Differential Co-Expression Analyses Allow the Identification of Critical Signalling Pathways Altered during Tumour Transformation and Progression


Abstract
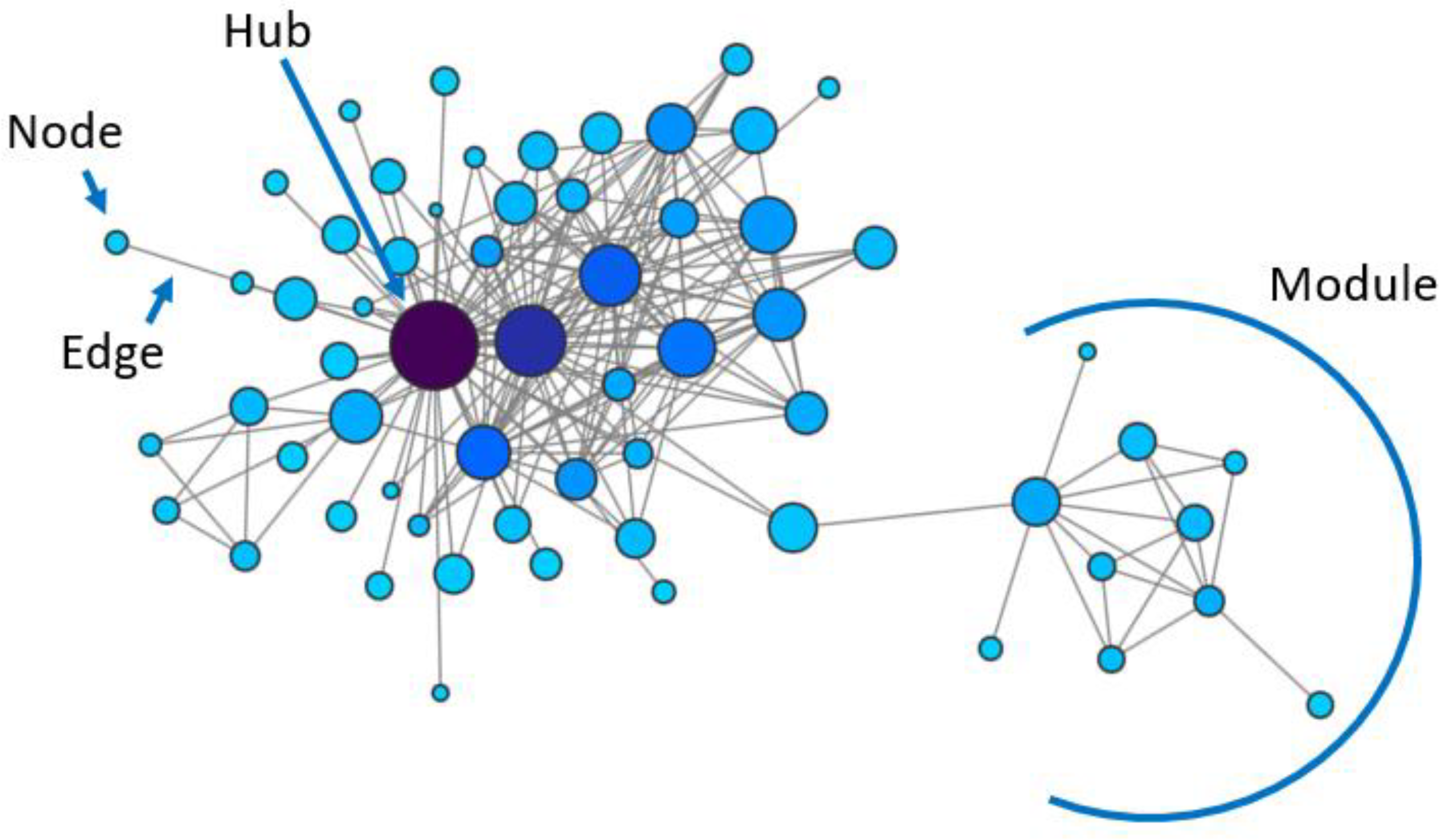
:1. Biological Networks
2. Gene Co-Expression Networks
3. Methods for Differential Co-Expression Analysis
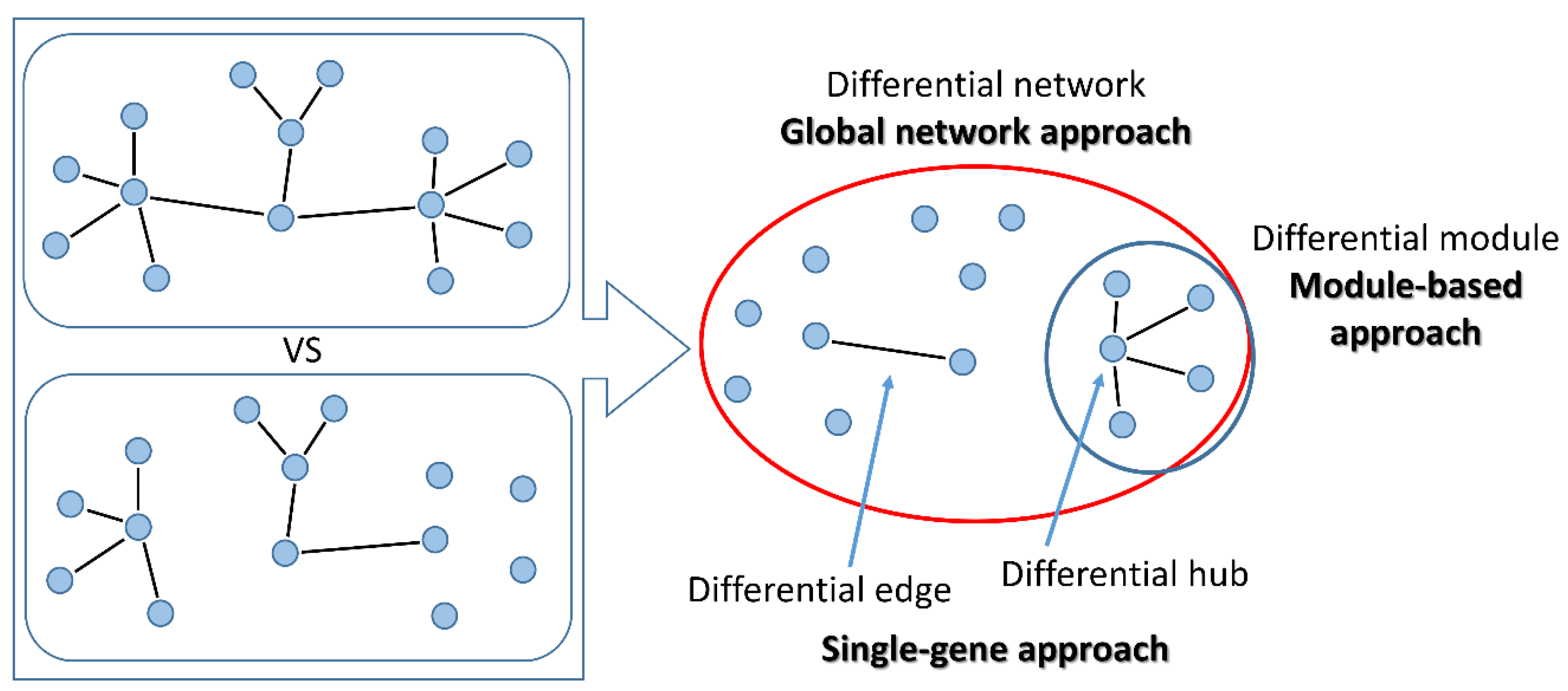
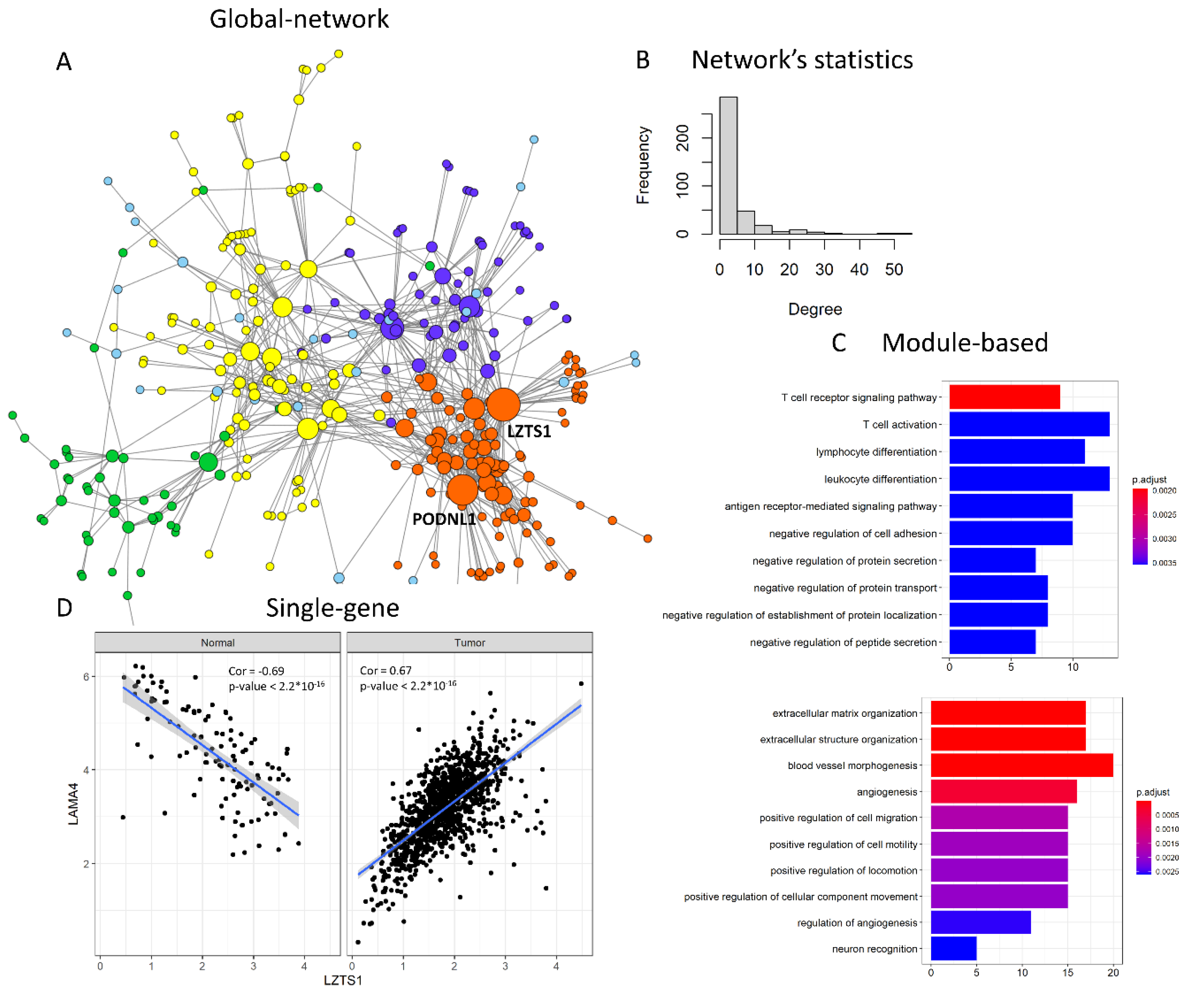
- “global network” approaches aim at reconstructing the whole differential co-expression network between two or multiple conditions. Global features of the differential network can be studied, such as edge distribution, modularity or entropy.
- “module-based” approaches aim at identifying groups of co-regulated genes that are differentially interconnected under specific conditions. Usually, differences in connectivity within a module are analysed, but also methods for the identification of pairs of DC modules (connectivity between modules) have been proposed. Additionally, modules can be either identified unbiasedly from data, or pre-specified based on prior knowledge (here defined as “pathway-based” methods).
- “single-gene” approaches study the change in co-expression between pairs of genes or between a gene and its neighbours in the network. These approaches are particularly suited to select experimentally testable hypotheses. In principle, all “global network” methods can be used to drive “gene-specific” outputs using node-centred metrics that summarise the relevance of a gene within the differential network. On the other side, single genes thus identified can be used as seeds to build differential modules with neighbouring genes within the network.
4. Differential Co-Expression Networks in Cancer
4.1. Global Topological Features of Cancer Networks Show Increasingly High Entropy
4.2. Pathways Dysregulated in Cancer
4.3. Differentially Co-Regulated Genes
4.4. Regulatory Mechanisms
5. Conclusions and Perspectives
Funding
Conflicts of Interest
Abbreviations
| CNV | Copy Number Variation |
| DC | Differentially Co-expressed |
| GRN | Gene Regulatory Network |
| KD | Knock-Down |
| PPI | Protein–Protein Interaction |
| SNP | Single Nucleotide Polymorphism |
| TF | Transcription Factor |
References
- Barabási, A.-L.; Oltvai, Z.N. Network Biology: Understanding the Cell’s Functional Organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Ingber, D.E. A Non-Genetic Basis for Cancer Progression and Metastasis: Self-Organizing Attractors in Cell Regulatory Networks. Breast Dis. 2006, 26, 27–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Srivas, R.; Shen, J.P.; Yang, C.C.; Sun, S.M.; Li, J.; Gross, A.M.; Jensen, J.; Licon, K.; Bojorquez-Gomez, A.; Klepper, K.; et al. A Network of Conserved Synthetic Lethal Interactions for Exploration of Precision Cancer Therapy. Mol. Cell 2016, 63, 514–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conte, F.; Fiscon, G.; Licursi, V.; Bizzarri, D.; D’Antò, T.; Farina, L.; Paci, P. A Paradigm Shift in Medicine: A Comprehensive Review of Network-Based Approaches. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194416. [Google Scholar] [CrossRef] [PubMed]
- Derisi, J.L.; Iyer, V.R.; Brown, P. Exploring the Metabolic and Genetic Control of Gene Expression on a Genomic Scale. Science 1997, 278, 680–686. [Google Scholar] [CrossRef] [Green Version]
- Jansen, R.; Greenbaum, D.; Gerstein, M. Relating Whole-Genome Expression Data with Protein-Protein Interactions. Genome Res. 2002, 12, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Ge, H.; Liu, Z.; Church, G.M.; Vidal, M. Correlation between Transcriptome and Interactome Mapping Data from Saccharomyces Cerevisiae. Nat. Genet. 2001, 29, 482–486. [Google Scholar] [CrossRef]
- Kemmeren, P.; Van Berkum, N.L.; Vilo, J.; Bijma, T.; Donders, R.; Brazma, A.; Holstege, F.C.P. Protein Interaction Verification and Functional Annotation by Integrated Analysis of Genome-Scale Data. Mol. Cell 2002, 9, 1133–1143. [Google Scholar] [CrossRef]
- Holding, A.N.; Giorgi, F.M.; Donnelly, A.; Cullen, A.E.; Nagarajan, S.; Selth, L.A.; Markowetz, F. VULCAN Integrates ChIP-Seq with Patient-Derived Co-Expression Networks to Identify GRHL2 as a Key Co-Regulator of ERa at Enhancers in Breast Cancer. Genome Biol. 2019, 20, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Salwinski, L.; Miller, C.S.; Smith, A.J.; Pettit, F.K.; Bowie, J.U.; Eisenberg, D. The Database of Interacting Proteins: 2004 Update. Nucleic Acids Res. 2004, 32, D449–D451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Licata, L.; Briganti, L.; Peluso, D.; Perfetto, L.; Iannuccelli, M.; Galeota, E.; Sacco, F.; Palma, A.; Nardozza, A.P.; Santonico, E.; et al. MINT, the Molecular Interaction Database: 2012 Update. Nucleic Acids Res. 2012, 40, D857–D861. [Google Scholar] [CrossRef] [PubMed]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; del-Toro, N.; et al. The MIntAct Project—IntAct as a Common Curation Platform for 11 Molecular Interaction Databases. Nucleic Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oughtred, R.; Stark, C.; Breitkreutz, B.-J.; Rust, J.; Boucher, L.; Chang, C.; Kolas, N.; O’Donnell, L.; Leung, G.; McAdam, R.; et al. The BioGRID Interaction Database: 2019 Update. Nucleic Acids Res. 2019, 47, D529–D541. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein–Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Obayashi, T.; Kagaya, Y.; Aoki, Y.; Tadaka, S.; Kinoshita, K. COXPRESdb v7: A Gene Coexpression Database for 11 Animal Species Supported by 23 Coexpression Platforms for Technical Evaluation and Evolutionary Inference. Nucleic Acids Res. 2019, 47, D55–D62. [Google Scholar] [CrossRef]
