Effect of Sepatronium Bromide (YM-155) on DNA Double-Strand Breaks Repair in Cancer Cells


{kind=link}
Abstract
:1. Introduction
2. Selected DNA Damage Response Pathways Relevant to the YM-155’s Mode of Action
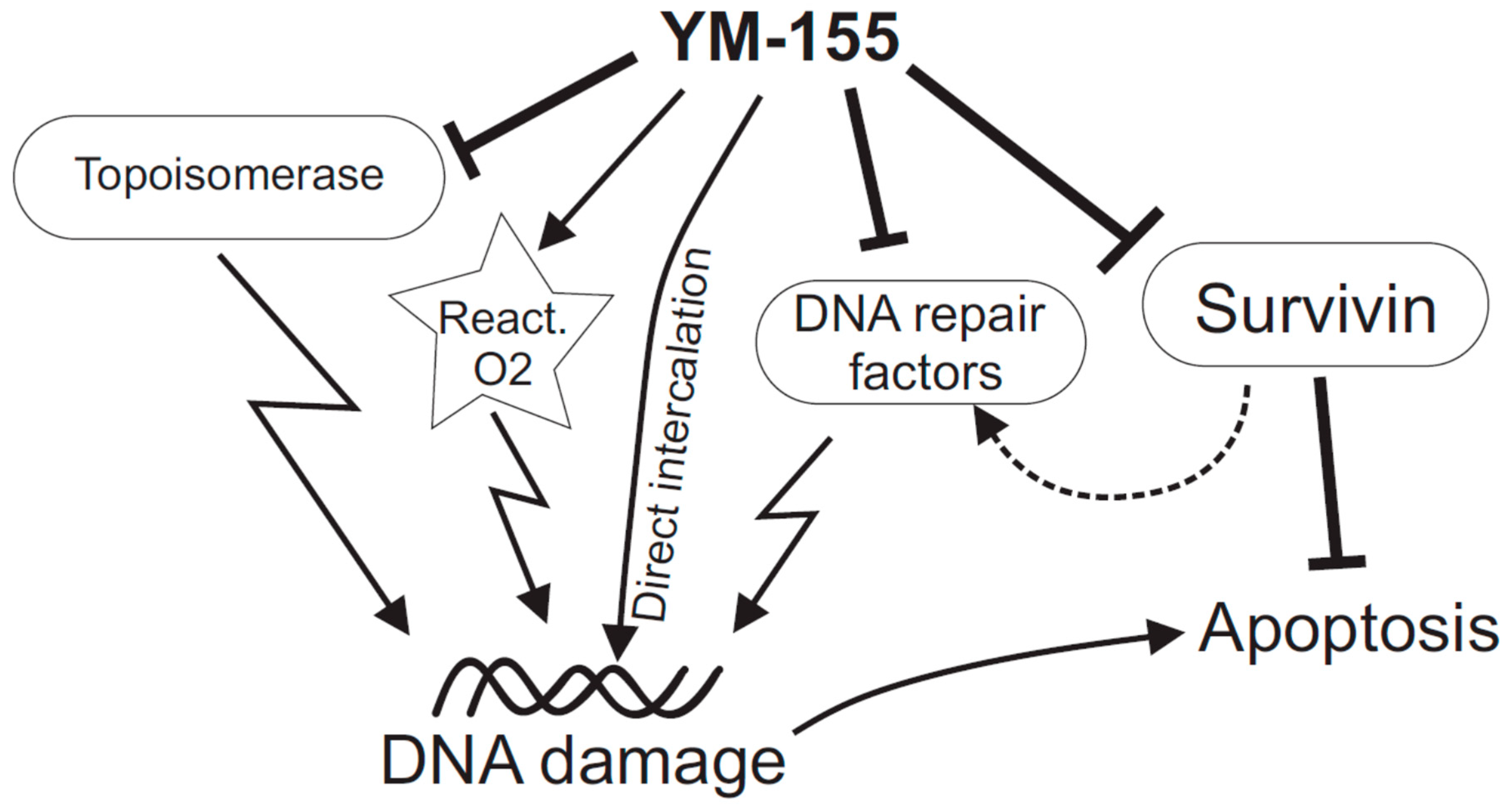
3. YM-155 Effects on DNA Integrity
3.1. Radio- and Chemosensitizing Effects of YM-155 Are Survivin-Independent
3.2. YM-155 Treatment Causes DNA Damage in Cells
3.3. Reported YM-155 Toxicity and Cell Cycle Arrest Is p53-Independent
3.4. DNA Damage as the Primary Mode of Action of YM-155
3.5. DNA Damage Pathways Affected by YM-155
3.6. Other Possible Molecular Mechanisms behind the DNA Damage Caused by YM-155
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Deveraux, Q.L.; Stennicke, H.R.; Salvesen, G.S.; Reed, J.C. Endogenous inhibitors of caspases. J. Clin. Immunol. 1999, 19, 388–398. [Google Scholar] [CrossRef]
- Deveraux, Q.L.; Reed, J.C. IAP family proteins-Suppressors of apoptosis. Genes Dev. 1999, 13, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.J.; Lopez-Chavez, A.; Citrin, D.; Janik, J.E.; Morris, J.C. Impacting tumor cell-fate by targeting the inhibitor of apoptosis protein survivin. Mol. Cancer 2011, 10, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, H.; Suri, P.; Gupta, J.C.; Talwar, G.P.; Dubey, S. Survivin: A unique target for tumor therapy. Cancer Cell Int. 2016, 16, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheatley, S.P.; Altieri, D.C. Survivin at a glance. J. Cell Sci. 2019, 132, jcs.223826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakahara, T.; Takeuchi, M.; Kinoyama, I.; Minematsu, T.; Shirasuna, K.; Matsuhisa, A.; Kita, A.; Tominaga, F.; Yamanaka, K.; Kudoh, M.; et al. YM155, a novel small-molecule survivin suppressant, induces regression of established human hormone-refractory prostate tumor xenografts. Cancer Res. 2007, 67, 8014–8021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauch, A.; Hennig, D.; Schäfer, C.; Wirth, M.; Marx, C.; Heinzel, T.; Schneider, G.; Krämer, O.H. Survivin and YM155: How faithful is the liaison? Biochim. Biophys. Acta-Rev. Cancer 2014, 1845, 202–220. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Mita, A.; Lewis, L.D.; Garrett, C.R.; Till, E.; Daud, A.I.; Patnaik, A.; Papadopoulos, K.; Takimoto, C.; Bartels, P.; et al. Phase I and pharmacokinetic study of YM155, a small-molecule inhibitor of survivin. J. Clin. Oncol. 2008, 26, 5198–5203. [Google Scholar] [CrossRef] [Green Version]
- De Vries, E.G.E.; De Jong, S. Exploiting the apoptotic route for cancer treatment: A single hit will rarely result in a home run. J. Clin. Oncol. 2008, 26, 5151–5153. [Google Scholar] [CrossRef]
- Iwasa, T.; Okamoto, I.; Suzuki, M.; Nakahara, T.; Yamanaka, K.; Hatashita, E.; Yamada, Y.; Fukuoka, M.; Ono, K.; Nakagawa, K. Radiosensitizing effect of YM155, a novel small-molecule survivin suppressant, in non-small cell lung cancer cell lines. Clin. Cancer Res. 2008, 14, 6496–6504. [Google Scholar] [CrossRef] [Green Version]
