Solid Lipid Curcumin Particles Protect Medium Spiny Neuronal Morphology, and Reduce Learning and Memory Deficits in the YAC128 Mouse Model of Huntington’s Disease

 ,
,  , , , ,
, , , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
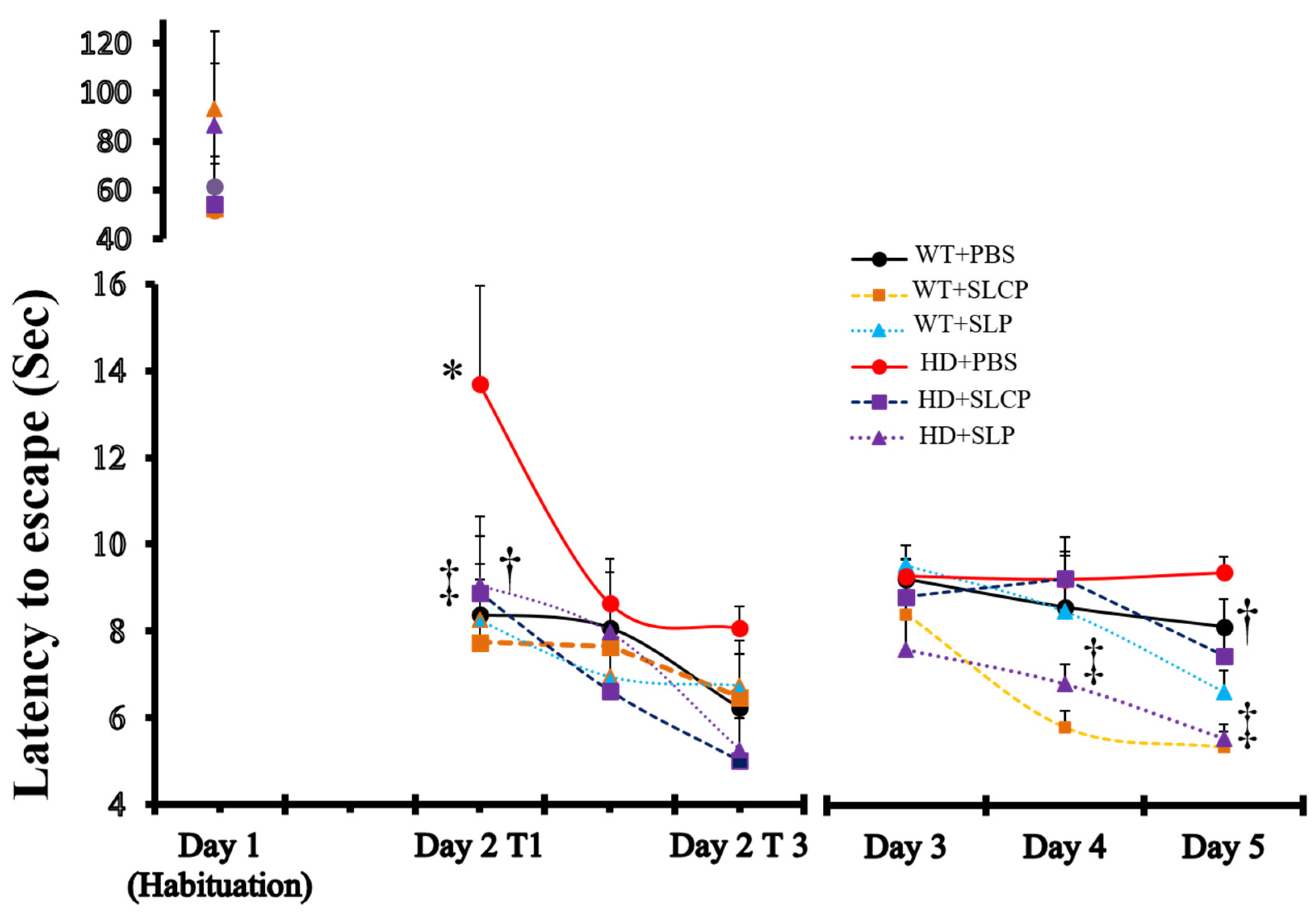
2.1. Active Avoidance
2.2. Golgi-Cox
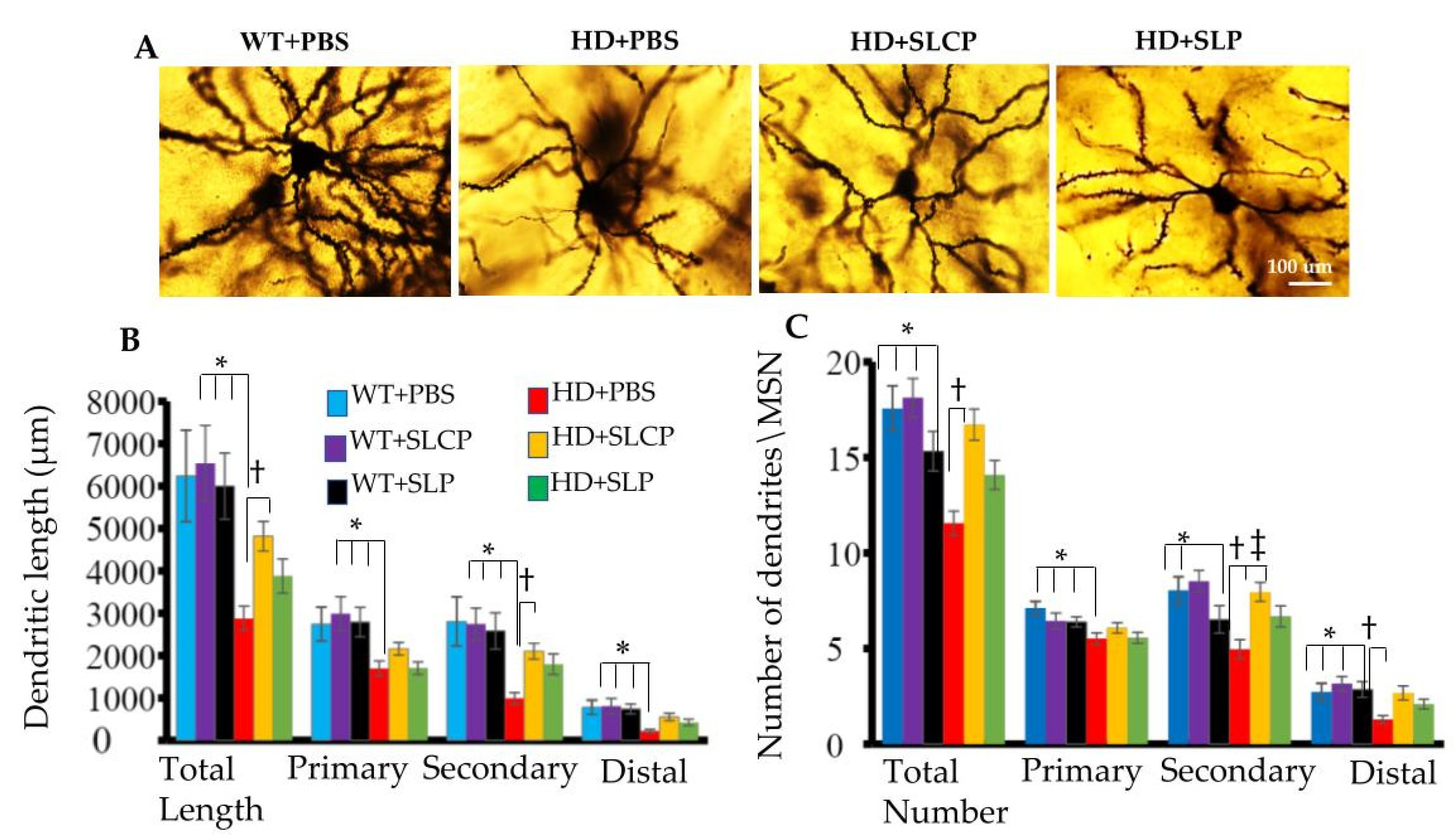
2.2.1. Dendritic Arborization
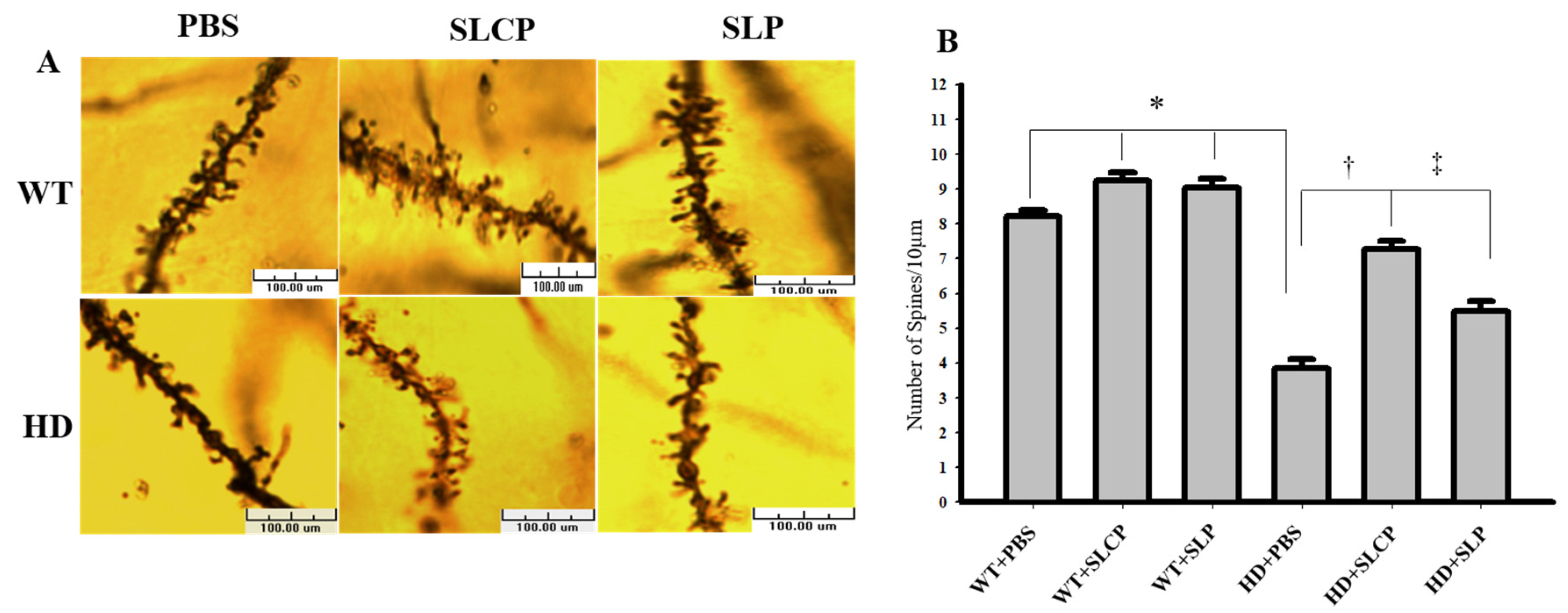
2.2.2. Density of Dendritic Spines
2.3. Western Blots
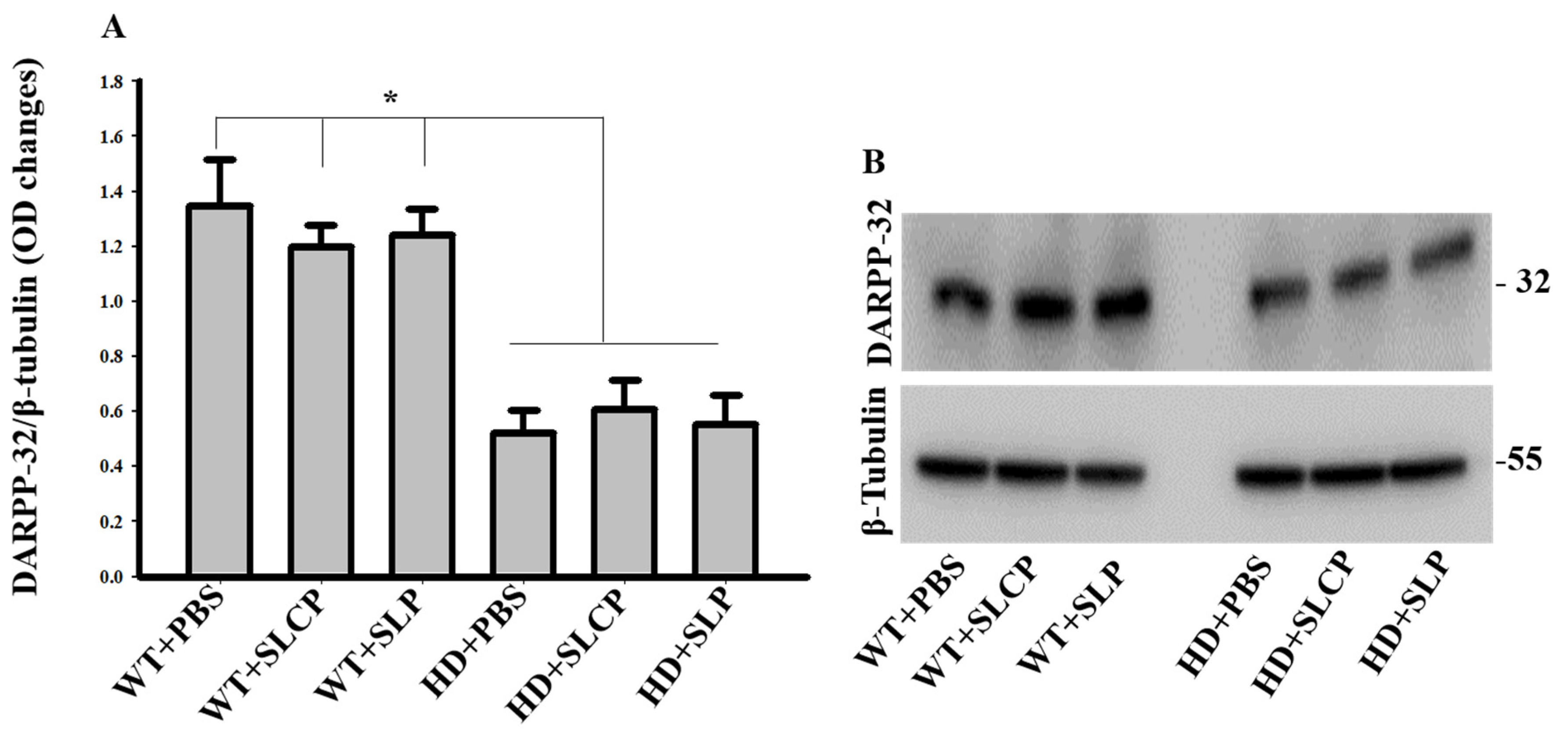
2.3.1. DARPP-32
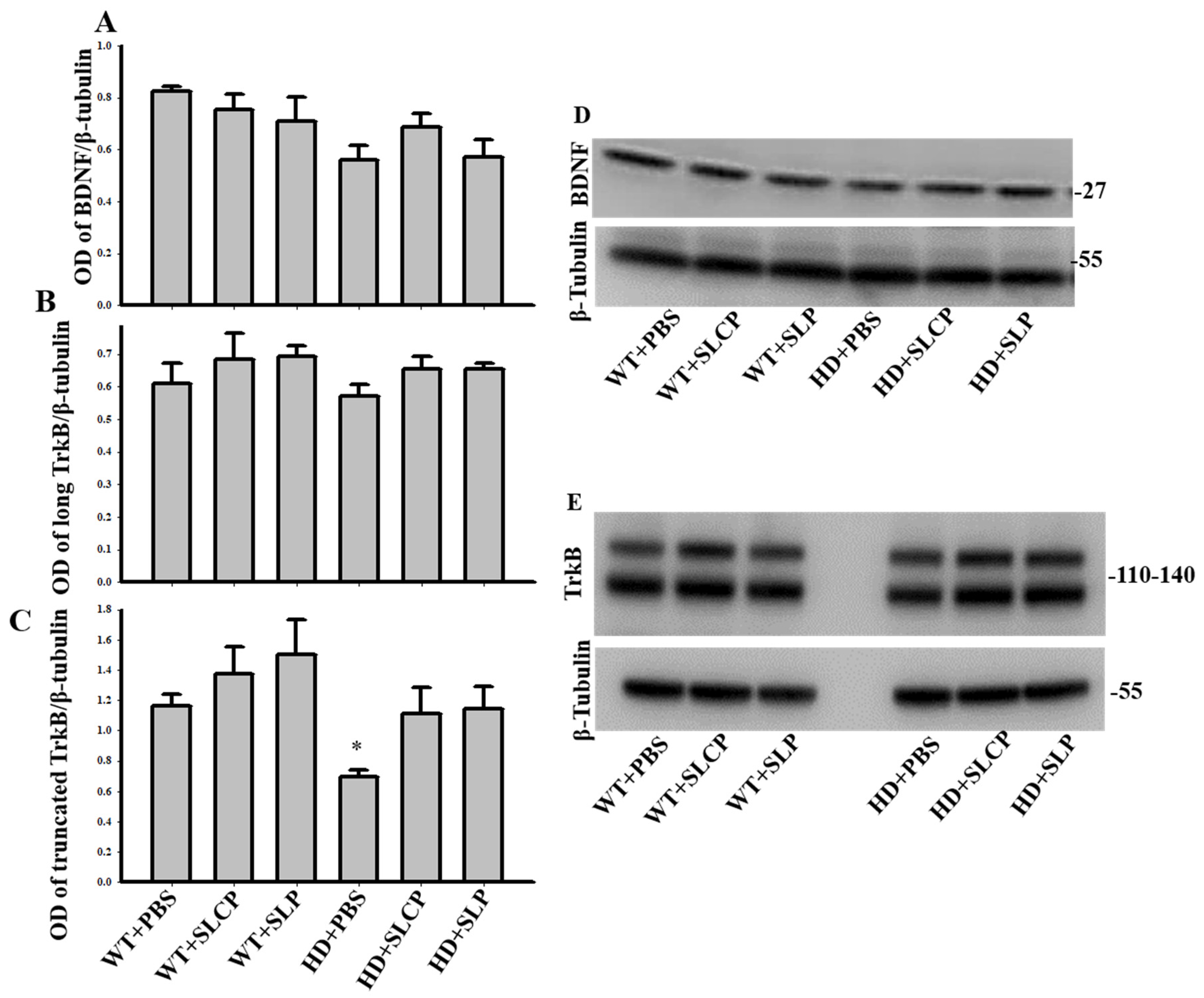
2.3.2. BDNF
2.3.3. TrkB
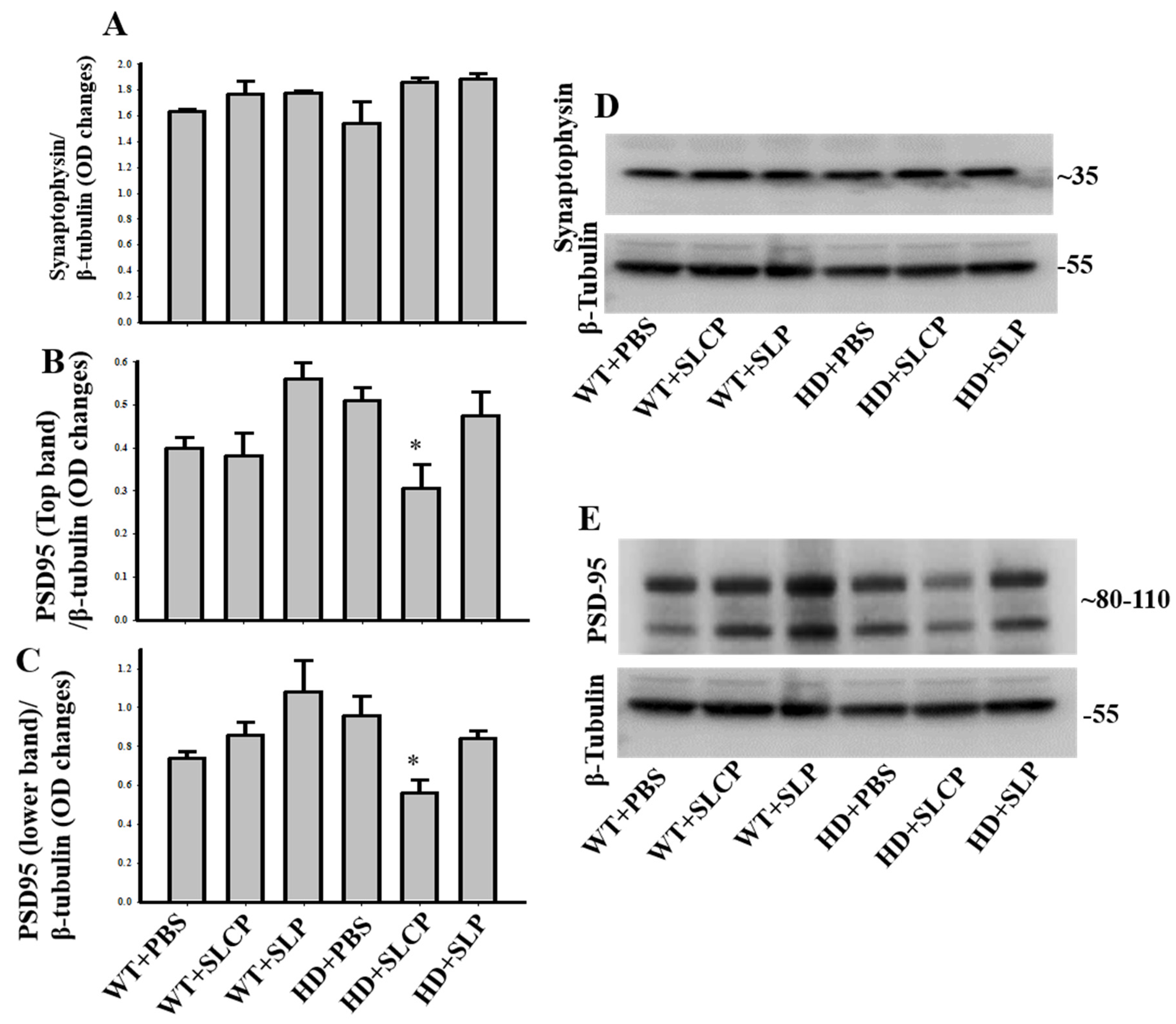
2.3.4. Synaptophysin
2.3.5. PSD-95
3. Discussion
4. Materials and Methods
4.1. Animals and Treatment
4.2. Active Avoidance
4.3. Golgi-Cox Staining
4.4. Western Blot (WB)
4.5. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ANOVA | Analysis of variance |
| BCA | Bicinchoninic acid assay |
| BDNF | Brain-derived neurotrophic factor |
| DARPP32 | Dopamine- and cAMP-regulated phosphoprotein of 32 kDa |
| EDTA | Ethylene-di-amino-tetra-acetic-acid |
| HD | Huntington’s disease |
| HRP | Horseradish peroxidase |
| MSN | Medium spiny neuron |
| OD | Optical density |
| PBS | Phosphate-buffered saline |
| PSD-95 | Postsynaptic density protein 95 |
| PVDF | Polyvinylidene difluoride |
| QA | Quinolinic acid |
| RIPA | Radioimmunoprecipitation assay |
| SDS | Sodium dodecyl sulfate |
| TBS | Tris-buffered saline |
| TrkB | Tropomyosin-related kinase B |
| YAC | Yeast artificial chromosome |
References