- Zhu, Q.; Wong, A.K.; Krishnan, A.; Aure, M.R.; Tadych, A.; Zhang, R.; Corney, D.C.; Greene, C.S.; Bongo, L.A.; Kristensen, V.N.; et al. Targeted Exploration and Analysis of Large Cross-Platform Human Transcriptomic Compendia. Nat. Methods 2015, 12, 211–214. [Google Scholar] [CrossRef] [Green Version]
- Jeong, H.; Mason, S.P.; Barabási, A.-L.; Oltvai, Z.N. Lethality and Centrality in Protein Networks. Nature 2001, 411, 41. [Google Scholar] [CrossRef] [Green Version]
- Pržulj, N.; Wigle, D.A.; Jurisica, I. Functional Topology in a Network of Protein Interactions. Bioinformatics 2004, 20, 340–348. [Google Scholar] [CrossRef]
- Furlong, L.I. Human Diseases through the Lens of Network Biology. Trends Genet. 2013, 29, 150–159. [Google Scholar] [CrossRef]
- Malod-dognin, N.; Petschnigg, J.; Windels, S.F.L.; Povh, J.; Hemmingway, H.; Ketteler, R. Towards a Data-Integrated Cell. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, P.F.; Bates, P.A. Global Topological Features of Cancer Proteins in the Human Interactome. Bioinformatics 2006, 22, 2291–2297. [Google Scholar] [CrossRef] [PubMed]
- Feldman, I.; Rzhetsky, A.; Vitkup, D. Network Properties of Genes Harboring Inherited Disease Mutations. Proc. Natl. Acad. Sci. USA 2008, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ala, U.; Piro, R.M.; Grassi, E.; Damasco, C.; Silengo, L.; Oti, M.; Provero, P.; Di Cunto, F. Prediction of Human Disease Genes by Human-Mouse Conserved Coexpression Analysis. PLoS Comput. Biol. 2008, 4, e1000043. [Google Scholar] [CrossRef]
- Magger, O.; Waldman, Y.Y.; Ruppin, E.; Sharan, R. Enhancing the Prioritization of Disease-Causing Genes through Tissue Specific Protein Interaction Networks. PLoS Comput. Biol. 2012, 8, e1002690. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Zhang, J.; Liu, Q.; Wang, J.; Wu, F.X. Prediction of Disease-Related Genes Based on Weighted Tissue-Specific Networks by Using DNA Methylation. BMC Med. Genom. 2014, 7, S4. [Google Scholar] [CrossRef] [Green Version]
- Rives, A.W.; Galitski, T. Modular Organization of Cellular Networks. Proc. Natl. Acad. Sci. USA 2003, 100, 1128–1133. [Google Scholar] [CrossRef] [Green Version]
- Eisen, M.B.; Spellman, P.T.; Brown, P.O.; Botstein, D. Cluster Analysis and Display of Genome-Wide Expression Patterns. Proc. Natl. Acad. Sci. USA 1998, 95, 14863–14868. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Han, L.; Yuan, Y.; Li, J.; Hei, N.; Liang, H. Gene Co-Expression Network Analysis Reveals Common System-Level Properties of Prognostic Genes across Cancer Types. Nat. Commun. 2014, 5, 3231. [Google Scholar] [CrossRef] [Green Version]
- Luscombe, N.M.; Babu, M.M.; Yu, H. Genomic Analysis of Regulatory Network Dynamics Reveals Large Topological Changes. Lett. Nat. 2004, 431, 714–717. [Google Scholar] [CrossRef]
- Neph, S.; Stergachis, A.B.; Reynolds, A.; Sandstrom, R.; Borenstein, E.; Stamatoyannopoulos, J.A. Circuitry and Dynamics of Human Transcription Factor Regulatory Networks. Cell 2012, 150, 1274–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayer, D.E.; Eisenman, R.N. A Switch from Myc:Max to Mad:Max Heterocomplexes Accompanies Monocyte/Macrophage Differentiation. Genes Dev. 1993, 7, 2110–2119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Fuente, A. From “differential Expression” to “Differential Networking”—Identification of Dysfunctional Regulatory Networks in Diseases. Trends Genet. 2010, 26, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y. Genome-Wide Co-Expression Based Prediction of Differential Expressions. Bioinformatics 2008, 24, 666–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, N.J.; Reverter, A.; Dalrymple, B.P. A Differential Wiring Analysis of Expression Data Correctly Identifies the Gene Containing the Causal Mutation. PLoS Comput. Biol. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, H.A.; Bhattacharyya, D.K.; Kalita, J.K. (Differential) Co-Expression Analysis of Gene Expression: A Survey of Best Practices. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 17, 1154–1173. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, S.; Mehta, M.; Kuo, D.; Sung, M.K.; Chuang, R.; Jaehnig, E.J.; Bodenmiller, B.; Licon, K.; Copeland, W.; Shales, M.; et al. Rewiring of Genetic Networks in Response to DNA Damage. Science 2010, 330, 1385–1389. [Google Scholar] [CrossRef] [Green Version]
- Basha, O.; Shpringer, R.; Argov, C.M.; Yeger-Lotem, E. The DifferentialNet Database of Differential Protein-Protein Interactions in Human Tissues. Nucleic Acids Res. 2018, 46, D522–D526. [Google Scholar] [CrossRef]
- van Dam, S.; Võsa, U.; van der Graaf, A.; Franke, L.; de Magalhães, J.P. Gene Co-Expression Analysis for Functional Classification and Gene-Disease Predictions. Brief. Bioinform. 2018, 19, 575–592. [Google Scholar] [CrossRef]
- Ha, M.J.; Baladandayuthapani, V.; Do, K.A. DINGO: Differential Network Analysis in Genomics. Bioinformatics 2015, 31, 3413–3420. [Google Scholar] [CrossRef] [Green Version]
- Ho, Y.-Y.; Cope, L.; Dettling, M.; Parmigiani, G. Statistical Methods for Identifying Differentially Expressed Gene Combinations; Ochs, M.F., Ed.; Humana Press: Totowa, NJ, USA, 2007; pp. 171–191. [Google Scholar] [CrossRef]
