The Role of Astrocytes in the Modulation ofK+-Cl−-Cotransporter-2 Function

Abstract
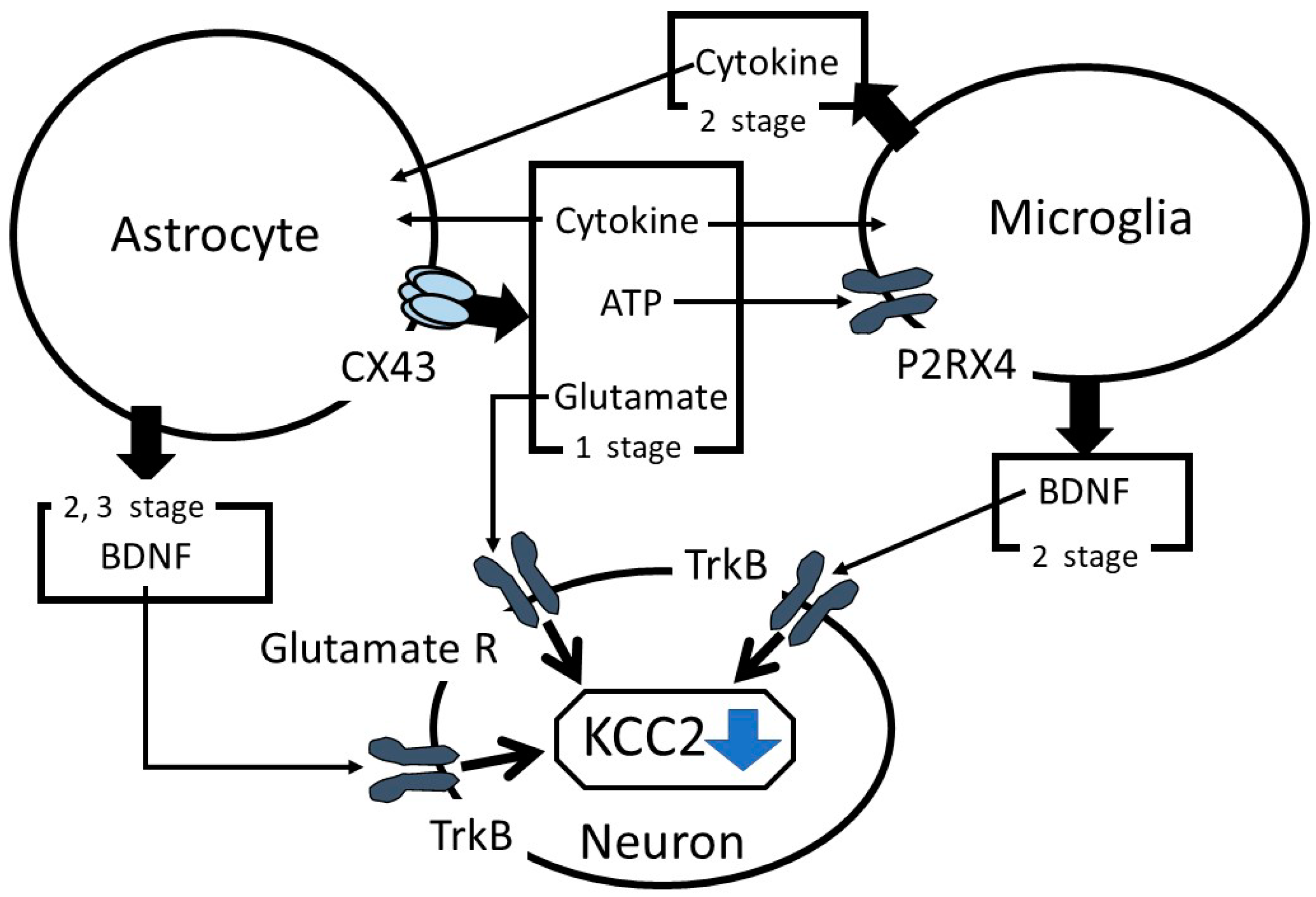
:1. Introduction
2. Modulation of KCC2 Function by Several Cascade Reaction
3. Astrocytes Maintain Balance of the Sensory System between Excitation and Inhibition
4. Astrocytes Release Proinflammatory Cytokines and Chemokines after Injury
5. Astrocyte Modulates Purinergic Signaling
6. Upregulation of Matrix Metalloproteases in Astrocytes after Injury
7. The Alteration of Zinc Ion Signaling in Astrocytes and the Central Nervous System after Injury
8. The Effects of KCC2 Downregulation Depends on the Region of Central Nervous System
9. Discussion
10. Future Perspectives
Funding
Conflicts of Interest
Abbreviations
| BDNF | Brain-derived neurotrophic factor |
| CX | Connexin |
| ERK | Extracellular signal-regulated kinase |
| GABA | γ-aminobutyric acid |
| GlyT | Glycine transporter |
| IL | Interleukin |
| KCC2 | K+-Cl−-cotransporter-2 |
| MAPK | Mitogen-activated protein kinase |
| MMP | Matrix metalloproteinase |
| mZnR | Metabotropic zinc sensing receptor |
| NF-κB | Nuclear factor-κB |
| NMDA | N-methyl-D-aspartate |
| P2RX4 | P2X purinoreceptor 4 |
| PKC | Protein Kinase C |
| TNF | Tumor necrosis factor |
| TrkB | Tropomyosin receptor kinase B |
| ZIP | Zrt/Irt-like protein |
| ZnT | Zinc transporter |
References
- Coull, J.A.M.; Boudreau, D.; Bachand, K.; Prescott, S.A.; Nault, F.; De Koninck, P.; De Koninck, Y. Trans-synaptic shift in anion gradient in spinal lamina 1 Neurons as a mechanism of neuropathic pain. Nature 2003, 242, 938–942. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, V.; Woodin, M.A. Regulation of neuronal chloride homeostasis by neuromodulaters. J. Physiol. 2016, 594, 2593–2605. [Google Scholar] [CrossRef] [PubMed]
- Kaila, K.; Price, T.J.; Payne, J.A.; Puskarjov, M.; Voipio, J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat. Rev. Neurosci. 2014, 15, 637–654. [Google Scholar] [CrossRef] [Green Version]
- Moore, Y.E.; Conway, L.C.; Wobst, H.J.; Brandon, N.J.; Deeb, T.Z.; Moss, S.J. Developmental regulation of KCC2 phosphorylation has long-term impacts on cognitive function. Front. Mol. Neurosci. 2019, 12, 173. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, B.; Chen, D.; Hou, X.; Wang, T.; Wang, J.; Zou, W.; Song, Z.; Huang, C.; Guo, Q.; Weng, Y. Normalizing HDAC2 levels in the spinal cord alleviates thermal and mechanical hyperalgesia after peripheral nerve injury and promotes GAD65 and KCC2 expression. Front. Neurosci. 2019, 13, 346. [Google Scholar] [CrossRef] [Green Version]
- Kitayama, T.; Morita, K.; Motoyama, N.; Dohi, T. Down-regulation of zinc transporter-1 in astrocytes induces neuropathic pain via the brain-derived neurotrophic factor—K+-Cl− cotransporter-2 signaling pathway in the mouse spinal cord. Neurochem. Int. 2016, 101, 120–131. [Google Scholar] [CrossRef]
- López-Álvarez, V.M.; Cobianchi, S.; Navarro, X. Chronic electrical stimulation reduces hyperalgesia and associated spinal changes induced by peripheral nerve injury. Neuromodulation 2019, 22, 509–518. [Google Scholar] [CrossRef] [Green Version]
