Multifaceted Functions of Platelets in Cancer: From Tumorigenesis to Liquid Biopsy Tool and Drug Delivery System




Abstract
:1. Introduction
2. Role of Platelets in the Early Events of Tumorigenesis
3. Platelets and T Cell Immunity
4. Role of Platelets in Metastasis
5. Platelet-Derived Vesicles and Their Implication to Cancer
6. Antiplatelet Drugs and Cancer
6.1. Aspirin
6.2. P2Y12 Receptor Antagonists
6.3. Thrombin Receptor Antagonists
6.4. Glycoprotein IIb/IIIa Antagonists
7. Platelet Omics as a Diagnostic and Prognostic Tool in Cancer
7.1. Platelet Proteomics
7.2. Platelet Transcriptome
8. Tumor-Educated Platelets as a Liquid Biopsy Tool in Cancer
9. Platelet as a Drug Delivery Tool in Cancer
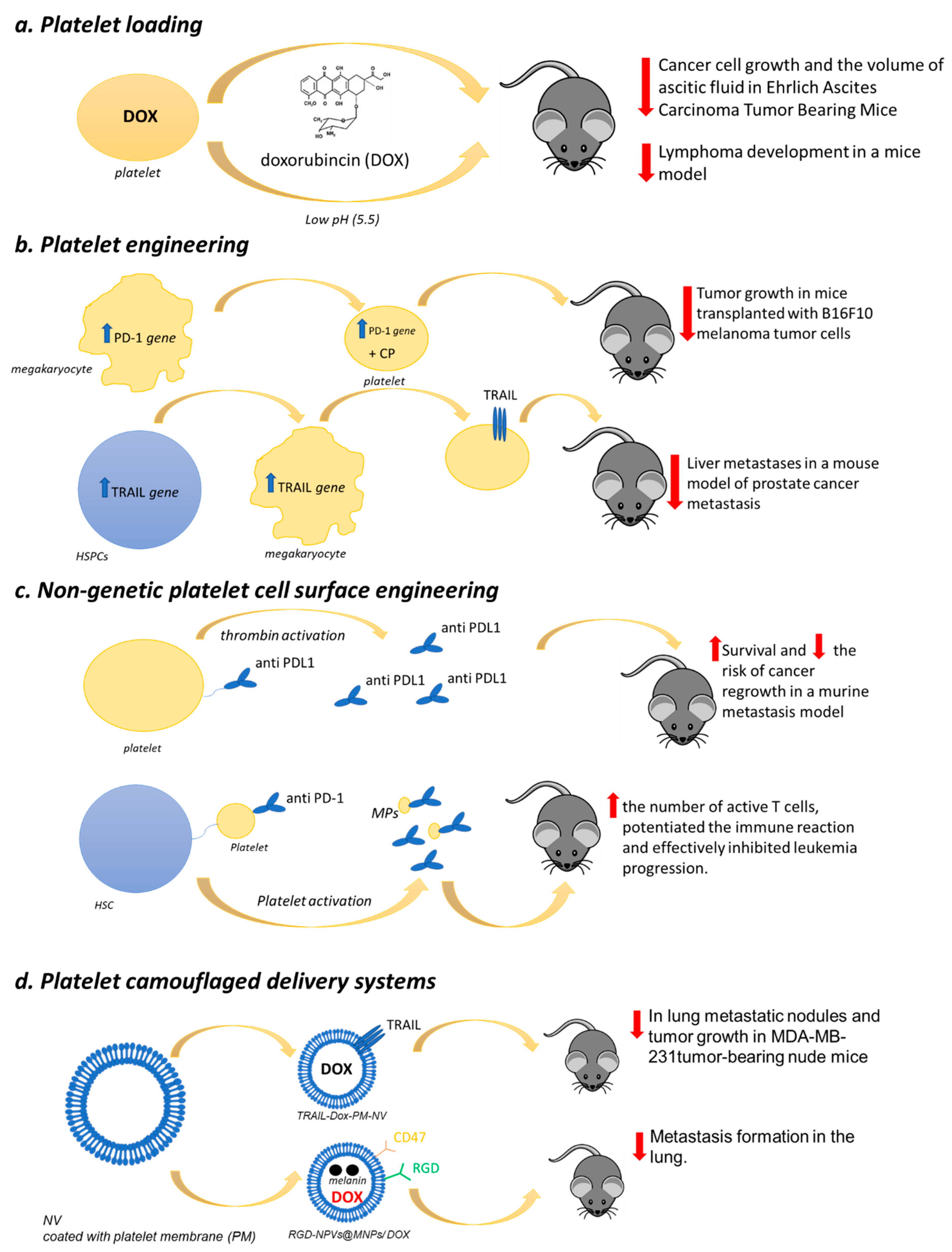
9.1. Platelet Loading
9.2. Platelet Engineering
9.3. Nongenetic Platelet Cell Surface Engineering
9.4. Platelet-Camouflaged Delivery Systems
10. Conclusive Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Ruggeri, Z.M. Platelets in atherothrombosis. Nat. Med. 2002, 8, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Van der Meijden, P.E.J.; Heemskerk, J.W.M. Platelet biology and functions: New concepts and clinical perspectives. Nat. Rev. Cardiol. 2019, 16, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Gawaz, M.; Langer, H.; May, A.E. Platelets in inflammation and atherogenesis. J. Clin. Investig. 2005, 115, 3378–3384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gay, L.J.; Felding-Habermann, B. Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 2011, 11, 123–134. [Google Scholar] [CrossRef]
- Contursi, A.; Sacco, A.; Grande, R.; Dovizio, M.; Patrignani, P. Platelets as crucial partners for tumor metastasis: From mechanistic aspects to pharmacological targeting. Cell. Mol. Life Sci. 2017, 74, 3491–3507. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Fowkes, F.G.; Belch, J.F.; Ogawa, H.; Warlow, C.P.; Meade, T.W. Effect of daily aspirin on long-term risk of death due to cancer: Analysis of individual patient data from randomised trials. Lancet 2011, 377, 31–41. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Wilson, M.; Elwin, C.E.; Norrving, B.; Algra, A.; Warlow, C.P.; Meade, T.W. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet 2010, 376, 1741–1750. [Google Scholar] [CrossRef]
- Patrignani, P.; Patrono, C. Aspirin and cancer. J. Am. Coll. Cardiol. 2016, 68, 967–976. [Google Scholar] [CrossRef]
- Patrignani, P.; Filabozzi, P.; Patrono, C. Selective cumulative inhibition of platelet thromboxane production by low-dose aspirin in healthy subjects. J. Clin. Investig. 1982, 69, 1366–1372. [Google Scholar] [CrossRef]
- Patrignani, P.; Patrono, C. Cyclooxygenase inhibitors: From pharmacology to clinical read-outs. Biochim. Biophys. Acta 2015, 1851, 422–432. [Google Scholar] [CrossRef]
- Patrignani, P.; Tacconelli, S.; Piazuelo, E.; Di Francesco, L.; Dovizio, M.; Sostres, C.; Marcantoni, E.; Guillem-Llobat, P.; Del Boccio, P.; Zucchelli, M.; et al. Reappraisal of the clinical pharmacology of low-dose aspirin by comparing novel direct and traditional indirect biomarkers of drug action. J. Thromb. Haemost. 2014, 12, 1320–1330. [Google Scholar] [CrossRef] [Green Version]
- Baigent, C.; Sudlow, C.; Collins, R.; Peto, R. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. Br. Med. J. 2002, 324, 71–86. [Google Scholar]
- Patrignani, P.; Sacco, A.; Sostres, C.; Bruno, A.; Dovizio, M.; Piazuelo, E.; Di Francesco, L.; Contursi, A.; Zucchelli, M.; Schiavone, S.; et al. Low-Dose Aspirin Acetylates Cyclooxygenase-1 in Human Colorectal Mucosa: Implications for the Chemoprevention of Colorectal Cancer. Clin. Pharmacol. Ther. 2017, 102, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Cha, Y.I.; DuBois, R.N. NSAIDs and cancer prevention: Targets downstream of COX-2. Annu Rev. Med. 2007, 58, 239–252. [Google Scholar] [CrossRef]