- McKenzie, A.T.; Katsyv, I.; Song, W.M.; Wang, M.; Zhang, B. DGCA: A Comprehensive R Package for Differential Gene Correlation Analysis. BMC Syst. Biol. 2016, 10, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siska, C.; Bowler, R.; Kechris, K. The Discordant Method: A Novel Approach for Differential Correlation. Bioinformatics 2016, 32, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, T.H.; Chiu, Y.C.; Hsu, P.Y.; Lu, T.P.; Lai, L.C.; Tsai, M.H.; Huang, T.H.M.; Chuang, E.Y.; Chen, Y. Differential Network Analysis Reveals the Genome-Wide Landscape of Estrogen Receptor Modulation in Hormonal Cancers. Sci. Rep. 2016, 6, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, J.A.; Ye, S.; Kendziorski, C. R/Ebcoexpress: An Empirical Bayesian Framework for Discovering Differential Co-Expression. Bioinformatics 2012, 28, 1939–1940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, J.H.; Lazarus, R.; Carey, V.J.; Raby, B.A. Quantifying Differential Gene Connectivity between Disease States for Objective Identification of Disease-Relevant Genes. BMC Syst. Biol. 2011, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, D.; Gu, Q.; Ma, J. Identifying Gene Regulatory Network Rewiring Using Latent Differential Graphical Models. Nucleic Acids Res. 2016, 44, 1–11. [Google Scholar] [CrossRef]
- Gill, R.; Datta, S.; Datta, S. A Statistical Framework for Differential Network Analysis from Microarray Data. BMC Bioinform. 2010, 11, 95. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Li, H.; Riggins, R.B.; Zhan, M.; Xuan, J.; Zhang, Z.; Hoffman, E.P.; Clarke, R.; Wang, Y. Differential Dependency Network Analysis to Identify Condition-Specific Topological Changes in Biological Networks. Bioinformatics 2009, 25, 526–532. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.D.; Cai, T.T.; Li, H. Direct Estimation of Differential Networks. Biometrika 2014, 101, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Ji, J.; He, D.; Feng, Y.; He, Y.; Xue, F.; Xie, L. JDINAC: Joint Density-Based Non-Parametric Differential Interaction Network Analysis and Classification Using High-Dimensional Sparse Omics Data. Bioinformatics 2017, 33, 3080–3087. [Google Scholar] [CrossRef]
- Zhang, X.F.; Ou-Yang, L.; Zhao, X.M.; Yan, H. Differential Network Analysis from Cross-Platform Gene Expression Data. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Xin, M.; Feldmann, K.A.; Wang, X. Machine Learning-Based Differential Network Analysis: A Study of Stress-Responsive Transcriptomes in Arabidopsis. Plant Cell 2014, 26, 520–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Yu, H.; Liu, B.H.; Zhao, Z.; Liu, L.; Ma, L.X.; Li, Y.X.; Li, Y.Y. DCGL v2.0: An R Package for Unveiling Differential Regulation from Differential Co-Expression. PLoS ONE 2013, 8, e79729. [Google Scholar] [CrossRef] [PubMed]
- Lui, T.W.H.; Tsui, N.B.Y.; Chan, L.W.C.; Wong, C.S.C.; Siu, P.M.F.; Yung, B.Y.M. DECODE: An Integrated Differential Co-Expression and Differential Expression Analysis of Gene Expression Data. BMC Bioinform. 2015, 16, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Mo, W.; Fu, X.; Han, X.T.; Yang, G.Y.; Zhang, J.G.; Guo, F.H.; Huang, Y.; Mao, Y.M.; Li, Y.; Xie, Y. A Stochastic Model for Identifying Differential Gene Pair Co-Expression Patterns in Prostate Cancer Progression. BMC Genom. 2009, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Gan, C.Y.; Wang, W.; Liao, L.D.; Li, C.Q.; Xu, L.Y.; Li, E.M. Identification of LncRNA-Associated Differential Subnetworks in Oesophageal Squamous Cell Carcinoma by Differential Co-Expression Analysis. J. Cell. Mol. Med. 2020, 24, 4804–4818. [Google Scholar] [CrossRef]
- Hu, R.; Qiu, X.; Glazko, G. A New Gene Selection Procedure Based on the Covariance Distance. Bioinformatics 2009, 26, 348–354. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Wang, J.; Jiang, Y.; Liang, Y.; Xu, D. BFDCA: A Comprehensive Tool of Using Bayes Factor for Differential Co-Expression Analysis. J. Mol. Biol. 2017, 429, 446–453. [Google Scholar] [CrossRef] [Green Version]
- Ray, S.; Lall, S.; Bandyopadhyay, S. OPEN CODC: A Copula-Based Model to Identify Differential Coexpression. NPJ Syst. Biol. Appl. 2020, 1–13. [Google Scholar] [CrossRef]
- Zhang, J.; Ji, Y.; Zhang, L. Extracting Three-Way Gene Interactions from Microarray Data. Bioinformatics 2007, 23, 2903–2909. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Saito, M.; Bisikirska, B.C.; Alvarez, M.J.; Lim, W.K.; Rajbhandari, P.; Shen, Q.; Nemenman, I.; Basso, K.; Margolin, A.A.; et al. Genome-Wide Identification of Post-Translational Modulators of Transcription Factor Activity in Human B Cells. Nat. Biotechnol. 2009, 27, 829–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amar, D.; Safer, H.; Shamir, R. Dissection of Regulatory Networks That Are Altered in Disease via Differential Co-Expression. PLoS Comput. Biol. 2013, 9, e1002955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesson, B.M.; Breitling, R.; Jansen, R.C. DiffCoEx: A Simple and Sensitive Method to Find Differentially Coexpressed Gene Modules. BMC Bioinform. 2010, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Zhang, Z.; Dai, E.N.; Tian, J.X.; Xin, J.Z.; Xu, L. Modeling Osteosarcoma Progression by Measuring the Connectivity Dynamics Using an Inference of Multiple Differential Modules Algorithm. Mol. Med. Rep. 2017, 16, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Werner-Washburne, M.; Lane, T. A Multiple Network Learning Approach to Capture System-Wide Condition-Specific Responses. Bioinformatics 2011, 27, 1832–1838. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Moreno-Moral, A.; Rotival, M.; Bottolo, L.; Petretto, E. Multi-Tissue Analysis of Co-Expression Networks by Higher-Order Generalized Singular Value Decomposition Identifies Functionally Coherent Transcriptional Modules. PLoS Genet. 2014, 10. [Google Scholar] [CrossRef] [Green Version]
- Watson, M. CoXpress: Differential Co-Expression in Gene Expression Data. BMC Bioinform. 2006, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Fukushima, A. DiffCorr: An R Package to Analyze and Visualize Differential Correlations in Biological Networks. Gene 2013, 518, 209–214. [Google Scholar] [CrossRef] [Green Version]
- Amar, D.; Shamir, R. Constructing Module Maps for Integrated Analysis of Heterogeneous Biological Networks. Nucleic Acids Res. 2014, 42, 4208–4219. [Google Scholar] [CrossRef]