- Morita, K.; Motoyama, N.; Kitayama, T.; Morioka, N.; Kifune, K.; Dohi, T. Spinal antiallodynia action of glycine transporter inhibitors in neuropathic pain models in mice. J. Pharmacol. Exp. Ther. 2008, 326, 633–645. [Google Scholar] [CrossRef] [Green Version]
- Okada-Ogawa, A.; Nakaya, Y.; Imamura, Y.; Kobayashi, M.; Shinoda, M.; Kita, K.; Sessle, B.J.; Iwata, K. Involvement of medullary GABAergic system in extraterritonal neuropathic pain mechanisms associated with inferior alveolar nerve transection. Exp. Neurol. 2015, 267, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Tsuruga, K.; Hashimoto, T.; Kato, R.; Uchida, Y.; Hase, T.; Morimoto, Y. Planter Injection of formalin in rat reduces the expression of a potassium chloride cotransporter KCC2 in the spinal cord and a kinase inhibitor suppresses this reduction. Biomed. Res. 2016, 37, 243–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Locke, S.; Yousefpour, N.; Mannarino, M.; Xing, S.; Yashmin, F.; Bourassa, V.; Ribeiro-da-Silva, A. Peripheral and central nervous system alterations in a rat model of inflammatory arthritis. Pain 2000, 161, 1483–1496. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Q.; Li, H.; Wei, J.; Qu, L.; Wu, L.A. Expression changes of K+-Cl− co-transporter 2 and Na+-K+-Cl− co-transporter 1 in mouse trigeminal subnucleus caudalis following pulpal inflammation. Brain Res. Bull. 2010, 81, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Jolivalt, C.G.; Lee, C.A.; Ramos, K.M.; Calcutt, N.A. Allodynia and hyperalgesia in diabetic rats are mediated by GABA and depletion of spinal potassium-chloride co-transporters. Pain 2008, 140, 48–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coull, J.A.M.; Beggs, S.; Boudreau, D.; Boivin, D.; Tsuda, M.; Inoue, K.; Gravel, C.; Salter, M.W.; Koninck, Y.D. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 2005, 438, 1017–1021. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zhang, Y.Q.; Qadri, Y.J.; Serhan, C.N.; Ji, R.R. Microglia in pain: Detrimental and protective roles in pathogenesis and resolution of pain. Neuron 2018, 100, 1292–1311. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K.; Tsuda, M. Microglia in neuropathic pain: Cellular and molecular mechanisms and therapeutic potential. Nat. Rev. Neurosci. 2018, 19, 138–152. [Google Scholar] [CrossRef]
- Tozaki-Saitoh, H.; Tsuda, M. Microglia-neuron interactions in the models of neuropathic pain. Biochem. Pharmacol. 2019, 169, 113614. [Google Scholar] [CrossRef]
- Salter, M.W.; Stevens, B. Microglia emerge as central players in brain disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef]
- Zhao, H.; Alam, A.; Chen, Q.A.; Eusman, M.; Pal, A.; Eguchi, S.; Wu, L.; Ma, D. The role of microglia in the pathobiology of neuropathic pain development: What do we know? Br. J. Anaesth. 2017, 118, 504–516. [Google Scholar] [CrossRef] [Green Version]