- Zuccato, C.; Valenza, M.; Cattaneo, E. Molecular mechanisms and potential therapeutical targets in Huntington’s disease. Physiol. Rev. 2010, 90, 905–981. [Google Scholar] [CrossRef]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s Disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Goehler, H.; Lalowski, M.; Stelzl, U.; Waelter, S.; Stroedicke, M.; Worm, U.; Droege, A.; Lindenber, K.S.; Knoblich, M.; Haenig, C.; et al. A protein interaction network links GIT1, an enhancer of huntingtin aggregation, to Huntington’s disease. Mol. Cell 2004, 15, 853–865. [Google Scholar] [CrossRef]
- Colin, E.; Zala, D.; Liot, G.; Rangone, H.; Borrell-Pagès, M.; Li, X.-J.; Saudou, F.; Humbert, S. Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. EMBO J. 2008, 27, 2124–2134. [Google Scholar] [CrossRef] [Green Version]
- Hoffner, G.; Kahlem, P.; Dijan, P. Perinuclear localization of huntingtin as a consequence of its binding to microtubules through an interaction with β-tubulin: Relevance to Huntington’s disease. J. Cell Sci. 2002, 115, 941–948. [Google Scholar]
- Difiglia, M.; Sapp, E.; Chase, K.; Schwarz, C.; Meloni, A.; Young, C.; Martin, E.; Vonsattel, J.P.; Carraway, R.; Reeves, S.A.; et al. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron 1995, 14, 1075–1081. [Google Scholar] [CrossRef] [Green Version]
- Zala, D.; Colin, E.; Rangone, H.; Liot, G.; Humbert, S.; Saudou, F. Phosphorylation of mutant huntingtin at S421 restores anterograde and retrograde transport in neurons. Hum. Mol. Genet. 2008, 17, 3837–3846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, L.R.; Charrin, B.C.; Borrell-Pagès, M.; Dompierre, J.P.; Rangone, H.; Codelières, F.P.; Mey, J.D.; MacDonald, M.E.; Lessmann, V.; Humbert, S.; et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 2004, 118, 127–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modregger, J.; Schmidt, A.A.; Ritter, B.; Huttner, W.B.; Plomann, M. Characterization