- Chakravarti, A.; Zhai, G.G.; Zhang, M.; Malhotra, R.; Latham, D.E.; Delaney, M.A.; Robe, P.; Nestler, U.; Song, Q.; Loeffler, J. Survivin enhances radiation resistance in primary human glioblastoma cells via caspase-independent mechanisms. Oncogene 2004, 23, 7494–7506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capalbo, G.; Dittmann, K.; Weiss, C.; Reichert, S.; Hausmann, E.; Rödel, C.; Rödel, F. Radiation-Induced Survivin Nuclear Accumulation is Linked to DNA Damage Repair. Int. J. Radiat. Oncol. Biol. Phys. 2010, 77, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Glaros, T.G.; Stockwin, L.H.; Mullendore, M.E.; Smith, B.; Morrison, B.L.; Newton, D.L. The “survivin suppressants” NSC 80467 and YM155 induce a DNA damage response. Cancer Chemother. Pharmacol. 2012, 70, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Holmes, D. Cancer drug’s survivin suppression called into question. Nat. Med. 2012, 18, 842–843. [Google Scholar] [CrossRef]
- Lindahl, T.; Barnes, D.E. Repair of Endogenous DNA Damage. In Cold Spring Harbor Symposia on Quantitative Biology; The Cold Spring Harbor Laboratory: Cold Spring, NY, USA, 2000; Volume 65, pp. 127–133. [Google Scholar] [CrossRef]
- Friedberg, E.C. A brief history of the DNA repair field. Cell Res. 2008, 18, 3–7. [Google Scholar] [CrossRef] [Green Version]
- Eltabbakh, G.H.; Awtrey, C.S. Current treatment for ovarian cancere. Expert Opin. Pharmacother. 2001, 2, 109–124. [Google Scholar] [CrossRef]
- Yang, J.; Xu, Z.P.; Huang, Y.; Hamrick, H.E.; Duerksen-Hughes, P.J.; Yu, Y.N. ATM and ATR: Sensing DNA damage. World J. Gastroenterol. 2004, 10, 155–160. [Google Scholar] [CrossRef]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Gaillard, H.; García-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–280. [Google Scholar] [CrossRef]
- Bartek, J.; Mistrik, M.; Bartkova, J. Thresholds of replication stress signaling in cancer development and treatment. Nat. Struct. Mol. Biol. 2012, 19, 5–7. [Google Scholar] [CrossRef]
- Mazzio, E.A.; Lewis, C.A.; Elhag, R.; Soliman, K.F. Effects of sepantronium bromide (YM-155) on the whole transcriptome of MDA-MB-231 cells: Highlight on impaired ATR/ATM fanconi anemia DNA damage response. Cancer Genom. Proteom. 2018, 15, 249–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toledo, L.I.; Altmeyer, M.; Rask, M.B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. XATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. γH2AX and cancer. Nat. Rev. Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Bétermier, M.; Bertrand, P.; Lopez, B.S. Is Non-Homologous End-Joining Really an Inherently Error-Prone Process? PLoS Genet. 2014, 10, e1004086. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, A.; Tapryal, N.; Venkova, T.; Horikoshi, N.; Pandita, R.K.; Sarker, A.H.; Sarkar, P.S.; Pandita, T.K.; Hazra, T.K. Classical non-homologous end-joining pathway utilizes nascent RNA for error-free double-strand break repair of transcribed genes. Nat. Commun. 2016, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Zhou, B.S.; Elledge, S.J. DNADamageResponse. Nature 2000, 408, 433–439. [Google Scholar] [CrossRef]
