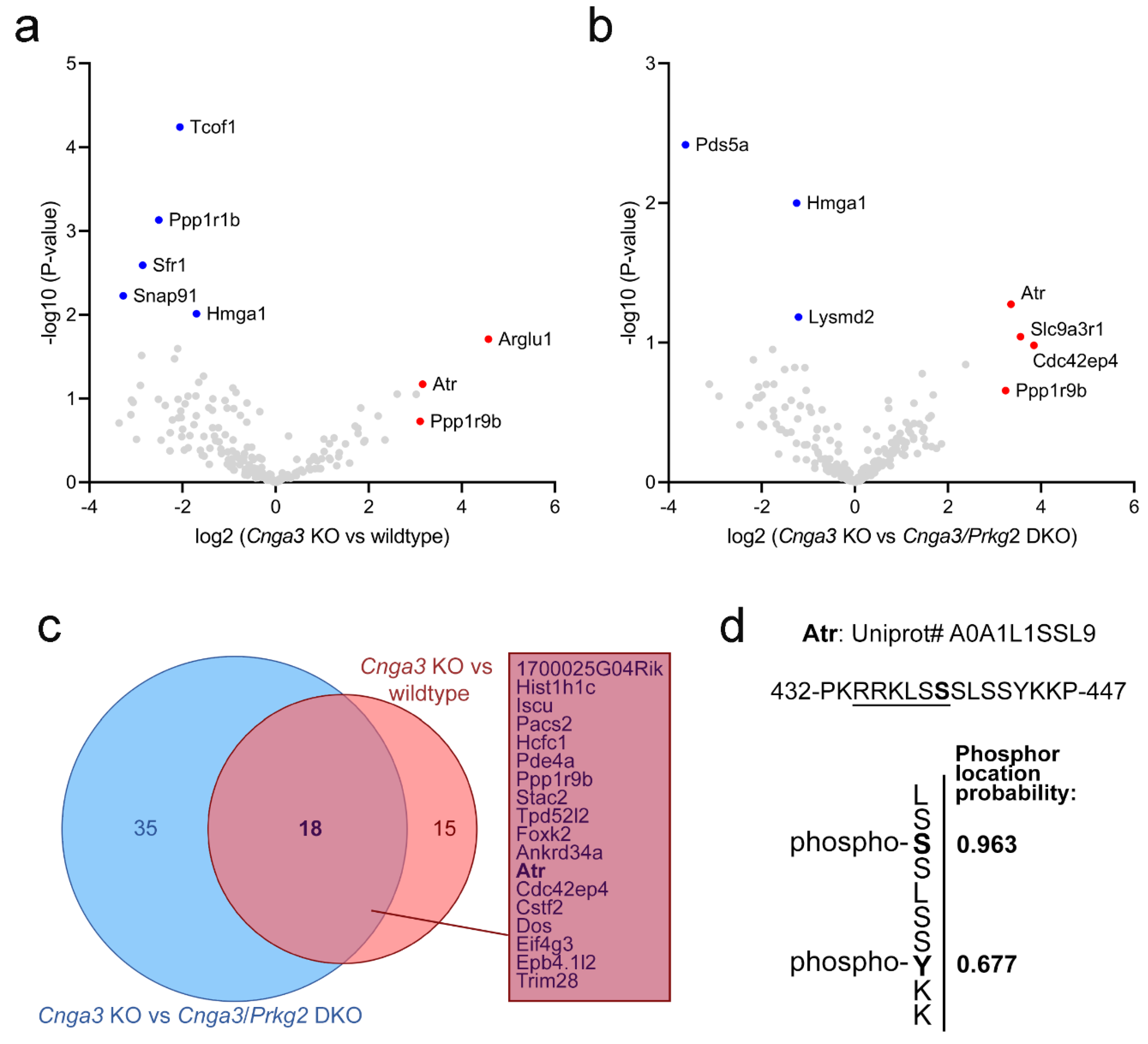
Since Prkg2 is a cGMP-dependent serine/threonine-specific kinase, it is tempting to speculate that this neuroprotective effect is mediated by the lack of its kinase activity. We, therefore, designed an experiment to identify potential changes in the pattern of phosphoproteins in
Cnga3 KO mice. In particular, we used titanium ion (Ti
4+) functional magnetic microparticles (Ti-IMAC) to enrich phosphorylated peptides [
16] from wildtype,
Cnga3 KO, and
Cnga3/
Prkg2 DKO retinal protein lysates for subsequent label free quantification (LFQ) using mass spectrometry (
Figure 4). We reasoned that relevant proteins whose phosphorylation pattern depends on elevated activity of Prkg2 would show an enrichment in
Cnga3 KO over wildtype retina and a depletion from
Cnga3/
Prkg2 DKO retinal phospho-lysates. As visualized by the volcano plots in
Figure 4a,b, several proteins were found to be enriched in phospho-lysates of
Cnga3 KO retina compared to wildtype or
Cnga3/
Prkg2 DKO. Among a total of 18 proteins showing the aforementioned enrichment/depletion pattern, we found the serine/threonine-specific protein kinase Atr (Atm-Rad3-related protein). Phosphorylated Atr peptides were found in
Cnga3 KO retinal lysates, but were not detected in lysates from
Cnga3/
Prkg2 DKO or wildtype retina.