of Endophilin B1b, a brain-specific membrane-associated lysophosphatidic acid acyl transferase with properties distinct from endophilin A1. J. Biol. Chem. 2003, 278, 4160–4167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, A.; Severin, F.; Lommer, B.; Shevchenko, A.; Zerial, M. Huntingtin–HAP40 complex is a novel Rab5 effector that regulates early endosome motility and is up-regulated in Huntington’s disease. J. Cell Biol. 2006, 172, 605–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slow, E.J.; Van Raamsdonk, J.; Rogers, D.; Coleman, S.H.; Graham, R.K.; Deng, Y.; Oh, R.; Bissada, N.; Hossain, S.M.; Yang, Y.Z.; et al. Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum. Mol. Genet. 2003, 12, 1555–1567. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, J.M.; Pearson, J.; Slow, E.J.; Hossain, S.M.; Leavitt, B.R.; Hayden, M.R. Cognitive dysfunction precedes neuropathology and motor abnormalities in the YAC128 mouse model of Huntington’s disease. J. Neurosci. 2005, 25, 4169–4180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; Maiti, P.; Ma, Q.; Zuo, X.; Jones, M.R.; Cole, G.M.; Frautschy, S.A. Clinical development of curcumin in neurodegenerative disease. Expert Rev. Neurother. 2015, 15, 629–637. [Google Scholar] [CrossRef]
- Maiti, P.; Manna, J.; Veleri, S.; Frautschy, S. Molecular chaperone dysfunction in neurodegenerative diseases and effects of curcumin. BioMed Res. Int. 2014, 2014, 495091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Begum, A.N.; Jones, M.R.; Lim, G.P.; Morihara, T.; Kim, P.; Heath, D.D.; Rock, C.L.; Pruitt, M.A.; Yang, F.; Hudspeth, B.; et al. Curcumin