- Padi, M.; Quackenbush, J. Detecting Phenotype-Driven Transitions in Regulatory Network Structure. NPJ Syst. Biol. Appl. 2018, 4. [Google Scholar] [CrossRef] [Green Version]
- Ray, S.; Maulik, U. Identifying Differentially Coexpressed Module during HIV Disease Progression: A Multiobjective Approach. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, G.; Kuang, R.; Pandey, G.; Steinbach, M.; Myers, C.L.; Kumar, V. Subspace Differential Coexpression Analysis: Problem Definition and a General Approach. Pac. Symp. Biocomput. 2010, 1, 145–156. [Google Scholar]
- Jiang, J.; Yin, X.Y.; Song, X.W.; Xie, D.; Xu, H.J.; Yang, J.; Sun, L.R. EgoNet Identifies Differential Ego-Modules and Pathways Related to Prednisolone Resistance in Childhood Acute Lymphoblastic Leukemia. Hematology 2018, 23, 221–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, C.; McDowell, I.C.; Zhao, S.; Brown, C.D.; Engelhardt, B.E. Context Specific and Differential Gene Co-Expression Networks via Bayesian Biclustering. PLoS Comput. Biol. 2016, 12, e1004791. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Brown, J.B.; Orsini, L.; Pan, Z.; Hu, G.; He, S. MODA: MOdule Differential Analysis for Weighted Gene Co-Expression Network. bixRiv 2016, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Schadt, E.E.; Kaplan, L.M.; Zhao, H. COSINE: COndition-SpecIfic Sub-NEtwork Identification Using a Global Optimization Method. Bioinformatics 2011, 27, 1290–1298. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Shang, X.; Li, X.; Liu, W.; Li, Z. Efficient Mining Differential Co-Expression Biclusters in Microarray Datasets. Gene 2013, 518, 59–69. [Google Scholar] [CrossRef]
- Dong, L.Y.; Zhou, W.Z.; Ni, J.W.; Wei, X.; Hu, W.H.; Yu, C.; Li, H.Y. Identifying the Optimal Gene and Gene Set in Hepatocellular Carcinoma Based on Differential Expression and Differential Co-Expression Algorithm. Oncol. Rep. 2017, 37, 1066–1074. [Google Scholar] [CrossRef]
- Lanciano, T.; Bonchi, F.; Gionis, A. Explainable Classification of Brain Networks via Contrast Subgraphs. In Proceedings of the 26th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, San Diego, CA, USA, 23–27 August 2020; KDD ’20. Association for Computing Machinery: New York, NY, USA, 2020; pp. 3308–3318. [Google Scholar] [CrossRef]
- Freudenberg, J.M.; Sivaganesan, S.; Wagner, M.; Medvedovic, M. A Semi-Parametric Bayesian Model for Unsupervised Differential Co-Expression Analysis. BMC Bioinform. 2010, 11. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Kendziorski, C. Statistical Methods for Gene Set Co-Expression Analysis. Bioinformatics 2009, 25, 2780–2786. [Google Scholar] [CrossRef] [Green Version]
- Rahnenführer, J.; Domingues, F.S.; Maydt, J.; Lengauer, T. Calculating the Statistical Significance of Changes in Pathway Activity from Gene Expression Data. Stat. Appl. Genet. Mol. Biol. 2004, 3. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, J.; Deng, H.W. Identifying Gene Interaction Enrichment for Gene Expression Data. PLoS ONE 2009, 4, e0008064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Siqueira Santos, S.; De Almeida Galatro, T.F.; Watanabe, R.A.; Oba-Shinjo, S.M.; Marie, S.K.N.; Fujita, A. CoGA: An R Package to Identify Differentially Co-Expressed Gene Sets by Analyzing the Graph Spectra. PLoS ONE 2015, 10, e0135831. [Google Scholar] [CrossRef]
- Cho, S.; Kim, J.; Kim, J.H. Identifying Set-Wise Differential Co-Expression in Gene Expression Microarray Data. BMC Bioinform. 2009, 10, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Shi, X.; Zhang, Y.; Xu, Y.; Jiang, Y.; Zhang, C.; Feng, L.; Yang, H.; Shang, D.; Sun, Z.; et al. ESEA: Discovering the Dysregulated Pathways Based on Edge Set Enrichment Analysis. Sci. Rep. 2015, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hung, J.H.; Whitfield, T.W.; Yang, T.H.; Hu, Z.; Weng, Z.; DeLisi, C. Identification of Functional Modules That Correlate with Phenotypic Difference: The Influence of Network Topology. Genome Biol. 2010, 11. [Google Scholar] [CrossRef] [Green Version]
- Jung, S. KEDDY: A Knowledge-Based Statistical Gene Set Test Method to Detect Differential Functional Protein–Protein Interactions. Bioinformatics 2019, 35, 619–627. [Google Scholar] [CrossRef]
- Tian, Y.; Zhang, B.; Hoffman, E.P.; Clarke, R.; Zhang, Z.; Shih, I.M.; Xuan, J.; Herrington, D.M.; Wang, Y. Knowledge-Fused Differential Dependency Network Models for Detecting Significant Rewiring in Biological Networks. BMC Syst. Biol. 2014, 8, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.; Wu, B.; Chen, L.; Zhao, H. A Statistical Method for Identifying Differential Gene-Gene Co-Expression Patterns. Bioinformatics 2004, 20, 3146–3155. [Google Scholar] [CrossRef] [Green Version]
- Fazlollahi, M.; Muroff, I.; Lee, E.; Causton, H.C.; Bussemaker, H.J. Identifying Genetic Modulators of the Connectivity between Transcription Factors and Their Transcriptional Targets. Proc. Natl. Acad. Sci. USA 2016, 113, E1835–E1843. [Google Scholar] [CrossRef] [Green Version]
- Lareau, C.A.; White, B.C.; Montgomery, C.G.; McKinney, B.A. DcVar: A Method for Identifying Common Variants That Modulate Differential Correlation Structures in Gene Expression Data. Front. Genet. 2015, 6, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayano, M.; Takigawa, I.; Shiga, M.; Tsuda, K.; Mamitsuka, H. Efficiently Finding Genome-Wide Three-Way Gene Interactions from Transcript- and Genotype-Data. Bioinformatics 2009, 25, 2735–2743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liany, H.; Rajapakse, J.C.; Karuturi, R.K.M. MultiDCoX: Multi-Factor Analysis of Differential Co-Expression. BMC Bioinform. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Cui, Y.; Yu, G.; Li, R.; Ressom, H.W. Incorporating Prior Biological Knowledge for Network-Based Differential Gene Expression Analysis Using Differentially Weighted Graphical LASSO. BMC Bioinform. 2017, 18, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Park, H.J.; Kim, S.; Li, W. Model-Based Analysis of Competing-Endogenous Pathways (MACPath) in Human Cancers. PLoS Comput. Biol. 2018, 14, 1–16. [Google Scholar] [CrossRef]