- Tsuda, M. Microglia in the spinal cord and neuropathic pain. J. Diabetes Investig. 2016, 7, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Moy, J.K.; Szabo-Pardi, T.; Tillu, D.V.; Megat, S.; Pradhan, G.; Kume, M.; Asiedu, M.N.; Burton, M.D.; Dussor, G.; Price, T.J. Temporal and sex differences in the role of BDNF/TrkB signaling in hyperalgesic priming in mice and rats. Neurobiol. Pain 2018, 19, 100024. [Google Scholar] [CrossRef] [PubMed]
- Mapplebeck, J.C.S.; Lorenzo, L.E.; Lee, K.Y.; Gauthier, C.; Muley, M.M.; De Koninck, Y.; Prescott, S.A.; Salter, M.W. Chloride dysregulation through downregulation of KCC2 mediates neuropathic pain in both sexes. Cell Rep. 2019, 28, 590–596. [Google Scholar] [CrossRef]
- Watanabe, M.; Zhang, J.; Mansuri, M.S.; Duan, J.; Karimy, J.K.; Delpire, E.; Alper, S.L.; Lifton, R.P.; Fukuda, A.; Kahle, K.T. Developmentally regulated KCC2 phosphorylation is essential for dynamic GABA-mediated inhibition and survival. Sci. Signal. 2019, 12, eaaw9315. [Google Scholar] [CrossRef] [PubMed]
- Pisella, L.I.; Gaiarsa, J.L.; Diabira, D.; Zhang, J.; Khalilov, I.; Duan, J.; Kahle, K.T.; Medina, I. Impaired regulation of KCC2 phosphorylation leads to neuronal network dysfunction and neurodevelopmental pathology. Sci. Signal. 2019, 12, eaay0300. [Google Scholar] [CrossRef] [PubMed]
- Kfir, A.; Awasthi, R.; Ghosh, S.; Kundu, S.; Paul, B.; Lamprecht, R.; Barkai, E. A cellular mechanism of learning-induced enhancement of synaptic inhibition: PKC-dependent upregulation of KCC2 activation. Sci. Rep. 2020, 10, 962. [Google Scholar] [CrossRef] [PubMed]
- Banke, T.G.; Gegelashvili, G. Tonic activation of group 1 mGluRs modulates inhibitory synaptic strength by regulating KCC2 activity. J. Physiol. 2008, 586, 4925–4934. [Google Scholar] [CrossRef]
- Lin, C.R.; Cheng, J.K.; Wu, C.H.; Chen, K.H.; Liu, C.K. Epigenetic suppression of potassium-chloride co-transporter 2 expression in inflammatory pain induced by complete Freund’s adjuvant (CFA). Eur. J. Pain 2017, 21, 309–321. [Google Scholar] [CrossRef]
- Ford, A.; Castonguay, A.; Cottet, M.; Little, J.W.; Chen, Z.; Symons-Liguori, A.M.; Doyle, T.; Egan, T.M.; Vanderah, T.W.; De Koninck, Y.; et al. Engagement of the GABA to KCC2 signaling pathway contributes to the analgesic effects of A3AR agonists in neuropathic pain. J. Neurosci. 2015, 35, 6057–6067. [Google Scholar] [CrossRef] [Green Version]
- Buldyrev, I.; Tanner, N.M.; Hsieh, H.Y.; Dodd, E.G.; Nguyen, L.T.; Balkowiec, A. Calcitonin gene-related peptide enhances release of native brain-derived neurotrophic factor from trigeminal ganglion neurons. J. Neurochem. 2006, 99, 1338–1350. [Google Scholar] [CrossRef] [Green Version]
- Jantzie, L.L.; Winer, J.L.; Corbett, C.J.; Robinson, S. Erythropoietin modulates cerebral and serum degradation products from excess calpain activation following prenatal hypoxia-ischemia. Dev. Neurosci. 2016, 38, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Zhang, D.; Chen, Y.; Cao, Z.; Fan, Z. Dexamethasone protects against arsanilic acid-induced rat vestibular dysfunction through the BDNF and JNK 1/2 signaling pathways. Mol. Med. Rep. 2019, 19, 1781–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildebrand, M.E.; Xu, J.; Dedek, A.; Li, Y.; Sengar, A.S.; Beggs, S.; Lombroso, P.J.; Salter, M.W. Potentiation of synaptic GluN2B NMDAR currents by Fyn kinase is gated through BDNF-mediated disinhibition in spinal pain processing. Cell Rep. 2016, 17, 2753–2765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.H.; Deeb, T.Z.; Walker, J.A.; Davies, P.A.; Moss, S.J. NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor-mediated currents. Nat. Neurosci. 