structure-function, bioavailability, and efficacy in models of neuroinflammation and Alzheimer’s disease. J. Pharmacol. Exp. Ther. 2008, 326, 196–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Padi, S.S.V.; Naidu, P.S.; Kumar, A. Possible neuroprotective mechanisms of curcumin in attenuating 3-nitropropionic acid-induced neurotoxicity. Methods Find. Exp. Clin. Pharmacol. 2007, 29, 19–26. [Google Scholar] [CrossRef]
- Hickey, M.A.; Zhu, C.; Medvedeva, V.; Lerner, R.P.; Patassini, S.; Franich, N.R.; Maiti, P.; Frautschy, S.A.; Zeitlin, S.; Levine, M.S.; et al. Improvement of neuropathology and transcriptional deficits in CAG 140 knock-in mice supports a beneficial effect of dietary curcumin in Huntington’s disease. Mol. Neurodegener. 2012, 7, 12. [Google Scholar] [CrossRef] [Green Version]
- Sandhir, R.; Yadav, A.; Mehrotra, A.; Sunkaria, A.; Singh, A.; Sharma, S. Curcumin nanoparticles attenuate neurochemical and neurobehavioral deficits in experimental model of Huntington’s disease. Neuromol. Med. 2014, 16, 106–118. [Google Scholar] [CrossRef]
- Gupta, S.C.; Patchva, S.; Aggarwal, B.B. Therapeutic roles of curcumin: Lessons learned from clinical trials. AAPS J. 2013, 15, 195–218. [Google Scholar] [CrossRef] [Green Version]
- Maiti, P.; Paladugu, L.; Dunbar, G.L. Solid lipid curcumin particles provide greater anti-amyloid, anti-inflammatory and neuroprotective effects than curcumin in the 5xFAD mouse model of Alzheimer’s disease. BMC Neurosci. 2018, 19, 7. [Google Scholar] [CrossRef] [Green Version]