- Yamamoto, H.; Ngan, C.Y.; Monden, M. Cancer cells survive with survivin. Cancer Sci. 2008, 99, 1709–1714. [Google Scholar] [CrossRef]
- Cohen, C.; Lohmann, C.M.; Cotsonis, G.; Lawson, D.; Santoianni, R. Survivin expression in ovarian carcinoma: Correlation with apoptotic markers and prognosis. Mod. Pathol. 2003, 16, 574–583. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, N.; Yamauchi, T.; Hiramoto, M.; Yuri, M.; Naito, M.; Takeuchi, M.; Yamanaka, K.; Kita, A.; Nakahara, T.; Kinoyama, I.; et al. Interleukin enhancer-binding factor 3/NF110 is a target of YM155, a suppressant of survivin. Mol. Cell. Proteom. 2012, 11, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Mir, R.; Stanzani, E.; Martinez-Soler, F.; Villanueva, A.; Vidal, A.; Condom, E.; Ponce, J.; Gil, J.; Tortosa, A.; Giménez-Bonafé, P. YM155 sensitizes ovarian cancer cells to cisplatin inducing apoptosis and tumor regression. Gynecol. Oncol. 2014, 132, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Sales, L.; De Sousa, G.R.; Ferreira-Silva, G.; Castro-Gamero, A.M.; Ionta, M.; De Oliveira, J.C. YM155 induces apoptosis in p53-deficient T-acute lymphoblastic leukemia cells independent of survivin inhibition. Anticancer Drugs 2016, 28, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Sim, M.Y.; Huynh, H.; Go, M.L.; Yuen, J.S.P. Action of YM155 on clear cell renal cell carcinoma does not depend on surviving expression levels. PLoS ONE 2017, 12, e0178168. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Cheng, H.; Lu, J.; Zhan, L.; Zheng, J.; Cai, J.; Yang, X.; Xu, L.; Zhu, H.; Zhang, C.; et al. Small-molecule survivin inhibitor YM155 enhances radiosensitization in esophageal squamous cell carcinoma by the abrogation of G2checkpoint and suppression of homologous recombination repair. J. Hematol. Oncol. 2014, 7, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terzoudi, G.I.; Manola, K.N.; Pantelias, G.E.; Iliakis, G. Checkpoint abrogation in G2 compromises repair of chromosomal breaks in ataxia telangiectasia cells. Cancer Res. 2005, 65, 11292–11296. [Google Scholar] [CrossRef] [Green Version]
- Benada, J.; Macurek, L. Targeting the checkpoint to kill cancer cells. Biomolecules 2015, 5, 1912–1937. [Google Scholar] [CrossRef] [Green Version]
- Hong, M.; Ren, M.; Silva, J.; Kennedy, T.; Choi, J.; Cowell, J.K.; Hao, Z. Sepantronium is a DNA damaging agent that synergizes with PLK1 inhibitor volasertib. Am. J. Cancer Res. 2014, 4, 135–147. [Google Scholar]
- Rudolph, D.; Steegmaier, M.; Hoffmann, M.; Grauert, M.; Baum, A.; Quant, J.; Haslinger, C.; Garin-Chesa, P.; Adolf, G.R. BI 6727, a polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin. Cancer Res. 2009, 15, 3094–3102. [Google Scholar] [CrossRef] [Green Version]