2011, 14, 736–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balapattabi, K.; Little, J.T.; Farmer, G.E.; Cunningham, J.T. High salt loading increases brain derived neurotrophic factor in supraoptic vasopressin neurons. J. Neuroendocrinol. 2018, 30, e12639. [Google Scholar] [CrossRef]
- Zelenka, M.; Schafers, M.; Sommer, C. Intraneural injection of interleukin-1beta and tumor necrosis factor-alpha into rat sciatic nerve at physiological doses induces signs of neuropathic pain. Pain 2005, 116, 257–263. [Google Scholar] [CrossRef]
- Souza, D.G.; Bellaver, B.; Bobermin, L.D.; Souza, D.O.; Quincozes-Santos, A. Anti-aging effects of guanosine in glial cells. Purinergic Signal. 2016, 12, 697–706. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.J.; Park, M.H.; Koh, J.Y. Copper activates TrkB in cortical neurons in a metalloproteinase-dependent manner. J. Neurosci. Res. 2007, 85, 2160–2166. [Google Scholar] [CrossRef]
- Wu, H.; Shao, A.; Zhao, M.; Chen, S.; Yu, J.; Zhou, J.; Liang, F.; Shi, L.; Dixon, B.J.; Wang, Z.; et al. Melatonin attenuates neuronal apoptosis through up-regulation of K(+)-Cl(−) cotransporter KCC2 expression following traumatic brain injury in rats. J. Pineal Res. 2016, 61, 241–250. [Google Scholar] [CrossRef]
- Chorin, E.; Vinograd, O.; Fleidervish, I.; Gilad, D.; Herrmann, S.; Sekler, I.; Aizenman, E.; Hershfinkel, M. Upregulation of KCC2 activity by zinc-mediated neurotransmission via the mZnR/GPR39 receptor. J. Neurosci. 2011, 31, 12916–12926. [Google Scholar] [CrossRef] [Green Version]
- Gu, W.; Zhang, W.; Lei, Y.; Cui, Y.; Chu, S.; Gu, X.; Ma, Z. Activation of spinal alpha-7 nicotinic acetylcholine receptor shortens the duration of remifentanil-induced postoperative hyperalgesia by upregulating KCC2 in the spinal dorsal horn in rats. Mol. Pain 2017, 13. [Google Scholar] [CrossRef]
- Taylor, A.M.; Castonguay, A.; Ghogha, A.; Vayssiere, P.; Pradhan, A.A.; Xue, L.; Mehrabani, S.; Wu, J.; Levitt, P.; Olmstead, M.C.; et al. Neuroimmune regulation of GABAergic neurons within the ventral tegmental area during withdrawal from chronic morphine. Neuropsychopharmacology 2016, 41, 949–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trang, T.; Beggs, S.; Wan, X.; Salter, M.W. P2X4-receptor-mediated synthesis and release of brain-derived neurotrophic factor in microglia is dependent on calcium and p38-mitogen-activated protein kinase activation. J. Neurosci. 2009, 29, 3518–3528. [Google Scholar] [CrossRef] [PubMed]