The MaxQuant analysis software [
17] allows for determination of the most likely phosphorylation locations in the identified phosho-peptides. For the Atr peptides, a high phospho location probability of 0.963 was determined for serine 439 (Ser439) and a localisation probability of 0.677 for tyrosine 444 (Y444). An example mass spectrometry (MS) spectrum showing fragments used for identification and phosphor-localization is depicted in
Supplementary Figure S2.
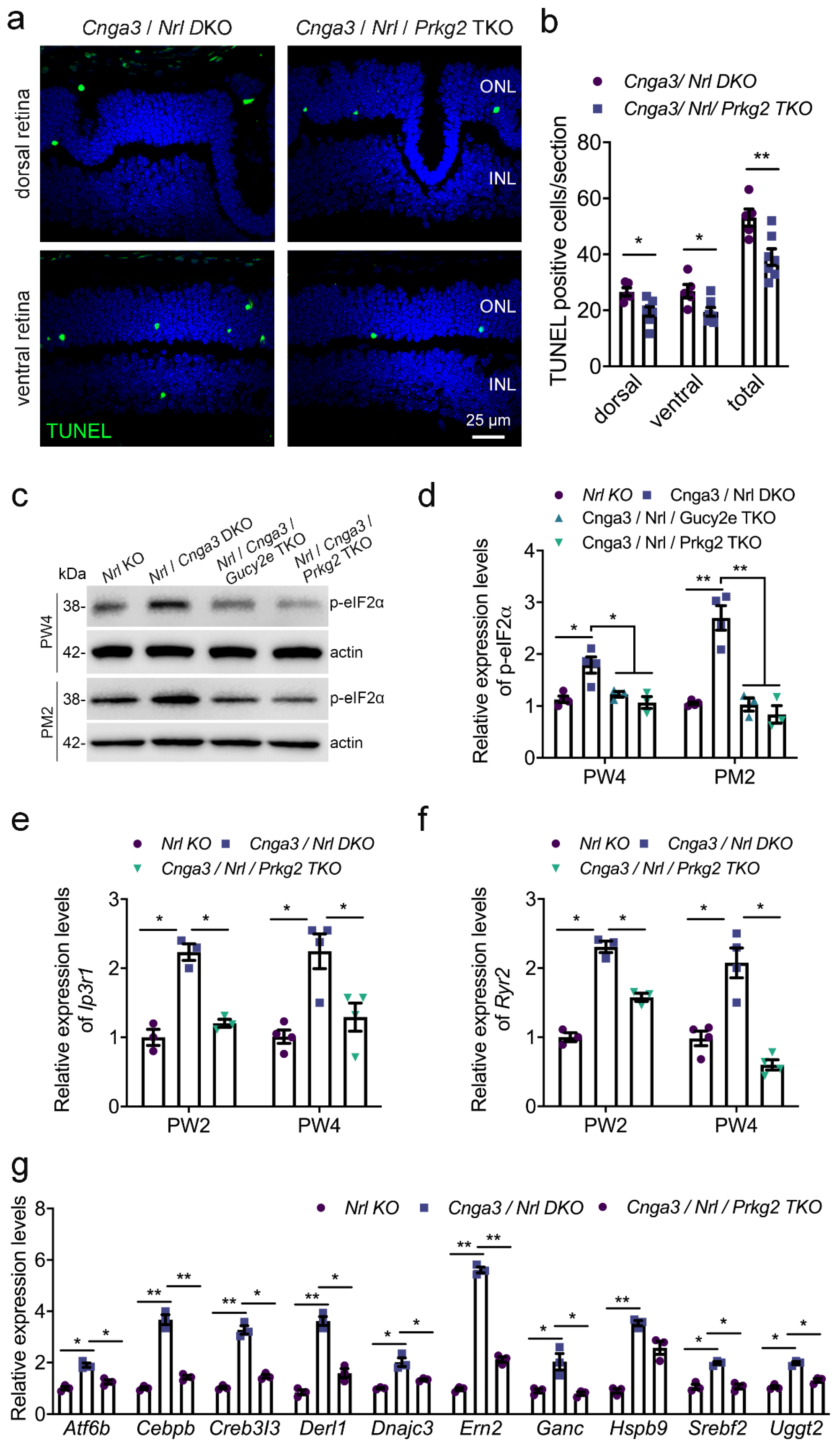
It is well established that increased ER stress contributes to degeneration and cell death of
Cnga3 KO cone photoreceptors [
18,
19,
20]. Thus, we next evaluated how
Prkg2-deletion influences ER stress markers in
Cnga3 KO cones. To facilitate the bulk analysis of cone photoreceptor markers, we generated a
Cnga3/
Prkg2 double-deficient mouse line on the cone-dominant
Nrl KO background [
20].
Cnga3/
Nrl DKO mice display a cone degeneration similar to the
Cnga3 single KO [
13,
20]. We first compared the level of photoreceptor cell death in
Cnga3/
Nrl DKO and
Cnga3/
Nrl/
Prkg2 triple knockout (TKO) mice. As shown in
Figure 5a,b, deletion of
Prkg2 lead to a significant reduction of the Terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) signal, indicative of a reduced cell death rate. We next analyzed the effect of Prkg2 knockout on the levels of phosphorylation of eukaryotic initiation factor 2 (p-eIF2a) which is an established ER stress marker already shown to be upregulated in
Cnga3-deficient cone photoreceptors [