- Medema, R.H.; Lin, C.C.; Yang, J.C.H. Polo-like kinase 1 inhibitors and their potential role in anticancer therapy, with a focus on NSCLC. Clin. Cancer Res. 2011, 17, 6459–6466. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.M.; Chang, Y.C.; Liu, C.Y.; Lee, J.Y.C.; Chan, H.H.; Kuo, C.W.; Lin, K.Y.; Tsai, S.L.; Chen, S.H.; Li, C.F.; et al. YM155 down-regulates survivin and XIAP, modulates autophagy and induces autophagy-dependent DNA damage in breast cancer cells. Br. J. Pharmacol. 2015, 172, 214–234. [Google Scholar] [CrossRef] [Green Version]
- Chang, B.H.; Johnson, K.; LaTocha, D.; Rowley, J.S.J.; Bryant, J.; Burke, R.; Smith, R.L.; Loriaux, M.; Müschen, M.; Mullighan, C.; et al. YM155 potently kills acute lymphoblastic leukemia cells through activation of the DNA damage pathway. J. Hematol. Oncol. 2015, 8, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyner, J.W.; Jemal, A.M.; Thayer, M.; Druker, B.J.; Chang, B.H. Targeting survivin and p53 in pediatric acute lymphoblastic leukemia. Leukemia 2012, 26, 623–632. [Google Scholar] [CrossRef]
- Véquaud, E.; Séveno, C.; Loussouarn, D.; Engelhart, L.; Campone, M.; Juin, P.; Barillé-Nion, S. YM155 potently triggers cell death in breast cancer cells through an autophagy-NF-kB network. Oncotarget 2015, 6, 13476–13486. [Google Scholar] [CrossRef] [PubMed]
- Sarosiek, K.A.; Ni Chonghaile, T.; Letai, A. Mitochondria: Gatekeepers of response to chemotherapy. Trends Cell Biol. 2013, 23, 612–619. [Google Scholar] [CrossRef] [Green Version]
- Janku, F.; McConkey, D.J.; Hong, D.S.; Kurzrock, R. Autophagy as a target for anticancer therapy. Nat. Rev. Clin. Oncol. 2011, 8, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Perkins, N.D. The diverse and complex roles of NF-κB subunits in cancer. Nat. Rev. Cancer 2012, 12, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Iwasa, T.; Okamoto, I.; Takezawa, K.; Yamanaka, K.; Nakahara, T.; Kita, A.; Koutoku, H.; Sasamata, M.; Hatashita, E.; Yamada, Y.; et al. Marked anti-tumour activity of the combination of YM155, a novel survivin suppressant, and platinum-based drugs. Br. J. Cancer 2010, 103, 36–42. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, M.; Paull, K.; Monks, A.; Hose, C.; Lee, J.S.; Weinstein, J.; Grever, M.; Bates, S.; Fojo, T. Generation of a drug resistance profile by quantitation of mdr-1/P- glycoprotein in the cell lines of the National Cancer Institute Anticancer Drug Screen. J. Clin. Investig. 1995, 95, 2205–2214. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2010, 461, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Fu, S.; Xu, X.; Chen, L.; Xu, J.; Li, B.; Qu, Y.; Yu, H.; Lu, S.; Li, W. The mechanism of radiosensitization by YM155, a novel small molecule inhibitor of survivin expression, is associated with DNA damage repair. Cell. Physiol. Biochem. 2015, 37, 1219–1230. [Google Scholar] [CrossRef]
- Véquaud, E.; Desplanques, G.; Jézéquel, P.; Juin, P.; Barillé-Nion, S. Survivin contributes to DNA repair by homologous recombination in breast cancer cells. Breast Cancer Res. Treat. 2016, 155, 53–63. [Google Scholar] [CrossRef] [Green Version]