- Nahar, P.P.; Slitt, A.L.; Seeram, N.P. Anti-inflammatory effects of novel standardized solid lipid curcumin formulations. J. Med. Food 2015, 18, 786–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dadhaniya, P.; Patel, C.; Muchhara, J.; Bhadja, N.; Mathuria, N.; Vachhani, K.; Soni, M.G. Safety assessment of a solid lipid curcumin particle preparation: Acute and subchronic toxicity studies. Food Chem. Toxicol. 2011, 49, 1834–1842. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Palanivelu, K. The effect of curcumin (turmeric) on Alzheimer’s disease: An overview. Ann. Indian Acad. Neurol. 2008, 11, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: Problems and promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef]
- Gota, V.S.; Maru, G.B.; Soni, T.G.; Gandhi, T.R.; Kochar, N.; Agarwal, M.G. Safety and pharmacokinetics of a solid lipid curcumin particle formulation in osteosarcoma patients and healthy volunteers. J. Agric. Food Chem. 2010, 58, 2095–2099. [Google Scholar] [CrossRef]
- Ueda, Y.; Wang, M.F.; Irei, A.V.; Sarukura, N.; Sakai, T.; Hsu, T.F. Effect of dietary lipids on longevity and memory in the SAMP8 mice. J. Nutr. Sci. Vitaminol. 2011, 57, 36–41. [Google Scholar] [CrossRef] [Green Version]
- Mehrotra, A.; Sood, A.; Sandhir, R. Mitochondrial modulators improve lipid composition and attenuate memory deficits in experimental model of Huntington’s disease. Mol. Cell. Biochem. 2015, 410, 281–292. [Google Scholar] [CrossRef]
- Monroy, A.; Lithgow, G.J.; Alavez, S. Curcumin and neurodegenerative diseases. Biofactors 2019, 39, 122–132. [Google Scholar] [CrossRef]
- Flores, G. Curcuma longa L. extract improves the cortical neural connectivity during the aging process. Neural Regen. Res. 2017, 12, 875–880. [Google Scholar] [CrossRef]