20]. In line with a neuroprotective effect of
Prkg2-deletion, the levels of p-eIF2a were reduced to a level similar to
Cnga3/
Nrl/
Gucy2e TKO mice (
Figure 5c,d), which are protected from cGMP-mediated cytotoxicity due to the lack of the cGMP-synthesizing retGC enzyme [
13]. Previous studies have demonstrated that CNG channel-deficient mice show increased expression/activity of ER Ca
2+-releasing channels, responsible for Ca
2+ efflux from the ER into the cytosol [
20,
21]. We, therefore, analyzed the gene expression of
Itpr1, which encodes the inositol 1,4,5-trisphosphate receptor type 1 (IP3R1), and
Ryr2, the gene encoding the type 2 ryanodine receptor (Ryr2). As depicted in
Figure 5e,f, both genes showed enhanced gene expression levels in the
Cnga3 KO context, which were normalized after additional knockout of
Prkg2. A similar effect was observed for the unfolded protein response (UPR)-related genes
Atf6b (activating transcription factor 6 beta),
Bax (Bcl2-associated X protein),
Cebpb (CCAAT/enhancer-binding protein beta),
Creb3l3 (cAMP-responsive element-binding protein 3-like 3),
Derl1 (degradation in ER protein 1),
Dnajc3 (DnaJ [Hsp40] homolog, subfamily C, member 3),
Ern2 (ER to nucleus signaling 2),
Ganc (glucosidase, alpha; neutral C),
Srebf2 (sterol regulatory element-binding factor 2), and
Uggt2 (UDP-glucose glycoprotein glucosyltransferase 2;
Figure 5e–g), which are upregulated in
Cnga3-deficient mice [
19,
21]. This suggests that
Prkg2 knockout counteracts ER stress and UPR and normalizes ER homeostasis.