- Ulmann, L.; Hatcher, J.P.; Hughes, J.P.; Chaumont, S.; Green, P.J.; Conquet, F.; Buell, G.N.; Reeve, A.J.; Chessell, I.P.; Rassendren, F. Up-regulation of P2X4 receptors in spinal microglia after peripheral nerve injury mediates BDNF release and neuropathic pain. J. Neurosci. 2008, 28, 11263–11268. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Yang, Y.; Ni, Z.; Dong, Y.; Cai, G.; Foncelle, A.; Ma, S.; Sang, K.; Tang, S.; Li, Y.; et al. Astroglia Kir4.1 in the lateral habenula drives neuronal bursts in depression. Nature 2018, 554, 323–327. [Google Scholar] [CrossRef] [Green Version]
- Jursky, F.; Nelson, N. Developmental expression of the glycine transporters GLYT1 and GLYT2 in mouse brain. J. Neurochem. 1996, 67, 336–344. [Google Scholar] [CrossRef]
- Roux, M.J.; Supplisson, S. Neuronal and glial glycine transporters have different stoichiometries. Neuron 2000, 25, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Min, R.; van der Knaap, M.S. Genetic defects disrupting glial ion and water homeostasis in the brain. Brain Pathol. 2018, 28, 372–387. [Google Scholar] [CrossRef] [Green Version]
- Lynch, J.W.; Zhang, Y.; Talwar, S.; Estrada-Mondragon, A. Glycine receptor drug discovery. Adv. Pharmacol. 2017, 79, 225–253. [Google Scholar]
- Zafra, F.; Aragón, C.; Olivares, L.; Danbolt, N.C.; Giménez, C.; Storm-Mathisen, J. Glycine transporters are differentially expressed among CNS cells. J. Neurosci. 1995, 15, 3952–3969. [Google Scholar] [CrossRef]
- Brasnjo, G.; Otis, T.S. Glycine transporters not only take out the garbage, they recycle. Neuron 2003, 40, 667–669. [Google Scholar] [CrossRef] [Green Version]
- Wafford, K.A.; Kathoria, M.; Bain, C.J.; Marshall, G.; Le Bourdellès, B.; Kemp, J.A.; Whiting, P.J. Identification of amino acids in the N-methyl-D-aspartate receptor NR1 subunit that contribute to the glycine binding site. Mol. Pharmacol. 1995, 47, 374–380. [Google Scholar] [PubMed]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Gris, D. Astrocytes maintain glutamate homeostasis in the CNS by controlling the balance between glutamate uptake and release. Cells 2019, 8, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, W.J.; Weng, H.R.; Dougherty, P.M. Plasticity in expression of the glutamate transporter GLT-1 and GLAST in spinal dorsal horn glial cells following partial sciatic nerve ligation. Mol. Pain 2009, 5, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falnikar, A.; Hala, T.J.; Poulsen, D.J.; Lepore, A.C. GLT1 overexpression reverses established neuropathic pain-related behavior and attenuates chronic dorsal horn neuron activation following cervical spinal cord injury. Glia 2016, 64, 396–406. [Google Scholar] [CrossRef]
- Spagnuolo, C.; Moccia, S.; Russo, G.L. Anti-inflammatory effects of flavonoids in neurodegenerative disorders. Eur. J. Med. Chem. 2018, 153, 105–115. [Google Scholar] [CrossRef]
- Dai, W.L.; Yan, B.; Bao, Y.N.; Fan, J.F.; Liu, J.H. Suppression of peripheral NGF attenuates neuropathic pain induced by chronic constriction injury through the TAK1-MAPK/NF-κB signaling pathways. Cell Commun. Signal. 2020, 18, 66. [Google Scholar] [CrossRef] [Green Version]
- Barrientos, R.M.; Sprunger, D.B.; Campeau, S.; Watkins, L.R.; Rudy, J.W.; Maier, S.F. BDNF mRNA expression in rat hippocampus following contextual learning is blocked by intrahippocampal IL-1beta administration. J. Neuroimmunol. 2004, 155, 119–126. [Google Scholar] [CrossRef]