- Patrono, C.; Patrignani, P.; Rodríguez, L.A.G. Cyclooxygenase-selective inhibition of prostanoid formation: Transducing biochemical selectivity into clinical read-outs. J. Clin. Investig. 2001, 108, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Patrignani, P.; Patrono, C. Aspirin, platelet inhibition and cancer prevention. Platelets 2018, 29, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Dovizio, M.; Maier, T.J.; Alberti, S.; Di Francesco, L.; Marcantoni, E.; Münch, G.; John, C.M.; Suess, B.; Sgambato, A.; Steinhilber, D.; et al. Pharmacological inhibition of platelet-tumor cell cross-talk prevents platelet-induced overexpression of cyclooxygenase-2 in HT29 human colon carcinoma cells. Mol. Pharmacol. 2013, 84, 25–40. [Google Scholar] [CrossRef]
- Wang, D.; DuBois, R.N. Urinary PGE-M: A promising cancer biomarker. Cancer Prev. Res. 2013, 6, 507–510. [Google Scholar] [CrossRef] [Green Version]
- Duffield-Lillico, A.J.; Boyle, J.O.; Zhou, X.K.; Ghosh, A.; Butala, G.S.; Subbaramaiah, K.; Newman, R.A.; Morrow, J.D.; Milne, G.L.; Dannenberg, A.J. Levels of Prostaglandin E Metabolite and Leukotriene E4 Are Increased in the Urine of Smokers. Evidence that Celecoxib Shunts Arachidonic Acid into the 5-Lipoxygenase Pathway. Cancer Prev. Res. 2009, 2, 322–329. [Google Scholar] [CrossRef] [Green Version]
- McAdam, B.F.; Catella-Lawson, F.; Mardini, I.A.; Kapoor, S.; Lawson, J.A.; FitzGerald, G.A. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: The human pharmacology of a selective inhibitor of COX-2. Proc. Natl. Acad. Sci. USA 1999, 96, 272–277. [Google Scholar] [CrossRef] [Green Version]
- Boutaud, O.; Sosa, I.R.; Amin, T.; Oram, D.; Adler, D.; Hwang, H.S.; Crews, B.C.; Milne, G.; Harris, B.K.; Hoeksema, M.; et al. Inhibition of the Biosynthesis of Prostaglandin E2 by Low-Dose Aspirin: Implications for Adenocarcinoma Metastasis. Cancer Prev. Res. 2016, 9, 855–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosser, T.; Fries, S.; FitzGerald, G.A. Biological basis for the cardiovascular consequences of COX-2 inhibition: Therapeutic challenges and opportunities. J. Clin. Investig. 2006, 116, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Tacconelli, S.; Contursi, A.; Falcone, L.; Mucci, M.; D’Agostino, I.; Fullone, R.; Sacco, A.; Zucchelli, M.; Bruno, A.; Ballerini, P.; et al. Characterization of cyclooxygenase-2 acetylation and prostanoid inhibition by aspirin in cellular systems. Biochem. Pharmacol. 2020, 178, 114094. [Google Scholar] [CrossRef]
- Dovizio, M.; Bruno, A.; Tacconelli, S.; Patrignani, P. Mode of action of aspirin as a chemopreventive agent. Recent Results Cancer Res. 2013, 191, 39–65. [Google Scholar] [PubMed]
- Bibbins-Domingo, K.; U.S. Preventive Services Task Force. Aspirin Use for the Primary Prevention of Cardiovascular Disease and Colorectal Cancer: U.S. Preventive Services Task Force Recommendation Statement. Ann. Intern. Med. 2016, 164, 836–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehmer, S.P.; Maciosek, M.V.; Flottemesch, T.J.; LaFrance, A.B.; Whitlock, E.P. Aspirin for the Primary Prevention of Cardiovascular Disease and Colorectal Cancer: A Decision Analysis for the U.S. Preventive Services Task Force. Ann. Intern. Med. 2016, 164, 777–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patrono, C.; Baigent, C. Role of aspirin in primary prevention of cardiovascular disease. Nat. Rev. Cardiol. 2019, 16, 675–686. [Google Scholar] [CrossRef]
- Nagalla, S.; Shaw, C.; Kong, X.; Kondkar, A.A.; Edelstein, L.C.; Ma, L.; Chen, J.; McKnight, G.S.; López, J.A.; Yang, L.; et al. Platelet microRNA-mRNA coexpression profiles correlate with platelet reactivity. Blood 2011, 117, 5189–5197. [Google Scholar] [CrossRef]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [Green Version]
- Burnouf, T.; Goubran, H.A.; Chou, M.L.; Devos, D.; Radosevic, M. Platelet microparticles: Detection and assessment of their paradoxical functional roles in disease and regenerative medicine. Blood Rev. 2014, 28, 155–166. [Google Scholar] [CrossRef]
- Dovizio, M.; Bruno, A.; Contursi, A.; Grande, R.; Patrignani, P. Platelets and extracellular vesicles in cancer: Diagnostic and therapeutic inmplications. Cancer Metastasis Rev. 2018, 37, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothwell, P.M.; Wilson, M.; Price, J.F.; Belch, J.F.; Meade, T.W.; Mehta, Z. Effect of daily aspirin on risk of cancer metastasis: A study of incident cancers during randomised controlled trials. Lancet 2012, 379, 1591–1601. [Google Scholar] [CrossRef]