3. Discussion
Loss of vision is one of the most severe handicaps with high socioeconomic importance. Many blinding disorders are inherited and caused by mutation in one of more than 250 retinal disease genes (
https://sph.uth.edu/retnet/), which affect rod and cone photoreceptors function. Until recently, there was no hope for patients suffering from blinding inherited retinal disorders. This has changed with the approval of a first gene therapy for a specific condition,
RPE65-linked retinal dystrophy [
22]. Many more of these gene-specific treatments or more broadly applicable therapies are needed in order to cover all conditions caused by the >250 disease genes.
In addition to a loss of function, a degeneration of affected photoreceptors is commonly observed. In order to provide a long-lasting treatment effect, any therapy needs to halt the degeneration processes. This is true for disorders like retinitis pigmentosa, which are a class of inherited retinal dystrophies characterized by a progressive degeneration of rod photoreceptors. It also applies to conditions like achromatospia affecting cone photoreceptors and, thus, high acuity daylight vision. However, the mechanisms involved in rod and cone degeneration are only partially understood and a more complete understanding is needed to develop improved treatments [
8,
23,
24].
Here, we explored cone cell death mechanisms in the
Cnga3 KO mouse model of achromatopsia.
Cnga3 KO cone photoreceptors lack the ability to respond to light stimuli [
25] and cannot generate or transfer corresponding signals to higher order neurons and central visual centres. In addition to the loss of function, cones are degenerating over time, inducing ER stress and eventually dying [
6,
8,
19,
20]. Affected cone photoreceptors also accumulate exuberant levels of cGMP due to lack of the cone CNG channel and, thus, absence of the Ca
2+-mediated negative feedback inhibition of the cGMP-synthesizing retGC enzyme (see
Figure 1A and corresponding text for more details). Deletion or knockdown of
Gucy2e eliminates the cGMP accumulation and preserved the number of cones (
Figure 1 and [
13]), confirming that cGMP is crucially involved in the degeneration process.
The CNG channel itself is a major effector of cGMP and, if still present, was shown to be an important factor in mediating cGMP cytotoxicity [
24,
26]. However,
Cnga3 KO cones lack a cGMP-gated channel and, thus, cGMP cytotoxicity must be due to other cGMP effectors or an impairment of more generic cell homeostasis/metabolic processes. Other potential cGMP effectors are the cGMP-dependent serine/threonine kinases encoded by the two genes
Prkg1 and
Prkg2, which are involved in a variety of physiological and pathophysiological processes, but have no established physiological function in retinal photoreceptors [
27]. Indeed, the role of Prkg1 in survival of diseased rod photoreceptor has been already addressed [
28,
29]. Based on this, inhibition of Prkg1 is currently explored as a neuroprotective treatment to delay degeneration of photoreceptors in retinitis pigmentosa [
28,
30]. In contrast, the role of Prkg2 in photoreceptor degeneration has not been investigated so far.
Here, we discovered a crucial role of the Prkg2 in degenerating
Cnga3 KO cone photoreceptors. We found that AAV-mediated knockdown or genetic deletion of
Prkg2 rescues
Cnga3 KO cone photoreceptors from degeneration. While the effect on cone survival is clear, deletion of Prkg2 does not fully prevent all cone photoreceptors from dying. This suggests that, besides cGMP-Prkg2-mediated cytotoxicity, additional cell death mechanisms contribute to the degeneration of cones. Mechanistically, deletion of Prkg2 dampened the levels of ER stress and UPR markers, suggesting that cGMP-Prkg2 signalling contributes to both phenomena and crucially impacts viability of CNG channel-deficient cone photoreceptors [
18,
21,
31].
Using an approach based on Ti-IMAC enrichment of phosphorylated proteins in combination with
Cnga3/
Prkg2 DKO tissue as a reference, we were able to identify phosphoproteins that are upregulated in
Cnga3 KO retinal tissue and whose phosphorylation depends on Prkg2. In total we identified 18 phosphoproteins being enriched in
Cnga3 KO retina against both wildtype and
Cnga3/
Prkg2 DKO. Among those we found Atr, which belongs to the phosphatidylinositol 3-kinase-related kinase family and is a known DNA damage sensor, activating the DNA damage response (DDR) by phosphorylating downstream effector proteins [
32]. Of note, the MaxQuant analysis software identified Ser439 as the most likely phosphorylation site. Ser439 is part of a putative recognition motif (underlined sequence in
Figure 4d) that is similar to the sequence of previously postulated Prkg2 substrates [
33]. Given that the identified Ser439 phosphorylation has not been previously characterized, its relevance for the degeneration process in
Cnga3 KO cones remains unclear. At this stage we can only speculate that cGMP-Prkg2 signalling triggers cell death by activating DNA damage pathways. This intriguing hypothesis deserves further investigation in the future, in particular since recent evidence suggests a cross-talk between DDR and ER stress [
34].