- Teh, D.B.L.; Prasad, A.; Jiang, W.; Ariffin, M.Z.; Khanna, S.; Belorkar, A.; Wong, L.; Liu, X.; All, A.H. Transcriptome analysis reveals neuroprotective aspects of human reactive astrocytes induced by interleukin 1β. Sci. Rep. 2017, 25, 13988. [Google Scholar] [CrossRef] [Green Version]
- Saha, R.N.; Liu, X.; Pahan, K. Up-regulation of BDNF in astrocytes by TNF-alpha: A case for the neuroprotective role of cytokine. J. Neuroimmune Pharmacol. 2006, 1, 212–222. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.J.; Jiang, B.C.; Gao, Y.J. Chemokines in neuron-glial cell interaction and pathogenesis of neuropathic pain. Cell Mol. Life Sci. 2017, 74, 3275–3291. [Google Scholar] [CrossRef]
- Gao, Y.J.; Zhang, L.; Samad, O.A.; Suter, M.R.; Yasuhiko, K.; Xu, Z.Z.; Park, J.Y.; Lind, A.L.; Ma, Q.; Ji, R.R. JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J. Neurosci. 2009, 29, 4096–4108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosselin, R.D.; Varela, C.; Banisadr, G.; Mechighel, P.; Rostene, W.; Kitabgi, P.; Melik-Parsadaniantz, S. Constitutive expression of CCR2 chemokine receptor and inhibition by MCP-1/CCL2 of GABA-induced currents in spinal cord neurons. J. Neurochem. 2005, 95, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Park, C.K.; Xie, R.G.; Berta, T.; Nedergaard, M.; Ji, R.R. Connexin-43 induces chemokine release from spinal cord astrocytes to maintain late-phase neuropathic pain in mice. Brain 2014, 137, 2193–2209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tada, M.; Diserens, A.C.; Desbaillets, I.; de Tribolet, N. Analysis of cytokine receptor messenger RNA expression in human glioblastoma cells and normal astrocytes by reverse-transcription polymerase chain reaction. J. Neurosurg. 1994, 80, 1063–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fields, R.D.; Burnstock, G. Purinergic signalling in neuron-glia interactions. Nat. Rev. Neurosci. 2006, 7, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Halassa, M.M.; Florian, C.; Fellin, T.; Munoz, J.R.; Lee, S.Y.; Abel, T.; Haydon, P.G.; Frank, M.G. Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron 2009, 61, 213–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalo, U.; Verkhratsky, A.; Pankratov, Y. Ionotropic ATP receptors in neuronal-glial communication. Semin. Cell Dev. Biol. 2011, 22, 220–228. [Google Scholar] [CrossRef]
- Harada, K.; Kamiya, T.; Tsuboi, T. Gliotransmitter release from astrocytes: Functional, developmental, and pathological implications in the brain. Front. Neurosci. 2016, 9, 499. [Google Scholar] [CrossRef] [Green Version]
- Xing, L.; Yang, T.; Cui, S.; Chen, G. Connexin hemichannels in astrocytes: Role in CNS disorders. Front. Mol. Neurosci. 2019, 12, 23. [Google Scholar] [CrossRef] [Green Version]
- Tonkin, R.S.; Bowles, C.; Perera, C.J.; Keating, B.A.; Makker, P.G.S.; Duffy, S.S.; Lees, J.G.; Tran, C.; Don, A.S.; Fath, T.; et al. Attenuation of mechanical pain hypersensitivity by treatment with Peptide5, a connexin-43 mimetic peptide, involves inhibition of NLRP3 inflammasome in nerve-injured mice. Exp. Neurol. 2018, 300, 1–12. [Google Scholar] [CrossRef]