- Algra, A.M.; Rothwell, P.M. Effects of regular aspirin on long-term cancer incidence and metastasis: A systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol. 2012, 13, 518–527. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Price, J.F.; Fowkes, F.G.; Zanchetti, A.; Roncaglioni, M.C.; Tognoni, G.; Lee, R.; Belch, J.F.; Wilson, M.; Mehta, Z.; et al. Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: Analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet 2012, 379, 1602–1612. [Google Scholar] [CrossRef]
- Charman, W.N.; Charman, S.A.; Monkhouse, D.C.; Frisbee, S.E.; Lockhart, E.A.; Weisman, S.; Fitzgerald, G.A. Biopharmaceutical characterisation of a low-dose (75 mg) controlled-release aspirin formulation. Br. J. Clin. Pharmacol. 1993, 36, 470–473. [Google Scholar] [CrossRef] [Green Version]
- The Medical Research Council’s General Practice Research Framework. Thrombosis Prevention Trial: Randomised trial of lowintensity oral anticoagulation with warfarin and low-dose aspirin in the primary prevention of ischaemic heart disease in men at increased risk. Lancet 1998, 351, 233–241. [Google Scholar] [CrossRef]
- Haemmerle, M.; Stone, R.L.; Menter, D.G.; Afshar-Kharghan, V.; Sood, A.K. The Platelet Lifeline to Cancer: Challenges and Opportunities. Cancer Cell 2018, 33, 965–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Best, M.G.; Wesseling, P.; Wurdinger, T. Tumor-Educated Platelets as a Noninvasive Biomarker Source for Cancer Detection and Progression Monitoring. Cancer Res. 2018, 78, 3407–3412. [Google Scholar] [CrossRef] [Green Version]
- Sol, N.; In ‘t Veld, S.G.J.G.; Vancura, A.; Tjerkstra, M.; Leurs, C.; Rustenburg, F.; Schellen, P.; Verschueren, H.; Post, E.; Zwaan, K.; et al. Tumor-Educated Platelet RNA for the Detection and (Pseudo)progression Monitoring of Glioblastoma. Cell Rep. Med. 2020, 1, 100101. [Google Scholar] [CrossRef]
- Lu, Y.; Hu, Q.; Jiang, C.; Gu, Z. Platelet for drug delivery. Curr. Opin. Biotechnol. 2019, 58, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Dovizio, M.; Alberti, S.; Guillem-Llobat, P.; Patrignani, P. Role of platelets in inflammation and cancer: Novel therapeutic strategies. Basic Clin. Pharmacol. Toxicol. 2014, 114, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Sacco, A.; Bruno, A.; Contursi, A.; Dovizio, M.; Tacconelli, S.; Ricciotti, E.; Guillem- Llobat, P.; Salvatore, T.; Di Francesco, L.; Fullone, R.; et al. Platelet-Specific Deletion of Cyclooxygenase-1 Ameliorates Dextran Sulfate Sodium-Induced Colitis in Mice. J. Pharmacol. Exp. Ther. 2019, 370, 416–426. [Google Scholar] [CrossRef]
- Daniel, T.O.; Liu, H.; Morrow, J.D.; Crews, B.C.; Marnett, L.J. Thromboxane A2 is a mediator of cyclooxygenase-2-dependent endothelial migration and angiogenesis. Cancer Res. 1999, 59, 4574–4577. [Google Scholar]
- Wang, D.; DuBois, R.N. An inflammatory mediator, prostaglandin E2, in colorectal cancer. Cancer J. 2013, 19, 502–510. [Google Scholar] [CrossRef] [Green Version]
- Myung, S.J.; Rerko, R.M.; Yan, M.; Platzer, P.; Guda, K.; Dotson, A.; Lawrence, E.; Dannenberg, A.J.; Lovgren, A.K.; Luo, G.; et al. 15-Hydroxyprostaglandin dehydrogenase is an in vivo suppressor of colon tumorigenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 12098–12102. [Google Scholar] [CrossRef] [Green Version]
- Smartt, H.J.; Greenhough, A.; Ordóñez-Morán, P.; Talero, E.; Cherry, C.A.; Wallam, C.A.; Parry, L.; Al Kharusi, M.; Roberts, H.R.; Mariadason, J.M.; et al. β-catenin represses expression of the tumour suppressor 15-prostaglandin dehydrogenase in the normal intestinal epithelium and colorectal tumour cells. Gut 2012, 61, 1306–1314. [Google Scholar] [CrossRef]
- Moore, C.E.; Xie, J.; Gomez, E.; Herbert, T.P. Identification of cAMP-dependent kinase as a third in vivo ribosomal protein S6 kinase in pancreatic beta-cells. J. Mol. Biol. 2009, 389, 480–494. [Google Scholar] [CrossRef] [Green Version]