Further studies are also needed to get more insight into the timing of the pathophysiological events that eventually lead to cone cell death. From the upregulation of Prkg2 transcript at PW2, we can speculate that a transient upregulation of the cGMP-Prkg2 signalling around eye opening could trigger subsequent cell death processes. Since the progression of cone degeneration in the Cnga3 KO mouse is rather slow, we believe that multiple signaling processes running in parallel contribute to degeneration of cones; thus, Prkg2-dependent mechanisms are responsible only in part for the observed effects and do not dictate the kinetics of degeneration. Additionally, more focused experiments with Cnga3 KO mice on a cone-rich Nrl KO background are needed to clarify this issue.
Taken together, this study establishes Prkg2 as a novel therapeutic target for neuroprotection of degenerating cone photoreceptors in achromatopsia, although the therapeutic applicability of Prkg2 inhibition needs to be further validated. Another aspect that remains to be elucidated is whether this neuroprotective effect is specific for cones or if it is also relevant for rod photoreceptors. Moreover, Prkg2 inhibition could provide protection not only from cGMP-mediated cytotoxicity, but also from degeneration caused by other detrimental stimuli. Importantly, in the case of inherited retinal disorders like achromatopsia, a genetic mutation causes a functional defect. Thus, the underlying gene defect needs to be corrected or a healthy copy of the affected gene needs to be supplemented in order to achieve an improvement in cone function. However, such gene correction or supplementation therapies are only applicable if a sufficient number of morphologically intact and, thus, rescuable target cells remains. Therefore, advanced stage patients who lost the majority of their photoreceptors are excluded from gene-specific gene therapies. Based on the presented results it is tempting to speculate that a neuroprotective therapy by Prkg2 inhibition has the potential to delay degeneration of cones and to widen the window of opportunity for gene therapy in achromatopsia.
4. Materials and Methods
4.1. Animals
All animal experiments were performed according to the ARVO statement for the use of animals in ophthalmic and vision research and were approved by the local authorities (the Government of Upper Bavaria, Germany or the Institutional Animal Care and Use Committee of the University of Oklahoma Health Sciences Center, Oklahoma City, OK, USA). The project identification codes were ROB-55.2-2532.Vet_02-14-59 and 18-060-EAFI, respectively. All mice used were on a mixed C57-BL6/J-129/Sv genetic background.
Cnga3/
Prkg2 double knockout mice were generated by cross-breeding
Cnga3 KO mice [
25] with
Prkg2 KO mice [
14].
Cnga3/
Nrl DKO and
Cnga3/
Nrl/
Gucy2e TKO mice were generated by cross-breeding as described previously [
19,
35].
Cnga3/
Nrl/
Prkg2 TKO mice were generated by cross-breeding
Cnga3 KO mice with
Prkg2 KO mice [
14] and/or
Nrl KO [
36] mice. For in vivo imaging of cones
Cnga3 KO mice were also crossbred with RG-eGFP mice [
37] to generate RG-eGFP/
Cnga3 KO mice. Mice were maintained on a 12 h light/dark cycle. During the light cycle, cage illumination was approximately 7 foot-candles.
4.2. Eye Preparation, Immunofluorescence Labelling, and Confocal Microscopy
We prepared mouse eye cross-sections for immunohistochemical analysis as described previously [
35,
38]. Cell nuclei were stained with Hoechst 33,342 (Thermo Fisher Scientific, Langenselbold, Germany). The following primary antibodies were used: anti-cGMP (sheep, diluted 1:500, from Prof. Steinbusch, Maastricht University; anti-cGKI alpha (rabbit, 1:100, kindly rovided by F. Hofmann); anti-PRKG2 (rabbit, 1:50, Merck-Sigma Aldrich, Cat. Nr. HPA007386). In order to preabsorb against non-specific epitopes, the anti-PRKG2 antibody was preincubated for 3 h at 4 °C with brain and retina homogenate from a
Prkg2 KO mouse, then cleared by centrifugation and used at a final dilution of 1:50 for immunohistochemistry. Confocal images were collected at a Leica TCS SP8 spectral confocal laser scanning microscope (Leica Microsystems). The generated files were processed with LAS X software and the open-source software Fiji [
39,
40].