- Jane, E.P.; Premkumar, D.R.; Sutera, P.A.; Cavaleri, J.M.; Pollack, I.F. Survivin inhibitor YM155 induces mitochondrial dysfunction, autophagy, DNA damage and apoptosis in Bcl-xL silenced glioma cell lines. Mol. Carcinog. 2017, 56, 1251–1265. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, X.; Xu, R.; Ji, J.; Xu, Y.; Han, M.; Wei, Y.; Huang, B.; Chen, A.; Zhang, Q.; et al. YM155 decreases radiation-induced invasion and reverses epithelial-mesenchymal transition by targeting STAT3 in glioblastoma. J. Transl. Med. 2018, 16, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Park, C.M.; Park, M.J.; Kwak, H.J.; Lee, H.C.; Kim, M.S.; Lee, S.H.; Park, I.C.; Rhee, C.H.; Hong, S. Il Ionizing radiation enhances matrix metalloproteinase-2 secretion and invasion of glioma cells through Src/epidermal growth factor receptor-mediated p38/Akt and phosphatidylinositol 3-kinase/Akt signaling pathways. Cancer Res. 2006, 66, 8511–8519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Bezawy, R.; Cominetti, D.; Fenderico, N.; Zuco, V.; Beretta, G.L.; Dugo, M.; Arrighetti, N.; Stucchi, C.; Rancati, T.; Valdagni, R.; et al. miR-875-5p counteracts epithelial-to-mesenchymal transition and enhances radiation response in prostate cancer through repression of the EGFR-ZEB1 axis. Cancer Lett. 2017, 395, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Tong, D.; Liu, Q.; Liu, G.; Xu, J.; Lan, W.; Jiang, Y.; Xiao, H.; Zhang, D.; Jiang, J. Metformin inhibits castration-induced EMT in prostate cancer by repressing COX2/PGE2/STAT3 axis. Cancer Lett. 2017, 389, 23–32. [Google Scholar] [CrossRef]
- Lau, J.; Ilkhanizadeh, S.; Wang, S.; Miroshnikova, Y.A.; Salvatierra, N.A.; Wong, R.A.; Schmidt, C.; Weaver, V.M.; Weiss, W.A.; Persson, A.I. STAT3 Blockade Inhibits Radiation-Induced Malignant Progression in Glioma. Cancer Res. 2015, 75, 4302–4311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, G.E.; Radic, B.; Mayor-Ruiz, C.; Blomen, V.A.; Trefzer, C.; Kandasamy, R.K.; Huber, K.V.M.; Gridling, M.; Chen, D.; Klampfl, T.; et al. The solute carrier SLC35F2 enables YM155-mediated DNA damage toxicity. Nat. Chem. Biol. 2014, 10, 768–773. [Google Scholar] [CrossRef] [Green Version]
- Hong, M.; Ren, M.Q.; Silva, J.; Paul, A.; Wilson, W.D.; Schroeder, C.; Weinberger, P.; Janik, J.; Hao, Z. YM155 inhibits topoisomerase function. Anticancer Drugs 2017, 28, 142–152. [Google Scholar] [CrossRef] [Green Version]
- Hevener, K.E.; Verstak, T.A.; Lutat, K.E.; Riggsbee, D.L.; Mooney, J.W. Recent developments in topoisomerase-targeted cancer chemotherapy. Acta Pharm. Sin. B 2018, 8, 844–861. [Google Scholar] [CrossRef]
- Cuya, S.M.; Bjornsti, M.A.; van Waardenburg, R.C.A.M. DNA topoisomerase-targeting chemotherapeutics: What’s new? Cancer Chemother. Pharmacol. 2017, 80, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Delgado, J.L.; Hsieh, C.M.; Chan, N.L.; Hiasa, H. Topoisomerases as Anticancer Targets. Biochem. J. 2018, 475, 373–398. [Google Scholar] [CrossRef] [PubMed]