- Ruvinsky, I.; Sharon, N.; Lerer, T.; Cohen, H.; Stolovich-Rain, M.; Nir, T.; Dor, Y.; Zisman, P.; Meyuhas, O. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev. 2005, 19, 2199–2211. [Google Scholar] [CrossRef] [Green Version]
- Prescott, S.M. Is cyclooxygenase-2 the alpha and the omega in cancer? J. Clin. Investig. 2000, 105, 1511–1513. [Google Scholar] [CrossRef] [Green Version]
- Yoshimatsu, K.; Golijanin, D.; Paty, P.B.; Soslow, R.A.; Jakobsson, P.J.; DeLellis, R.A.; Subbaramaiah, K.; Dannenberg, A.J. Inducible microsomal prostaglandin E synthase is overexpressed in colorectal adenomas and cancer. Clin. Cancer Res. 2001, 7, 3971–3976. [Google Scholar] [PubMed]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef]
- Zelenay, S.; van der Veen, A.G.; Böttcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Rachidi, S.; Metelli, A.; Riesenberg, B.; Wu, B.X.; Nelson, M.H.; Wallace, C.; Paulos, C.M.; Rubinstein, M.P.; Garrett-Mayer, E.; Hennig, M.; et al. Platelets subvert T cell immunity against cancer via GARP-TGFβ axis. Sci Immunol. 2017, 2, eaai7911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcus, A.; Gowen, B.G.; Thompson, T.W.; Iannello, A.; Ardolino, M.; Deng, W.; Wang, L.; Shifrin, N.; Raulet, D.H. Recognition of tumors by the innate immune system and natural killer cells. Adv. Immunol. 2014, 122, 91–128. [Google Scholar] [PubMed] [Green Version]
- Maurer, S.; Kropp, K.N.; Klein, G.; Steinle, A.; Haen, S.P.; Walz, J.S.; Hinterleitner, C.; Märklin, M.; Kopp, H.G.; Salih, H.R. Platelet-mediated shedding of NKG2D ligands impairs NK cell immune-surveillance of tumor cells. Oncoimmunology 2017, 7, e1364827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Placke, T.; Örgel, M.; Schaller, M.; Jung, G.; Rammensee, H.G.; Kopp, H.G.; Salih, H.R. Platelet-derived MHC class I confers a pseudonormal phenotype to cancer cells that subverts the antitumor reactivity of natural killer immune cells. Cancer Res. 2012, 72, 440–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tímár, J.; Tóvári, J.; Rásó, E.; Mészáros, L.; Bereczky, B.; Lapis, K. Platelet-mimicry of cancer cells: Epiphenomenon with clinical significance. Oncology 2005, 69, 185–201. [Google Scholar] [CrossRef] [PubMed]
- Lonsdorf, A.S.; Krämer, B.F.; Fahrleitner, M.; Schönberger, T.; Gnerlich, S.; Ring, S.; Gehring, S.; Schneider, S.W.; Kruhlak, M.J.; Meuth, S.G.; et al. Engagement of αIIbβ3 (GPIIb/IIIa) with ανβ3 integrin mediates interaction of melanoma cells with platelets: A connection to hematogenous metastasis. J. Biol. Chem. 2012, 287, 2168–2178. [Google Scholar] [CrossRef] [Green Version]
- Felding-Habermann, B.; Habermann, R.; Saldívar, E.; Ruggeri, Z.M. Role of beta3 integrins in melanoma cell adhesion to activated platelets under flow. J. Biol. Chem. 1996, 271, 5892–5900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitrugno, A.; Williams, D.; Kerrigan, S.W.; Moran, N. A novel and essential role for FcγRIIa in cancer cell-induced platelet activation. Blood 2014, 123, 249–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boukerche, H.; Berthier-Vergnes, O.; Tabone, E.; Bailly, M.; Doré, J.F.; McGregor, J.L. Thrombospondin modulates melanoma--platelet interactions and melanoma tumour cell growth in vivo. Br. J. Cancer 1995, 72, 108–116. [Google Scholar] [CrossRef] [Green Version]
- Mammadova-Bach, E.; Zigrino, P.; Brucker, C.; Bourdon, C.; Freund, M.; De Arcangelis, A.; Abrams, S.I.; Orend, G.; Gachet, C.; Mangin, P.H. Platelet integrin controls lung metastasis through direct binding to cancer cell-derived ADAM9. JCI Insight 2016, 1, e88245. [Google Scholar] [CrossRef] [Green Version]
- Mannori, G.; Crottet, P.; Cecconi, O.; Hanasaki, K.; Aruffo, A.; Nelson, R.M.; Varki, A.; Bevilacqua, M.P. Differential colon cancer cell adhesion to E-, P-, and L-selectin: Role of mucintype glycoproteins. Cancer Res. 1995, 55, 4425–4431. [Google Scholar]
- Man, Y.; Goreke, U.; Kucukal, E.; Hill, A.; An, R.; Liu, S.; Bode, A.; Solis-Fuentes, A.; Nayak, L.V.; Little, J.A.; et al. Leukocyte adhesion to P-selectin and the inhibitory role of Crizanlizumab in sickle cell disease: A standardized microfluidic assessment. Blood Cells Mol. Dis. 2020, 83, 102424. [Google Scholar] [CrossRef]