- Hansen, M.; Everett, L.; Singh, L.; Hannenhalli, S. Mimosa: Mixture Model of Co-Expression to Detect Modulators of Regulatory Interaction. Algorithms Mol. Biol. 2010, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Shimamura, T.; Matsui, Y.; Kajino, T.; Ito, S.; Takahashi, T.; Miyano, S. GIMLET: Identifying Biological Modulators in Context-Specific Gene Regulation Using Local Energy Statistics. In Lecture Notes Computer Science (Including Its Subseries Lecture Notes in Artificial Intelligence (LNAI) and Lecture Notes in Bioinformatics); Springer: New York, NY, USA, 2019; Volume 10834, pp. 124–137. [Google Scholar] [CrossRef]
- Babur, Ö.; Demir, E.; Gönen, M.; Sander, C.; Dogrusoz, U. Discovering Modulators of Gene Expression. Nucleic Acids Res. 2010, 38, 5648–5656. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Bastian, M.; Heymann, S. Gephi: An Open Source Software for Exploring and Manipulating Networks; AAAI: New York, NY, USA, 2009. [Google Scholar]
- Bhuva, D.D.; Cursons, J.; Smyth, G.K.; Davis, M.J. Differential Co-Expression-Based Detection of Conditional Relationships in Transcriptional Data: Comparative Analysis and Application to Breast Cancer. Genome Biol. 2019, 20, 1–21. [Google Scholar] [CrossRef]
- Csardi, G.; Nepusz, T. The Igraph Software Package for Complex Network Research. InterJ. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. ClusterProfiler: An R Package for Comparing Biological Themes among Gene Clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichtblau, Y.; Zimmermann, K.; Haldemann, B.; Lenze, D.; Hummel, M.; Leser, U. Comparative Assessment of Differential Network Analysis Methods. Brief. Bioinform. 2017, 18, 837–850. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Valbuena, E.E.; Treviño, V. Metrics to Estimate Differential Co-Expression Networks. BioData Min. 2017, 10, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, J.; Bianconi, G.; Severini, S.; Teschendorff, A.E. Differential Network Entropy Reveals Cancer System Hallmarks. Sci. Rep. 2012, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandhu, R.; Georgiou, T.; Reznik, E.; Zhu, L.; Kolesov, I.; Senbabaoglu, Y.; Tannenbaum, A. Graph Curvature for Differentiating Cancer Networks. Sci. Rep. 2015, 5, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ayyildiz, D.; Gov, E.; Sinha, R.; Arga, K.Y. Ovarian Cancer Differential Interactome and Network Entropy Analysis Reveal New Candidate Biomarkers. Omi. A J. Integr. Biol. 2017, 21, 285–294. [Google Scholar] [CrossRef]
- Anglani, R.; Creanza, T.M.; Liuzzi, V.C.; Piepoli, A.; Panza, A.; Andriulli, A.; Ancona, N. Loss of Connectivity in Cancer Co-Expression Networks. PLoS ONE 2014, 9, e0087075. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; Severini, S. Increased Entropy of Signal Transduction in the Cancer Metastasis Phenotype. BMC Syst. Biol. 2010, 4. [Google Scholar] [CrossRef] [Green Version]
- Demetrius, L.; Manke, T. Robustness and Network Evolution—An Entropic Principle. Phys. Stat. Mech. Appl. 2005, 346, 682–696. [Google Scholar] [CrossRef]
- Cheng, F.; Liu, C.; Shen, B.; Zhao, Z. Investigating Cellular Network Heterogeneity and Modularity in Cancer: A Network Entropy and Unbalanced Motif Approach. BMC Syst. Biol. 2016, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carels, N.; Tilli, T.M.; Tuszynski, J.A. Optimization of Combination Chemotherapy Based on the Calculation of Network Entropy for Protein-Protein Interactions in Breast Cancer Cell Lines. EPJ Nonlinear Biomed. Phys. 2015, 3. [Google Scholar] [CrossRef] [Green Version]
- Schramm, G.; Kannabiran, N.; König, R. Regulation Patterns in Signaling Networks of Cancer. BMC Syst. Biol. 2010, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.W.H.; Cercone, N.; Jurisica, I. Comparative Network Analysis via Differential Graphlet Communities. Proteomics 2015, 15, 608–617. [Google Scholar] [CrossRef]
- Park, Y.; Lim, S.; Nam, J.W.; Kim, S. Measuring Intratumor Heterogeneity by Network Entropy Using RNA-Seq Data. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Banerji, C.R.S.; Severini, S.; Caldas, C.; Teschendorff, A.E. Intra-Tumour Signalling Entropy Determines Clinical Outcome in Breast and Lung Cancer. PLoS Comput. Biol. 2015, 11, 1–23. [Google Scholar] [CrossRef]
- Klein, C.A. Selection and Adaptation during Metastatic Cancer Progression. Nature 2013, 501, 365–372. [Google Scholar] [CrossRef]
- Basha, O.; Argov, C.M.; Artzy, R.; Zoabi, Y.; Hekselman, I.; Alfandari, L.; Chalifa-Caspi, V.; Yeger-Lotem, E. Differential Network Analysis of Multiple Human Tissue Interactomes Highlights Tissue-Selective Processes and Genetic Disorder Genes. Bioinformatics 2020. [Google Scholar] [CrossRef]
- Khosravi, P.; Gazestani, V.H.; Law, B.; Bader, G.D.; Sadeghi, M. Comparative Analysis of Co-Expression Networks Reveals Molecular Changes during the Cancer Progression. IFMBE Proc. 2015, 51, 1481–1487. [Google Scholar] [CrossRef]