- Noorafshan, A.; Karimi, F.; Kamali, A.M.; Karbalay-Doust, S.; Nami, M. Could curcumin protect the dendritic trees of the CA1 neurons from shortening and shedding induced by chronic sleep restriction in rats? Life Sci. 2018, 198, 65–70. [Google Scholar] [CrossRef]
- Dotti, C.G.; Esteban, J.A.; Ledesma, M.D. Lipid dynamics at dendritic spines. Front. Neuroanat. 2014, 8, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, R.; Brundin, P.; Li, J.Y. Synaptic dysfunction in Huntington’s disease: A new perspective. Cell. Mol. Life Sci. 2005, 62, 1901–1912. [Google Scholar] [CrossRef]
- Wiedenmann, B.; Franke, W.W.; Kuhn, C.; Moll, R.; Gould, V.E. Synaptophysin: A marker protein for neuroendocrine cells and neoplasms. Proc. Natl. Acad. Sci. USA 1986, 83, 3500–3504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, M. The postsynaptic NMDA-receptor—PSD-95 signaling complex in excitatory synapses of the brain. J. Cell Sci. 2001, 114, 1251. [Google Scholar] [PubMed]
- Fourie, C.; Kim, E.; Waldvogel, H.; Wong, J.M.; McGregor, A.; Faull, R.L.M.; Montgomery, J.M. Differential changes in postsynaptic density proteins in postmortem Huntington’s disease and Parkinson’s disease human brains. J. Neurodegener. Dis. 2014, 2014, 938530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Cowan, C.M.; Zhang, L.Y.J.; Hayden, M.R.; Raymond, L.A. Interaction of postsynaptic density protein-95 with NMDA receptors influences excitotoxicity in the yeast artificial chromosome mouse model of Huntington’s disease. J. Neurosci. 2009, 29, 10928–10938. [Google Scholar] [CrossRef] [Green Version]
- Pike, L.J. Rafts defined: A report on the Keystone Symposium on Lipid Rafts and Cell Function. J. Lipid Res. 2006, 47, 1597–1598. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Zhang, J.; Miyazawa, S.; Liu, Q.; Farzan, M.R.; Yao, W.D. Association of membrane rafts and postsynaptic density: Proteomics, biochemical, and ultrastructural analyses. J. Neurochem. 2011, 119, 64–77. [Google Scholar] [CrossRef] [Green Version]