- Gong, L.; Cai, Y.; Zhou, X.; Yang, H. Activated platelets interact with lung cancer cells through P-selectin glycoprotein ligand-1. Pathol. Oncol. Res. 2012, 18, 989–996. [Google Scholar] [CrossRef]
- Alves, C.S.; Burdick, M.M.; Thomas, S.N.; Pawar, P.; Konstantopoulos, K. The dual role of CD44 as a functional P-selectin ligand and fibrin receptor in colon carcinoma cell adhesion. Am. J. Physiol. Cell Physiol. 2008, 294, C907–C916. [Google Scholar] [CrossRef]
- Larrucea, S.; Butta, N.; Rodriguez, R.B.; Alonso-Martin, S.; Arias-Salgado, E.G.; Ayuso, M.S.; Parrilla, R. Podocalyxin enhances the adherence of cells to platelets. Cell. Mol. Life Sci. 2007, 64, 2965–2974. [Google Scholar] [CrossRef]
- Suzuki-Inoue, K.; Kato, Y.; Inoue, O.; Kaneko, M.K.; Mishima, K.; Yatomi, Y.; Yamazaki, Y.; Narimatsu, H.; Ozaki, Y. Involvement of the snake toxin receptor CLEC-2, in podoplanin-mediated platelet activation, by cancer cells. J. Biol. Chem. 2007, 282, 25993–26001. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.W.; Hsieh, P.W.; Chang, Y.T.; Lu, M.H.; Huang, T.F.; Chong, K.Y.; Liao, H.R.; Cheng, J.C.; Tseng, C.P. Identification of a novel platelet antagonist that binds to CLEC-2 and suppresses podoplanin-induced platelet aggregation and cancer metastasis. Oncotarget 2015, 6, 42733–42748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, Y.; Kaneko, M.K.; Kuno, A.; Uchiyama, N.; Amano, K.; Chiba, Y.; Hasegawa, Y.; Hirabayashi, J.; Narimatsu, H.; Mishima, K.; et al. Inhibition of tumor cell-induced platelet aggregation using a novel anti-podoplanin antibody reacting with its platelet-aggregation-stimulating domain. Biochem. Biophys. Res. Commun. 2006, 349, 1301–1307. [Google Scholar] [CrossRef] [PubMed]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signalling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillem-Llobat, P.; Dovizio, M.; Bruno, A.; Ricciotti, E.; Cufino, V.; Sacco, A.; Grande, R.; Alberti, S.; Arena, V.; Cirillo, M.; et al. Aspirin prevents colorectal cancer metastasis in mice by splitting the crosstalk between platelets and tumor cells. Oncotarget 2016, 7, 32462–32477. [Google Scholar] [CrossRef] [PubMed]
- Ungerer, M.; Rosport, K.; Bültmann, A.; Piechatzek, R.; Uhland, K.; Schlieper, P.; Gawaz, M.; Münch, G. Novel antiplatelet drug revacept (Dimeric Glycoprotein VI-Fc) specifically and efficiently inhibited collagen-induced platelet aggregation without affecting general hemostasis in humans. Circulation 2011, 123, 1891–1899. [Google Scholar] [CrossRef] [PubMed]
- Lucotti, S.; Cerutti, C.; Soyer, M.; Gil-Bernabé, A.M.; Gomes, A.L.; Allen, P.D.; Smart, S.; Markelc, B.; Watson, K.; Armstrong, P.C.; et al. Aspirin blocks formation of metastatic intravascular niches by inhibiting platelet-derived COX-1/thromboxane A2. J. Clin. Investig. 2019, 129, 1845–1862. [Google Scholar] [CrossRef] [Green Version]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Nieuwland, R.; van der Pol, E.; Gardiner, C.; Sturk, A. Platelet-Derived Microparticles. In Platelets, 3rd ed.; Academic Press: Cambridge, MA, USA, 2013; pp. 453–467. [Google Scholar]
- Lynch, S.F.; Ludlam, C.A. Plasma microparticles and vascular disorders. Br. J. Haematol. 2007, 137, 36–48. [Google Scholar] [CrossRef]
- Morel, O.; Toti, F.; Bakouboula, B.; Grunebaum, L.; Freyssinet, J.M. Procoagulant microparticles: ‘criminal partners’ in atherothrombosis and deleterious cellular exchanges. Pathophysiol. Haemost. Thromb. 2006, 35, 15–22. [Google Scholar] [CrossRef]
- Ratajczak, J.; Wysoczynski, M.; Hayek, F.; Janowska-Wieczorek, A.; Ratajczak, M.Z. Membrane-derived microvesicles: Important and underappreciated mediators of cell-to-cell communication. Leukemia 2006, 20, 1487–1495. [Google Scholar] [CrossRef]
- Tang, M.; Jiang, L.; Lin, Y.; Wu, X.; Wang, K.; He, Q.; Wang, X.; Li, W. Platelet microparticle-mediated transfer of miR- 939 to epithelial ovarian cancer cells promotes epithelial to mesenchymal transition. Oncotarget 2017, 8, 97464–97475. [Google Scholar] [CrossRef] [Green Version]