- Liu, Y.; Koyutürk, M.; Barnholtz-Sloan, J.S.; Chance, M.R. Gene Interaction Enrichment and Network Analysis to Identify Dysregulated Pathways and Their Interactions in Complex Diseases. BMC Syst. Biol. 2012, 6. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Lin, C.C.; Li, Y.Y.; Zhao, Z. Dynamic Protein Interaction Modules in Human Hepatocellular Carcinoma Progression. BMC Syst. Biol. 2013, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amgalan, B.; Lee, H. WMAXC: A Weighted Maximum Clique Method for Identifying Condition-Specific Sub-Network. PLoS ONE 2014, 9, e0104993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, L.; Chen, C.; Liu, C.H.; Zhang, M.; Liang, L. Revealing Differential Modules in Uveal Melanoma by Analyzing Differential Networks. Mol. Med. Rep. 2017, 15, 2261–2266. [Google Scholar] [CrossRef] [PubMed]
- Gulfidan, G.; Turanli, B.; Beklen, H.; Sinha, R.; Arga, K.Y. Pan-Cancer Mapping of Differential Protein-Protein Interactions. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Zhu, L.; Ding, Y.; Chen, C.-Y.; Wang, L.; Huo, Z.; Kim, S.; Sotiriou, C.; Oesterreich, S.; Tseng, G.C. MetaDCN: Meta-Analysis Framework for Differential Co-Expression Network Detection with an Application in Breast Cancer. Bioinformatics 2016, 33, 1121–1129. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Sun, P.; Qin, G. Identifying Condition-Specific Modules by Clustering Multiple Networks. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018, 15, 1636–1648. [Google Scholar] [CrossRef]
- Ma, X.; Gao, L.; Tan, K. Modeling Disease Progression Using Dynamics of Pathway Connectivity. Bioinformatics 2014, 30, 2343–2350. [Google Scholar] [CrossRef] [Green Version]
- Taylor, I.W.; Linding, R.; Warde-Farley, D.; Liu, Y.; Pesquita, C.; Faria, D.; Bull, S.; Pawson, T.; Morris, Q.; Wrana, J.L. Dynamic Modularity in Protein Interaction Networks Predicts Breast Cancer Outcome. Nat. Biotechnol. 2009, 27, 199–204. [Google Scholar] [CrossRef]
- Laaniste, L.; Srivastava, P.K.; Stylianou, J.; Syed, N.; Cases-Cunillera, S.; Shkura, K.; Zeng, Q.; Rackham, O.J.L.; Langley, S.R.; Delahaye-Duriez, A.; et al. Integrated Systems-Genetic Analyses Reveal a Network Target for Delaying Glioma Progression. Ann. Clin. Transl. Neurol. 2019, 6, 1616–1638. [Google Scholar] [CrossRef] [Green Version]
- Jin, N.; Wu, H.; Miao, Z.; Huang, Y.; Hu, Y.; Bi, X.; Wu, D.; Qian, K.; Wang, L.; Wang, C.; et al. Network-Based Survival-Associated Module Biomarker and Its Crosstalk with Cell Death Genes in Ovarian Cancer. Sci. Rep. 2015, 5, 1–12. [Google Scholar] [CrossRef]
- Zhou, J.; Chen, C.; Li, H.F.; Hu, Y.J.; Xie, H.L. Revealing Radiotherapy- and Chemoradiation-Induced Pathway Dynamics in Glioblastoma by Analyzing Multiple Differential Networks. Mol. Med. Rep. 2017, 16, 696–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hjaltelin, J.X.; Izarzugaza, J.M.G.; Jensen, L.J.; Russo, F.; Westergaard, D.; Brunak, S. Identification of Hyper-Rewired Genomic Stress Non-Oncogene Addiction Genes across 15 Cancer Types. NPJ Syst. Biol. Appl. 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.S.; Liu, B.Y.; Dai, W.T.; Zhou, W.X.; Li, Y.X.; Li, Y.Y. Differential Network Analysis Reveals Dysfunctional Regulatory Networks in Gastric Carcinogenesis. Am. J. Cancer Res. 2015, 5, 2605–2625. [Google Scholar] [PubMed]
- Wang, Y.; Jiang, T.; Li, Z.; Lu, L.; Zhang, D.; Wang, X.; Tan, J. Analysis of Differentially Co-Expressed Genes Based on Microarray Data of Hepatocellular Carcinoma. Neoplasma 2013, 60, 607–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, S.P.; Zhu, L.; Huang, D.S. Mining the Bladder Cancer-Associated Genes by an Integrated Strategy for the Construction and Analysis of Differential Co-Expression Networks. BMC Genom. 2015, 16, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Q.; Hu, T.; Andrew, A.S.; Karagas, M.R.; Moore, J.H. Bladder Cancer Specific Pathway Interaction Networks; The MIT Press: Cambridge, MA, USA, 2013; pp. 94–101. [Google Scholar]
- Xu, X.; Long, H.; Xi, B.; Ji, B.; Li, Z.; Dang, Y.; Jiang, C.; Yao, Y.; Yang, J. Molecular Network-Based Drug Prediction in Thyroid Cancer. Int. J. Mol. Sci. 2019, 20, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, S.; Pan, X.; Fang, W. Differential Co-Expression Analysis of a Microarray Gene Expression Profiles of Pulmonary Adenocarcinoma. Mol. Med. Rep. 2014, 10, 713–718. [Google Scholar] [CrossRef] [Green Version]
- Gill, R.; Datta, S.; Datta, S. Differential Network Analysis in Human Cancer Research. Curr. Pharm. Des. 2014, 20, 4–10. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q. A Powerful Nonparametric Method for Detecting Differentially Co-Expressed Genes: Distance Correlation Screening and Edge-Count Test. BMC Syst. Biol. 2018, 12, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Reznik, E.; Sander, C. Extensive Decoupling of Metabolic Genes in Cancer. PLoS Comput. Biol. 2015, 11, e1004176. [Google Scholar] [CrossRef] [Green Version]
- Asem, M.S.; Buechler, S.; Wates, R.B.; Miller, D.L.; Stack, M.S. Wnt5a Signaling in Cancer. Cancers 2016, 8, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Peng, Y.; Fan, S.; Li, Y.; Xiao, Z.X.; Li, C. A Double Dealing Tale of P63: An Oncogene or a Tumor Suppressor. Cell. Mol. Life Sci. 2018, 75, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Bach, D.H.; Park, H.J.; Lee, S.K. The Dual Role of Bone Morphogenetic Proteins in Cancer. Mol. Ther. Oncol. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinicropi-Yao, S.L.; Amann, J.M.; Lopez, D.L.Y.; Cerciello, F.; Coombes, K.R.; Carbone, D.P. Co-Expression Analysis Reveals Mechanisms Underlying the Varied Roles of NOTCH1 in NSCLC. J. Thorac. Oncol. 2019, 14, 223–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A CeRNA Hypothesis: The Rosetta Stone of a Hidden RNA Language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Ning, Q.; Zhang, G.; Sun, H.; Wang, Z.; Li, Y. Construction of Differential MRNA-LncRNA Crosstalk Networks Based on CeRNA Hypothesis Uncover Key Roles of LncRNAs Implicated in Esophageal Squamous Cell Carcinoma. Oncotarget 2016, 7, 85728–85740. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Wagner, E.K.; Hao, Y.; Rao, X.; Dai, H.; Han, J. Tissue-Specific Co-Expression of Long Non-Coding and Coding RNAs Associated with Breast Cancer. Sci. Rep. 2016, 6, 32731. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Li, C.X.; Lv, J.Y.; Li, Y.S.; Xiao, Y.; Shao, T.T.; Huo, X.; Li, X.; Zou, Y.; Han, Q.L.; et al. Prioritizing Candidate Disease MiRNAs by Topological Features in the MiRNA Target-Dysregulated Network: Case Study of Prostate Cancer. Mol. Cancer Ther. 2011, 10, 1857–1866. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.-C.; Mitra, R.; Cheng, F.; Zhongming, Z. Cross-Cancer Differential Co-Expression Network Reveals MicroRNA-Regulated Oncogenic Functional Modules. Mol. Biol. 2015, 11, 3244–3252. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Li, F.; Wu, T.; Xu, Y.; Yang, H.; Dong, Q.; Zheng, M.; Shang, D.; Zhang, C.; Zhang, Y.; et al. LncSubpathway: A Novel Approach for Identifying Dysfunctional Subpathways Associated with Risk LncRNAs by Integrating LncRNA and MRNA Expression Profiles and Pathway Topologies. Oncotarget 2017, 8, 15453–15469. [Google Scholar] [CrossRef] [Green Version]
- West, J.; Beck, S.; Wang, X.; Teschendorff, A.E. An Integrative Network Algorithm Identifies Age-Associated Differential Methylation Interactome Hotspots Targeting Stem-Cell Differentiation Pathways. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lareau, C.A.; White, B.C.; Oberg, A.L.; McKinney, B.A. Differential Co-Expression Network Centrality and Machine Learning Feature Selection for Identifying Susceptibility Hubs in Networks with Scale-Free Structure. BioData Min. 2015, 8, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, L.; Chen, M.; Zhang, C.K.; Cho, J.; Zhao, H. Guilt by Rewiring: Gene Prioritization through Network Rewiring in Genome Wide Association Studies. Hum. Mol. Genet. 2014, 23, 2780–2790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aran, D.; Sirota, M.; Butte, A.J. Systematic Pan-Cancer Analysis of Tumour Purity. Nat. Commun. 2015, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behan, F.M.; Iorio, F.; Picco, G.; Gonçalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of Cancer Therapeutic Targets Using CRISPR–Cas9 Screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef] [Green Version]
- McDonald, E.R.; de Weck, A.; Schlabach, M.R.; Billy, E.; Mavrakis, K.J.; Hoffman, G.R.; Belur, D.; Castelletti, D.; Frias, E.; Gampa, K.; et al. Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening. Cell 2017, 170, 577–592.e10. [Google Scholar] [CrossRef] [Green Version]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.-P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using Gene-Expression Signatures to Connect Small Molecules, Genes, and Disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [Green Version]
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R.; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-Generation Characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A Resource for Therapeutic Biomarker Discovery in Cancer Cells. Nucleic Acids Res. 2013, 41, 955–961. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL Database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef] [PubMed]
- Koedoot, E.; Fokkelman, M.; Rogkoti, V.M.; Smid, M.; van de Sandt, I.; de Bont, H.; Pont, C.; Klip, J.E.; Wink, S.; Timmermans, M.A.; et al. Uncovering the Signaling Landscape Controlling Breast Cancer Cell Migration Identifies Novel Metastasis Driver Genes. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavan, S.; Meyer-Schaller, N.; Diepenbruck, M.; Kalathur, R.K.R.; Saxena, M.; Christofori, G. A Kinome-Wide High-Content SiRNA Screen Identifies MEK5–ERK5 Signaling as Critical for Breast Cancer Cell EMT and Metastasis. Oncogene 2018, 37, 4197–4213. [Google Scholar] [CrossRef]
- Van Roosmalen, W.; Le Dévédec, S.E.; Golani, O.; Smid, M.; Pulyakhina, I.; Timmermans, A.M.; Look, M.P.; Zi, D.; Pont, C.; De Graauw, M.; et al. Tumor Cell Migration Screen Identifies SRPK1 as Breast Cancer Metastasis Determinant. J. Clin. Investig. 2015, 125, 1648–1664. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | Type of Network | Link | Reference |
|---|---|---|---|
| DIP | PPI | https://dip.doe-mbi.ucla.edu/dip/Main.cgi | [11] |
| MINT | PPI | https://mint.bio.uniroma2.it/ | [12] |
| IntAct | PPI | https://www.ebi.ac.uk/intact/ | [13] |
| BioGRID | PPI | https://thebiogrid.org/ | [14] |
| STRING | Various | https://string-db.org/ | [15] |
| COXPRESdb | Co-expression | https://coxpresdb.jp/ | [16] |
| SEEK | Co-expression | http://seek.princeton.edu/ | [17] |
| Method | Number of Conditions | Citation | Availability |
|---|---|---|---|
| DINGO | Multiple | [40] | CRAN R package (iDINGO) |
| Entropy | Two | [41] | Bioconductor R package (dcanr) |
| DGCA | Two | [42] | CRAN R package (DGCA) |
| Discordant | Two | [43] | Bioconductor R package (Discordant) |
| MAGIC | Two | [44] | Bioconductor R package (dcanr) MATLAB implementation at https://github.com/chiuyc/MAGIC |
| EBcoexpress | Two | [45] | Bioconductor R package (EBcoexpress) |
| GGM-based | Two | [46] | Bioconductor R package (dcanr) |
| LDGM | Two | [47] | Bioconductor R package (dcanr) Matlab implementation at https://github.com/ma-compbio/LDGM |
| Gill | Two | [48] | R package at http://www.somnathdatta.org/Supp/DNA |
| DDN | Two | [49] | MATLAB toolbox at http://www.cbil.ece.vt.edu/software.htm |
| Zhao | Two | [50] | |
| JDINAC | Two | [51] | R code at https://github.com/jijiadong/JDINAC |
| TDJGL | Two | [52] | R code at https://github.com/Zhangxf-ccnu/TDJGL |