- Hong, Y.; Zhao, T.; Li, X.J.; Li, S. Mutant huntingtin impairs BDNF release from astrocytes by disrupting conversion of Rab3a-GTP into Rab3a-GDP. J. Neurosci. 2016, 36, 8790–8801. [Google Scholar] [CrossRef]
- Zuccato, C.; Cattaneo, E. Role of brain-derived neurotrophic factor in Huntington’s disease. Prog. Neurobiol. 2007, 81, 294–330. [Google Scholar] [CrossRef]
- Dey, N.D.; Bombard, M.C.; Roland, B.P.; Davidson, S.; Lu, M.; Rossignol, J.; Sandstrom, M.I.; Skeel, R.L.; Lescaudron, L.; Dunbar, G.L. Genetically engineered mesenchymal stem cells reduce behavioral deficits in the YAC 128 mouse model of Huntington’s disease. Behav. Brain Res. 2010, 214, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Al-Gharaibeh, A.; Culver, R.; Stewart, A.N.; Srinageshwar, B.; Spelde, K.; Frollo, L.; Kolli, N.; Story, D.; Paladugu, L.; Anwar, S.; et al. Induced pluripotent stem cell-derived neural stem cell transplantations reduced behavioral deficits and ameliorated neuropathological changes in YAC128 mouse model of Huntington’s disease. Front. Neurosci. 2017, 11, 628. [Google Scholar] [CrossRef] [PubMed]
- Ginés, S.; Bosch, M.; Marco, S.; Gavaldà, N.; Díaz-Hernández, M.; Lucas, J.J.; Canals, J.M.; Alberch, J. Reduced expression of the TrkB receptor in Huntington’s disease mouse models and in human brain. Eur. J. Neurosci. 