- Michael, J.V.; Wurtzel, J.G.T.; Mao, G.F.; Rao, A.K.; Kolpakov, A.; Sabri, A.; Hoffman, N.E.; Rajan, S.; Tomar, D.; Madesh, M.; et al. Platelet microparticles infiltrating solid tumors transfer miRNAs that suppress tumor growth. Blood 2017, 130, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Barry, O.P.; Pratico, D.; Lawson, J.A.; FitzGerald, G.A. Transcellular activation of platelets and endothelial cells by bioactive lipids in platelet microparticles. J. Clin. Investig. 1997, 99, 2118–2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.K.; Song, K.S.; Park, Y.S.; Kang, Y.H.; Lee, Y.J.; Lee, K.R.; Ryu, K.W.; Bae, J.M.; Kim, S. Elevated levels of circulating platelet microparticles, VEGF, IL-6 and RANTES in patients with gastric cancer: Possible role of a metastasis predictor. Eur. J. Cancer 2003, 39, 184–191. [Google Scholar] [CrossRef]
- Grande, R.; Dovizio, M.; Marcone, S.; Szklanna, P.B.; Bruno, A.; Ebhardt, H.A.; Cassidy, H.; Ní Áinle, F.; Caprodossi, A.; Lanuti, P.; et al. Platelet-Derived Microparticles from Obese Individuals: Characterization of Number, Size, Proteomics, and Crosstalk with Cancer and Endothelial Cells. Front. Pharmacol. 2019, 10, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, A.; Kim, H.S.; Bojmar, L.; Gyan, K.E.; Cioffi, M.; Hernandez, J.; Zambirinis, C.P.; Rodrigues, G.; Molina, H.; Heissel, S.; et al. Extracellular Vesicle and Particle Biomarkers Define Multiple Human Cancers. Cell 2020, 182, 1044–1061.e18. [Google Scholar] [CrossRef] [PubMed]
- Thun, M.J.; Henley, S.J.; Patrono, C. Nonsteroidal anti-inflammatory drugs as anticancer agents: Mechanistic, pharmacologic, and clinical issues. J. Natl. Cancer Inst. 2002, 94, 252–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuzick, J.; Otto, F.; Baron, J.A.; Brown, P.H.; Burn, J.; Greenwald, P.; Jankowski, J.; La Vecchia, C.; Meyskens, F.; Senn, H.J.; et al. Aspirin and non-steroidal anti-inflammatory drugs for cancer prevention: An international consensus statement. Lancet Oncol. 2009, 10, 501–507. [Google Scholar] [CrossRef] [Green Version]
- Giovannucci, E.; Egan, K.M.; Hunter, D.J.; Stampfer, M.J.; Colditz, G.A.; Willett, W.C.; Speizer, F.E. Aspirin and the risk of colorectal cancer inwomen. N. Engl. J. Med. 1995, 333, 609–614. [Google Scholar] [CrossRef]
- Gann, P.H.; Manson, J.E.; Glynn, R.J.; Buring, J.E.; Hennekens, C.H. Low-dose aspirin and incidence of colorectal tumors in a randomized trial. J. Natl. Cancer Inst. 1993, 85, 1220–1224. [Google Scholar] [CrossRef]
- Cook, N.R.; Lee, I.M.; Gaziano, J.M.; Gordon, D.; Ridker, P.M.; Manson, J.E.; Hennekens, C.H.; Buring, J.E. Low-dose aspirin in the primary prevention ofcancer: The Women’s Health Study: A randomized controlled trial. JAMA 2005, 294, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron, J.A.; Cole, B.F.; Sandler, R.S.; Haile, R.W.; Ahnen, D.; Bresalier, R.; McKeown-Eyssen, G.; Summers, R.W.; Rothstein, R.; Burke, C.A.; et al. A randomized trial of aspirin to prevent colorectal adenomas. N. Engl. J. Med. 2003, 348, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Sandler, R.S.; Halabi, S.; Baron, J.A.; Budinger, S.; Paskett, E.; Keresztes, R.; Petrelli, N.; Pipas, J.M.; Karp, D.D.; Loprinzi, C.L.; et al. A randomized trial of aspirin to prevent colorectaladenomas in patients with previous colorectal cancer. N. Engl. J. Med. 2003, 348, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Benamouzig, R.; Deyra, J.; Martin, A.; Girard, B.; Jullian, E.; Piednoir, B.; Couturier, D.; Coste, T.; Little, J.; Chaussade, S.; et al. Daily soluble aspirin and prevention of colorectaladenoma recurrence: One-year results of the APACC trial. Gastroenterology 2003, 125, 328–336. [Google Scholar] [CrossRef]
- Benamouzig, R.; Uzzan, B.; Deyra, J.; Martin, A.; Girard, B.; Little, J.; Chaussade, S. Association pour la Prévention par l’Aspirine du Cancer Colorectal Study Group APACC). Prevention by daily soluble aspirin of colorectal adenoma recurrence: 4-year results of the APACC randomised trial. Gut 2012, 61, 255–261. [Google Scholar] [CrossRef] [Green Version]