4.3. TUNEL Assay
The terminal deoxynucleotidyltransferase dUTP nick end-labelling (TUNEL) was performed and analysed as described previously [
20,
35]. Confocal images were collected at an Olympus FV1000 confocal laser scanning microscope (Olympus, Melville, NY, USA).
4.4. Cloning and Production of AAV Vectors
For shRNA experiments a modified pSub201 plasmid [
41] was generated with a shRNA expression cassette and U6 promoter. Additionally, a phosphoglycerate kinase 1 (PKG) promoter-driven mCherry with SV 40 polyA and WPRE was inserted by standard restriction enzyme based cloning techniques. The resulting plasmid and shRNA hairpin sequences were pSub_U6_shRNAGucy2e_PGK_mcherry_SV40_WPRE, pSub_U6_shRNAPrkg2_PGK_mcherry_SV40_WPRE, and pSub_U6_shRNACtrl_PGK_mcherry_SV40_WPRE. Sequences are available upon request. AAV8-Y733F-pseudotyped AAV2 vectors carrying the corresponding shRNA/mCherry expression cassettes were produced, purified and characterized as previously described [
38].
4.5. Subretinal Injection and In Vivo Fundus Fluorescence Imaging
1 µL of titer-matched AAV8-shRNA vector containing approx. 5E10 total vector genomes was delivered via subretinal injection in 2-week-old RG-eGFP/
Cnga3 KO mice. Fundus fluorescence examination was performed with an adapted Spectralis HRA + OCT system from Heidelberg Engineering (Dossenheim, Germany) in combination with optic lenses, as described previously [
42].
4.6. Retinal Protein Preparation, SDS-PAGE, and Western Blot Analysis
Retinal protein preparation, SDS-PAGE, and western blot analysis were performed as described previously [
21,
35]. A Li-Cor Odyssey machine and Li-Cor software (Li-Cor Biosciences, Lincoln, NE, USA) were used for detection and densitometric analysis.
4.7. PCR and Quantitative RT-PCR
Total RNA preparation and reverse transcription were per- formed as described previously [
19,
21]. The quantitative reverse transcription-polymerase chain reaction (qRT-PCR) assays were performed with a real-time PCR detection system (iCycler; Bio-Rad, Hercules, CA, USA). The primers used and the ∆∆Ct method used are described in [
19,
21].
4.8. Ti-IMAC Phosphoenrichment and MS-Sample Preparation
After lysis, samples were incubated with 1 µL Benzonase for 1 h at 0 °C. Afterwards, 250 µL of MS-grade water was added and 2 retinas from the same date were pooled together. To precipitate the proteins, 7 mL of ice-cold acetone(aq) was added to each sample, followed by incubation at −20 °C overnight. After centrifugation at 10,000× g for 10 min, the supernatant was removed and the pellet was washed two times with 1 mL of 80% acetone each.
To the resulting pellets, 2 mL of digestion-buffer (50 mM TEAB, 1 mM MgCl2 in MS-grade water) and 2 mL of MS-grade water were added and the samples were resuspended by sonication (5 cycles at 50% power output, 5 min) on ice. Afterwards, the protein concentration of the samples was determined by conducting a Bradford assay.
A volume equivalent of 445 µg protein content was taken. For digestion, samples were processed in the same way as for the full-proteome analysis with the following amounts: TCEP(aq) (1M) was added to a final concentration of 41.8 mM, 255 µL of TEAB(aq) (1M) were added to neutralize the solution, iodoacetamide(aq) (1M) was added to a final concentration of 41.1 mM and 210 µL of TEAB(aq) (1M) were added to adjust the pH-value for digestion. Finally, 5 µg trypsin was added per sample (3 µg in case of the IA-triplicate samples).
After digestion, peptides were desalted by stage-tip purification (SDB-RPS material, see [
43]) and concentrated to dryness on a speed-vac.