| mlDNA | Two | [53] | CRAN R package (mlDNA, not maintained) |
| DCGL | Two | [54] | CRAN R package (DCGL) |
| DECODE | Two | [55] | CRAN R package (DECODE) |
| SIG method | Two | [56] | |
| Discordant | Two | [43] | Bioconductor R package (discordant) |
| DCN | Two | [57] | R package at https://github.com/weiliu123/DCN-package |
| TCDV | Two | [58] | |
| BFDCA | Two | [59] | R package at http://dx.doi.org/10.17632/jdz4vtvnm3.1 |
| CODC | Two | [60] | R package at https://github.com/Snehalikalall/CODC |
| z-score | Two/De novo | [61] | Bioconductor R package (dcanr) |
| MINDy | De novo | [62] | Bioconductor R package (dcanr) |
| Method | Module Definition | # of Conditions | Citation | Availability |
| DICER | Unbiased | Multiple | [63] | Bioconductor R package (dcanr) Java software at http://acgt.cs.tau.ac.il/dicer/ |
| DiffCoEx | Unbiased | Multiple | [64] | R package at https://github.com/ddeweerd/MODifieRDev.git Bioconductor R package (dcanr) |
| M-Modules | Unbiased | Multiple | [65] | |
| NIPD | Unbiased | Multiple | [66] | |
| C3D | Unbiased | Multiple | [67] | |
| CoXpress | Unbiased | Two | [68] | R package at http://coxpress.sourceforge.net/ |
| DiffCorr | Unbiased | Two | [69] | CRAN R package (DiffCorr) |
| ModMap | Unbiased | Two | [70] | Java executable at http://acgt.cs.tau.ac.il/modmap/ |
| ALPACA | Unbiased | Two | [71] | R package at https://github.com/meghapadi/ALPACA |
| BFDCA | Unbiased | Two | [59] | R package at http://dx.doi.org/10.17632/jdz4vtvnm3.1 |
| DiffCoMO | Unbiased | Two | [72] | |
| SCDA | Unbiased | Two | [73] | MATLAB implementation at http://vk.cs.umn.edu/SDC/ |
| CODC | Unbiased | Two | [60] | R package at https://github.com/Snehalikalall/CODC |
| EgoNet | Unbiased | Two | [74] | |
| BicMix | Unbiased | Two | [75] | R package at https://github.com/chuangao/BicMix |
| MODA | Unbiased | Two | [76] | Bioconductor R package (MODA) |
| COSINE | Unbiased | Two | [77] | CRAN R package (COSINE) |
| DECluster | Unbiased | Two | [78] | |
| DEDC | Unbiased | Two | [79] | |
| DCN | Unbiased | Two | [57] | R package at https://github.com/weiliu123/DCN-package |
| Contrast Subgraph | Unbiased | Two | [80] | |
| DCIM | Unbiased | De novo | [81] | |
| GSCA | A priori | Two | [82] | Bioconductor R package (GSCA) |
| ScorePAGE | A priori | Two | [83] | |
| IB-GSA | A priori | Two | [84] | |
| Gill | A priori | Two | [48] | R package at http://www.somnathdatta.org/Supp/DNA |
| CoGa | A priori | Two | [85] | |
| dCoxS | A priori pairs of gene sets | Two | [86] | R function at http://www.snubi.org/publication/dCoxS/ |
| MAGIC | A priori pairs of gene sets | Two | [44] | Bioconductor R package (dcanr) MATLAB implementation at https://github.com/chiuyc/MAGIC |
| ESEA | Structured pathway | Two | [87] | R package on CRAN (ESEA) |
| PWEA | Structured pathway | Two | [88] | R package on Bioconductor (ToPASeq) |
| KEDDY | Structured pathway | Two | [89] | Java implementation at https://sites.google.com/site/sjunggsm/keddy |
| kDDN | Structured pathway | Two | [90] | MATLAB implementation at http://www.cbil.ece.vt.edu/software.htm |
| Method | Description | # of Conditions | Citation | Availability |
|---|---|---|---|---|
| DEDC | Looks for the “best” DC gene | Two | [79] | |
| ECF | Given a pre-defined gene, selects others having differential co-expression with it | Two | [91] | CRAN R package (COSINE) |
| Gill | Given a pre-defined gene, tests whether its connectivity changes | Two | [48] | R package at http://www.somnathdatta.org/Supp/DNA |
| Method | Description | Citation | Availability |
|---|---|---|---|
| FTGI | Integrates co-expression and SNPs | [94] | Bioconductor R package (dcanr) |
| MultiDCox | Multivariate. Identifies variables correlated with differential co-expression | [95] | R package at https://github.com/lianyh/MultiDCoX |
| dcVar | Differential co-expression based on sequence variants | [93] | Linux command-line tool at http://insilico.utulsa.edu/dcVar.php |
| wgLASSO | Integrates co-expression with PPIs | [96] | |
| MACPath | Integrates co-expression with annotation of miRNA-responsive elements | [97] | Python code at https://github.com/thejustpark/MACPath |
| Method | Citation | Availability |
|---|---|---|
| z-score | [61] | Bioconductor R package (dcanr) |
| Mimosa | [98] | |
| GIMLET | [99] | R package at https://github.com/tshimam/GIMLET |
| MINDy | [62] | Bioconductor R package (dcanr) MINDy module in GenePattern |
| GEM | [100] | Implementation at https://sourceforge.net/projects/modulators |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Savino, A.; Provero, P.; Poli, V. Differential Co-Expression Analyses Allow the Identification of Critical Signalling Pathways Altered during Tumour Transformation and Progression. Int. J. Mol. Sci. 2020, 21, 9461. https://doi.org/10.3390/ijms21249461
Savino A, Provero P, Poli V. Differential Co-Expression Analyses Allow the Identification of Critical Signalling Pathways Altered during Tumour Transformation and Progression. International Journal of Molecular Sciences. 2020; 21(24):9461. https://doi.org/10.3390/ijms21249461
Chicago/Turabian StyleSavino, Aurora, Paolo Provero, and Valeria Poli. 2020. "Differential Co-Expression Analyses Allow the Identification of Critical Signalling Pathways Altered during Tumour Transformation and Progression" International Journal of Molecular Sciences 21, no. 24: 9461. https://doi.org/10.3390/ijms21249461
APA StyleSavino, A., Provero, P., & Poli, V. (2020). Differential Co-Expression Analyses Allow the Identification of Critical Signalling Pathways Altered during Tumour Transformation and Progression. International Journal of Molecular Sciences, 21(24), 9461. https://doi.org/10.3390/ijms21249461