- Logan, R.F.; Grainge, M.J.; Shepherd, V.C.; Armitage, N.C.; Muir, K.R.; ukCAP Trial Group. Aspirin and folic acid for the prevention of recurrent colorectal adenomas. Gastroenterology 2008, 134, 29–38. [Google Scholar] [CrossRef]
- Burn, J.; Bishop, D.T.; Chapman, P.D.; Elliott, F.; Bertario, L.; Dunlop, M.G.; Eccles, D.; Ellis, A.; Evans, D.G.; Fodde, R.; et al. International CAPP consortium. A randomized placebo-controlled prevention trial of aspirin and/or resistant starch in young people with familial adenomatous polyposis. Cancer Prev. Res. 2011, 4, 655–665. [Google Scholar] [CrossRef] [Green Version]
- Burn, J.; Gerdes, A.M.; Macrae, F.; Mecklin, J.P.; Moeslein, G.; Olschwang, S.; Eccles, D.; Evans, D.G.; Maher, E.R.; Bertario, L.; et al. Long- term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: An analysis from the CAPP2 randomised controlled trial. Lancet 2011, 378, 2081–2087. [Google Scholar] [CrossRef] [Green Version]
- Burn, J.; Sheth, H.; Elliott, F.; Reed, L.; Macrae, F.; Mecklin, J.P.; Möslein, G.; McRonald, F.E.; Bertario, L.; Evans, D.G.; et al. Cancer prevention with aspirin in hereditary colorectal cancer (Lynch syndrome), 10-year follow-up and registry-based 20-year data in the CAPP2 study: A double-blind, randomised, placebo-controlled trial. Lancet 2020, 395, 1855–1863. [Google Scholar] [CrossRef]
- Okada, S.; Morimoto, T.; Ogawa, H.; Sakuma, M.; Matsumoto, C.; Soejima, H.; Nakayama, M.; Doi, N.; Jinnouchi, H.; Waki, M.; et al. Effect of Aspirin on Cancer Chemoprevention in Japanese Patients with Type 2 Diabetes: 10-Year Observational Follow-up of a Randomized Controlled Trial. Diabetes Care 2018, 41, 1757–1764. [Google Scholar] [CrossRef] [Green Version]
- ASCEND Study Collaborative Group; Bowman, L.; Mafham, M.; Wallendszus, K.; Stevens, W.; Buck, G.; Barton, J.; Murphy, K.; Aung, T.; Haynes, R.; et al. Effects of Aspirin for Primary Prevention in Persons with Diabetes Mellitus. N. Engl. J. Med. 2018, 379, 1529–1539. [Google Scholar]
- McNeil, J.J.; Wolfe, R.; Woods, R.L.; Tonkin, A.M.; Donnan, G.A.; Nelson, M.R.; Reid, C.M.; Lockery, J.E.; Kirpach, B.; Storey, E.; et al. Effect of Aspirin on Cardiovascular Events and Bleeding in the Healthy Elderly. N. Engl. J. Med. 2018, 379, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- McNeil, J.J.; Gibbs, P.; Orchard, S.G.; Lockery, J.E.; Bernstein, W.B.; Cao, Y.; Ford, L.; Haydon, A.; Kirpach, B.; Macrae, F.; et al. Effect of aspirin on cancer incidence and mortality in older adults. J. Natl. Cancer Inst. 2020. [Google Scholar] [CrossRef] [PubMed]
- Coyle, C.; Cafferty, F.H.; Rowley, S.; MacKenzie, M.; Berkman, L.; Gupta, S.; Pramesh, C.S.; Gilbert, D.; Kynaston, H.; Cameron, D.; et al. ADD-ASPIRIN: A phase III, double-blind, placebo controlled, randomised trial assessing the effects of aspirin on disease recurrence and survival after primary therapy in common non-metastatic solid tumours. Contemp. Clin. Trials 2016, 51, 56–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzgerald, D.J.; Fitzgerald, G.A. Historical lessons in translational medicine: Cyclooxygenase inhibition and P2Y12 antagonism. Circ. Res. 2013, 112, 174–194. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, U.R.; Geiger, J.; Walter, U.; Eigenthaler, M. Flow cytometry analysis of intracellular VASP phosphorylation for the assessment of activating and inhibitory signal transduction pathways in human platelets—Definition and detection of ticlopidine/clopidogrel effects. Thromb. Haemost. 1999, 82, 1145–1152. [Google Scholar] [CrossRef] [Green Version]
- Ballerini, P.; Dovizio, M.; Bruno, A.; Tacconelli, S.; Patrignani, P. P2Y12 Receptors in Tumorigenesis and Metastasis. Front. Pharmacol. 2018, 9, 66. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Sun, Y.; Li, D.; Zhang, L.; Wang, K.; Zuo, Y.; Gartner, T.K.; Liu, J. Platelet P2Y12 is involved in murine pulmonary metastasis. PLoS ONE 2013, 8, e80780. [Google Scholar] [CrossRef] [Green Version]
- Sitia, G.; Aiolfi, R.; Di Lucia, P.; Mainetti, M.; Fiocchi, A.; Mingozzi, F.; Esposito, A.; Ruggeri, Z.M.; Chisari, F.V.; Iannacone, M.; et al. Antiplatelet therapy prevents hepatocellular carcinoma and improves survival in a mouse model of chronic hepatitis B. Proc. Natl. Acad. Sci. USA 2012, 109, E2165–E2172. [Google Scholar] [CrossRef] [Green Version]