2006, 23, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Das, G.; Reuhl, K.; Zhou, R. The Golgi–Cox method. In Neural Development; Humana Press: Totowa, NJ, USA, 2013; pp. 313–321. [Google Scholar]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gharaibeh, A.; Maiti, P.; Culver, R.; Heileman, S.; Srinageshwar, B.; Story, D.; Spelde, K.; Paladugu, L.; Munro, N.; Muhn, N.; et al. Solid Lipid Curcumin Particles Protect Medium Spiny Neuronal Morphology, and Reduce Learning and Memory Deficits in the YAC128 Mouse Model of Huntington’s Disease. Int. J. Mol. Sci. 2020, 21, 9542. https://doi.org/10.3390/ijms21249542
Gharaibeh A, Maiti P, Culver R, Heileman S, Srinageshwar B, Story D, Spelde K, Paladugu L, Munro N, Muhn N, et al. Solid Lipid Curcumin Particles Protect Medium Spiny Neuronal Morphology, and Reduce Learning and Memory Deficits in the YAC128 Mouse Model of Huntington’s Disease. International Journal of Molecular Sciences. 2020; 21(24):9542. https://doi.org/10.3390/ijms21249542
Chicago/Turabian StyleGharaibeh, Abeer, Panchanan Maiti, Rebecca Culver, Shiela Heileman, Bhairavi Srinageshwar, Darren Story, Kristin Spelde, Leela Paladugu, Nikolas Munro, Nathan Muhn, and et al. 2020. "Solid Lipid Curcumin Particles Protect Medium Spiny Neuronal Morphology, and Reduce Learning and Memory Deficits in the YAC128 Mouse Model of Huntington’s Disease" International Journal of Molecular Sciences 21, no. 24: 9542. https://doi.org/10.3390/ijms21249542
APA StyleGharaibeh, A., Maiti, P., Culver, R., Heileman, S., Srinageshwar, B., Story, D., Spelde, K., Paladugu, L., Munro, N., Muhn, N., Kolli, N., Rossignol, J., & Dunbar, G. L. (2020). Solid Lipid Curcumin Particles Protect Medium Spiny Neuronal Morphology, and Reduce Learning and Memory Deficits in the YAC128 Mouse Model of Huntington’s Disease. International Journal of Molecular Sciences, 21(24), 9542. https://doi.org/10.3390/ijms21249542