For each replicate, 50 µL MagReSyn® magnetic Ti-IMAC beads (ReSyn Biosciences, Gauteng, South Africa) were prepared according to the manufacturer’s instructions. For loading, the dried peptides were dissolved in 100 µL loading-buffer (1M glycolic acid, 80% MeCN, 5% TFA in MS-grade water) and incubated for 20 min at room temperature with end-over-top mixing with the prepared Ti-IMAC beads. Unbound sample was removed by washing two times with 100 µL loading buffer for 30 s. Subsequently, the beads were incubated four times with 100 µL wash buffer (80% MeCN, 1% TFA in MS-grade water) for 2 min each. Phosphopeptides were eluted by incubation three times with 80 µL elution buffer (1% NH4OH in MS-grade water) for 15 min each.
After elution, phosphopeptide-containing samples were acidified with FA(aq), desalted by stage-tip purification (SDB-RPS material, see [
43]) and concentrated to dryness on a speed-vac.
The phosphopeptide-samples were solved in 12 µL MS-solvent.
4.9. MS-Analysis of Phospho-Enriched Samples.
The samples were analyzed with an UltiMate 3000 RSLCnano liquid chromatography system (Dionex, Thermo Fisher Scientific) attached to a Q Exactive HF mass spectrometer (Thermo Fisher Scientific). They were concentrated on a µ-precolumn cartridge (PepMap100, C18, 5 µM, 100 Å, size 300 µm i.d. × 5 mm (Dionex, Thermo Fisher Scientific)) and further processed on an in house packed analytical column (ReproSil-Pur 120 C18-AQ, C18, 1.9 µM, 120 Å (Dr. A Maisch GmbH), packed into a 75 µm i.d. × 150 mm fused silica picotip emitter with a 8 µm tip (New Objective, Littleton, MA, USA).
The samples were processed via a 120 min multi-step analytical separation at a flow rate of 300 nL/min and a column temperature of 30 °C. Only LC-MS grade solvents were used (solvent A: water + 0.1% formic acid; solvent B: acetonitrile + 0.1% formic acid). The gradient with percentages of solvent B was programmed in the following way: 1% for 3 min; from 1% to 6% in 2 min; from 6% to 34% in 85 min; from 34% to 60% in 10 min; from 60% to 85% in 5 min; 85% for 7 min; from 85% to 1% in 3 min; 1% for 5 min.
Per analysis, 11 µL of the full proteome samples and 11.5 µL of the phosphopeptide samples were injected.
Mass spectrometric analysis was done with a full mass scan in the mass range between m/z 300 and 1750 at a resolution of 120,000, an AGC target of 3e6 charges and a maximum ion injection time of 20 ms in profile mode. Following this survey scan, the 15 most intense ions were selected, fragmented and measured in profile mode with the following parameters: resolution of 15,000; AGC target of 2e5 charges; maximum ion injection time of 100 ms; isolation window of 2 m/z, with an offset of +0.3 m/z; stepped normalized HCD energy of 20%, 25% and 30%. Signals with an unrecognized charge state or a charge state of 1, 7, 8 or higher were not picked for fragmentation. To avoid supersampling of the peptides, signals were excluded from the analysis for 25 s after being selected for isolation and fragmentation. The peptide match setting was set to “-” and the exclude isotope setting was set to “on”.
Data analysis of the phosphor-enriched samples was performed with MaxQuant version 1.5.1.0 (MPI for Biochemistry, Martinsried) with the following settings: variable modifications of acetyl (protein N-term), oxidation (M) and phosphorylation (STY); fixed modification of carbamidomethyl on cysteines. FDR-levels were set to 0.01. Fast LFQ was performed for quantification. The “match between runs”-feature was enabled. A mus musculus FASTA (proteome-ID: UP000000589) was used from uniprot. Trypsin was set as the protease and a maximum of 2 missed cleavages and a minimal peptide length of seven was chosen. Further data analysis was performed with Perseus. To do so, LFQ intensities obtained from MaxQuant analysis were log2-transformed and only proteins identified in at least three of the retinal tissue samples were retained.
4.10. Statistical Analysis
One-way analysis of variance and unpaired Student’s t test were used to evaluate significant differences between multiple groups and two groups, respectively. Differences were considered statistically significant when p < 0.05. Data were analysed and graphed using the GraphPad Prism software (GraphPad Software).

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