- Wani, T.H.; Surendran, S.; Jana, A.; Chakrabarty, A.; Chowdhury, G. Quinone-Based Antitumor Agent Sepantronium Bromide (YM155) Causes Oxygen-Independent Redox-Activated Oxidative DNA Damage. Chem. Res. Toxicol. 2018, 31, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Loor, G.; Kondapalli, J.; Schriewer, J.M.; Chandel, N.S.; Vanden Hoek, T.L.; Schumacker, P.T. Menadione triggers cell death through ROS-dependent mechanisms involving PARP activation without requiring apoptosis. Free Radic. Biol. Med. 2010, 49, 1925–1936. [Google Scholar] [CrossRef] [Green Version]
- Wani, T.H.; Surendran, S.; Mishra, V.S.; Chaturvedi, J.; Chowdhury, G.; Chakrabarty, A. Adaptation to chronic exposure to sepantronium bromide (YM155), a prototypical survivin suppressant is due to persistent DNA damage-response in breast cancer cells. Oncotarget 2018, 9, 33589–33600. [Google Scholar] [CrossRef]
- Cree, I.A.; Charlton, P. Molecular chess? Hallmarks of anti-cancer drug resistance. BMC Cancer 2017, 17, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Satoh, T.; Okamoto, I.; Miyazaki, M.; Morinaga, R.; Tsuya, A.; Hasegawa, Y.; Terashima, M.; Ueda, S.; Fukuoka, M.; Ariyoshi, Y.; et al. Phase I study of YM155, a novel survivin suppressant, in patients with advanced solid tumors. Clin. Cancer Res. 2009, 15, 3872–3880. [Google Scholar] [CrossRef] [Green Version]
- Skrott, Z.; Mistrik, M.; Andersen, K.K.; Friis, S.; Majera, D.; Gursky, J.; Ozdian, T.; Bartkova, J.; Turi, Z.; Moudry, P.; et al. Alcohol-abuse drug disulfiram targets cancer via p97 segregase adaptor NPL4. Nature 2017, 552, 194–199. [Google Scholar] [CrossRef]
- Majera, D.; Skrott, Z.; Chroma, K.; Merchut-Maya, J.M.; Mistrik, M.; Bartek, J. Targeting the NPL4 Adaptor of p97/VCP Segregase by Disulfiram as an Emerging Cancer Vulnerability Evokes Replication Stress and DNA Damage while Silencing the ATR Pathway. Cells 2020, 9, 469. [Google Scholar] [CrossRef] [Green Version]
- Skrott, Z.; Majera, D.; Gursky, J.; Buchtova, T.; Hajduch, M.; Mistrik, M.; Bartek, J. Disulfiram’s anti-cancer activity reflects targeting NPL4, not inhibition of aldehyde dehydrogenase. Oncogene 2019, 38, 6711–6722. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majera, D.; Mistrik, M. Effect of Sepatronium Bromide (YM-155) on DNA Double-Strand Breaks Repair in Cancer Cells. Int. J. Mol. Sci. 2020, 21, 9431. https://doi.org/10.3390/ijms21249431
Majera D, Mistrik M. Effect of Sepatronium Bromide (YM-155) on DNA Double-Strand Breaks Repair in Cancer Cells. International Journal of Molecular Sciences. 2020; 21(24):9431. https://doi.org/10.3390/ijms21249431
Chicago/Turabian StyleMajera, Dusana, and Martin Mistrik. 2020. "Effect of Sepatronium Bromide (YM-155) on DNA Double-Strand Breaks Repair in Cancer Cells" International Journal of Molecular Sciences 21, no. 24: 9431. https://doi.org/10.3390/ijms21249431
APA StyleMajera, D., & Mistrik, M. (2020). Effect of Sepatronium Bromide (YM-155) on DNA Double-Strand Breaks Repair in Cancer Cells. International Journal of Molecular Sciences, 21(24), 9431. https://doi.org/10.3390/ijms21249431