- Huang, C.; Han, X.; Li, X.; Lam, E.; Peng, W.; Lou, N.; Torres, A.; Yang, M.; Garre, J.M.; Tian, G.F.; et al. Critical role of connexin 43 in secondary expansion of traumatic spinal cord injury. J. Neurosci. 2012, 32, 3333–3338. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, L.; Batten, T.F.; Deuchars, J. P2X(2) receptor immunoreactivity in the dorsal vagal complex and area postrema of the rat. Neuroscience 2000, 99, 683–696. [Google Scholar] [CrossRef]
- Freitas, H.R.; Reis, R.A. Glutathione induces GABA release through P2X(7)R activation on Muller glia. Neurogenesis 2017, 4, e1283188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, Y.H.; Cheng, P.Y.; Tsai, R.Y.; Chen, Y.F.; Wong, C.S. Purinergic P2X receptor regulates N-methyl-D-aspartate receptor expression and synaptic excitatory amino acid concentration in morphine-tolerant rats. Anesthesiology 2010, 113, 1163–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawynok, J. Adenosine receptor targets for pain. Neuroscience 2016, 338, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Chopra, S.; Overall, C.M.; Dufour, A. Matrix metalloproteinases in the CNS: Interferons get nervous. Cell. Mol. Life Sci. 2019, 76, 3083–3095. [Google Scholar] [CrossRef]
- Kawasaki, Y.; Xu, Z.Z.; Wang, X.; Park, J.Y.; Zhuang, Z.Y.; Tan, P.H.; Gao, Y.J.; Roy, K.; Corfas, G.; Lo, E.H.; et al. Distinct roles of matrix metalloproteases in the early-and late-phase development of neuropathic pain. Nat. Med. 2008, 14, 331–336. [Google Scholar] [CrossRef]
- Suh, S.W.; Chen, J.W.; Motamedi, M.; Bell, B.; Listiak, K.; Pons, N.F.; Danscher, G.; Frederickson, C.J. Evidence that synaptically-released zinc contributes to neuronal injury after traumatic brain injury. Brain Res. 2000, 852, 268–273. [Google Scholar] [CrossRef]
- Baltaci, A.K.; Yuce, K. Zinc transporter proteins. Neurochem. Res. 2018, 43, 517–530. [Google Scholar] [CrossRef]
- Segawa, S.; Tatsumi, N.; Ohishi, A.; Nishida, K.; Nagasawa, K. Characterization of zinc uptake by mouse primary cultured astrocytes and microglia. Metallomics 2015, 7, 1067–1077. [Google Scholar] [CrossRef]
- Furuta, T.; Ohshima, C.; Matsumura, M.; Takebayashi, N.; Hirota, E.; Mawaribuchi, T.; Nishida, K.; Nagasawa, K. Oxidative stress upregulates zinc uptake activity via Zrt/Irt-like protein 1 (ZIP1) in cultured mouse astrocytes. Life Sci. 2016, 151, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Wang, H.; Watanabe, M.; Shimizu, K.; Zou, S.; LaGraize, S.C.; Wei, F.; Dubner, R.; Ren, K. Glial-cytokine-neuronal interactions underlying the mechanisms of persistent pain. J. Neurosci. 2007, 27, 6006–6018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mah, W.; Lee, S.M.; Lee, J.; Bae, J.Y.; Ju, J.S.; Lee, C.J.; Ahn, D.K.; Bae, Y.C. A role for the purinergic receptor P2X 3 in astrocytes in the mechanism of craniofacial neuropathic pain. Sci. Rep. 2017, 7, 13627. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Zhu, M.D.; Cao, D.L.; Bai, X.Q.; Gao, Y.J.; Wu, X.B. Chemokine CXCL13 activates p38 MAPK in the trigeminal ganglion after infraorbital nerve injury. Inflammation 2017, 40, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Tornberg, J.; Voikar, V.; Savilahti, H.; Rauvala, H.; Airaksinen, M.S. Behavioural phenotypes of hypomorphic KCC2-deficient mice. Eur. J. Neurosci. 2005, 21, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Hara, M.; Kobayakawa, K.; Matsumoto, Y.; Nakashima, Y. Astrocyte reactivity and astrogliosis after spinal cord injury. Neurosci. Res. 2018, 126, 39–43. [Google Scholar] [CrossRef]
- Raghavendra, V.; Tanga, F.; DeLeo, J.A. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J. Pharmacol. Exp. Ther. 2003, 306, 624–630. [Google Scholar] [CrossRef] [Green Version]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef]
- Kitayama, T. The Role of K+-Cl−-Cotransporter-2 in Neuropathic Pain. Neurochem. Res. 2018, 43, 110–115. [Google Scholar] [CrossRef]


{kind=link}
| Molecule | Mechanisms | References |
|---|---|---|
| Adenosine receptor, A3 | Phosphorylation control | Ford et al. [28] |
| Calcitonin gene-related receptor | BDNF-TrkB-KCC2 reaction | Buldyrev et al. [29] |
| Erythropoietin | BDNF-TrkB-KCC2 reaction | Jantzie et al. [30] |
| Glucocorticoid | BDNF-TrkB-KCC2 reaction | Luo et al. [31] |
| Glutamate receptor (group 1 metabotropic) | Phosphorylation control | Banke et al. [26] |
| Glutamate receptor (NMDA receptor) | Phosphorylation control | Hildebrand et al. [32] Lee et al. [33] |
| High salt loading | BDNF-TrkB-KCC2 reaction | Balapattabi et al. [34] |
| Interleukin-1β Tumor necrosis factor α | BDNF-TrkB-KCC2 reaction | Zelenka et al. [35] Souza et al. [36] |
| Interleukin-6 | BDNF-TrkB-KCC2 reaction | Kitayama et al. [6] |
| Matrix metalloproteinase 2 Matrix metalloproteinase 9 | BDNF-TrkB-KCC2 reaction | Hwang et al. [37] |
| Melatonin | BDNF-TrkB-KCC2 reaction | Wu et al. [38] |
| Metabotropic zinc sensing receptor (mZnR/GPR39) | Phosphorylation control | Chorin et al. [39] |
| Nicotinic acetylcholine receptor | BDNF-TrkB-KCC2 reaction | Gu et al. [40] |
| Opioid receptor (μ) | BDNF-TrkB-KCC2 reaction | Taylor et al. [41] |
| P2X purinergic receptor 4 | BDNF-TrkB-KCC2 reaction | Trang et al. [42] Ulmann et al. [43] |
| Zinc transporter 1 | BDNF-TrkB-KCC2 reaction | Kitayama et al. [6] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kitayama, T. The Role of Astrocytes in the Modulation ofK+-Cl−-Cotransporter-2 Function. Int. J. Mol. Sci. 2020, 21, 9539. https://doi.org/10.3390/ijms21249539
Kitayama T. The Role of Astrocytes in the Modulation ofK+-Cl−-Cotransporter-2 Function. International Journal of Molecular Sciences. 2020; 21(24):9539. https://doi.org/10.3390/ijms21249539
Chicago/Turabian StyleKitayama, Tomoya. 2020. "The Role of Astrocytes in the Modulation ofK+-Cl−-Cotransporter-2 Function" International Journal of Molecular Sciences 21, no. 24: 9539. https://doi.org/10.3390/ijms21249539
APA StyleKitayama, T. (2020). The Role of Astrocytes in the Modulation ofK+-Cl−-Cotransporter-2 Function. International Journal of Molecular Sciences, 21(24), 9539. https://doi.org/10.3390/ijms21249539