- Kohga, S.; Kinjo, M.; Tanaka, K.; Ogawa, H.; Ishimara, M.; Tanaka, N. Effects of 5-(2-chlorobenzyl)- 4,5,6,7-tetrahydrothieno[3,2-C]pyridine hydrochloride (Ticlopidine), a platelet aggregation inhibitor, on blood-borne metastasis. Cancer Res. 1981, 41, 4710–4714. [Google Scholar]
- Gareau, A.J.; Brien, C.; Gebremeskel, S.; Liwski, R.S.; Johnston, B.; Bezuhly, M. Ticagrelor inhibits platelet-tumor cell interactions and metastasis in human and murine breast cancer. Clin. Exp. Metastasis 2018, 35, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Miguel, A.; García-Rodríguez, L.A.; Gil, M.; Montoya, H.; Rodríguez-Martín, S.; de Abajo, F.J. Clopidogrel and Low-Dose Aspirin, Alone or Together, Reduce Risk of Colorectal Cancer. Clin. Gastroenterol. Hepatol. 2019, 17, 2024–2033.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, H.; Greenberg, D.L.; Fujikawa, K.W.; Chung, D.W.; Davie, E.W. Protease-activated receptor 1 is the primary mediator of thrombin-stimulated platelet procoagulant activity. Proc. Natl. Acad. Sci. USA 1999, 96, 11189–11193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojtukiewicz, M.Z.; Hempel, D.; Sierko, E.; Tucker, S.C.; Honn, K.V. Protease-activated receptors (PARs)--biology and role in cancer invasion and metastasis. Cancer Metastasis Rev. 2015, 34, 775–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Italiano, J.E., Jr.; Richardson, J.L.; Patel-Hett, S.; Battinelli, E.; Zaslavsky, A.; Short, S.; Ryeom, S.; Folkman, J.; Klement, G.L. Angiogenesis is regulated by a novel mechanism: Pro- and antiangiogenic proteins are organized into separate platelet alpha granules and differentially released. Blood 2008, 111, 1227–1233. [Google Scholar] [CrossRef] [Green Version]
- Adams, G.N.; Rosenfeldt, L.; Frederick, M.; Miller, W.; Waltz, D.; Kombrinck, K.; McElhinney, K.E.; Flick, M.J.; Monia, B.P.; Revenko, A.S. Colon Cancer Growth and Dissemination Relies upon Thrombin, Stromal PAR-1, and Fibrinogen. Cancer Res. 2015, 75, 4235–4243. [Google Scholar] [CrossRef] [Green Version]
- Chanakira, A.; Westmark, P.R.; Ong, I.M.; Sheehan, J.P. Tissue factor-factor VIIa complex triggers protease activated receptor 2-dependent growth factor release and migration in ovarian cancer. Gynecol. Oncol. 2017, 145, 167–175. [Google Scholar] [CrossRef] [Green Version]
- Boukerche, H.; Berthier-Vergnes, O.; Tabone, E.; Dore, J.F.; Leung, L.L.; McGregor, J.L. Platelet–melanoma cell interaction is mediated by the glycoprotein IIb–IIIa complex. Blood 1989, 74, 658–663. [Google Scholar] [CrossRef] [Green Version]
- Kononczuk, J.; Surazynski, A.; Czyzewska, U.; Prokop, I.; Tomczyk, M.; Palka, J.; Miltyk, W. αIIbβ3-integrin Ligands: Abciximab and Eptifibatide as Proapoptotic Factors in MCF-7 Human Breast Cancer Cells. Curr. Drug Targets 2015, 16, 1429–1437. [Google Scholar] [CrossRef]
- Parikka, M.; Nissinen, L.; Kainulainen, T.; Bruckner-Tuderman, L.; Salo, T.; Heino, J.; Tasanen, K. Collagen, X.VII promotes integrin-mediated squamous cell carcinoma transmigration—A novel role for alphaIIb integrin and tirofiban. Exp. Cell Res. 2006, 312, 1431–1438. [Google Scholar] [CrossRef]
- Zhao, F.; Li, L.; Guan, L.; Yang, H.; Wu, C.; Liu, Y. Roles for GP IIb/IIIa and αvβ3 integrins in MDA-MB-231 cell invasion and shear flow-induced cancer cell mechanotransduction. Cancer Lett. 2014, 344, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.; Dovizio, M.; Tacconelli, S.; Contursi, A.; Ballerini, P.; Patrignani, P. Antithrombotic Agents and Cancer. Cancers 2018, 10, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bizzozero, G. Su di un nuovo elemento morfologico del sangue dei mammiferi e della sua importanza nella trombosi e nella coagulazione. L’Osservatore 1881, 17, 785–787. [Google Scholar]
- Li, L.; Geraghty, O.C.; Mehta, Z.; Rothwell, P.M.; Oxford Vascular Study. Age-specific risks, severity, time course, and outcome of bleeding on long-term antiplatelet treatment after vascular events: A population-based cohort study. Lancet 2017, 390, 490–499. [Google Scholar] [CrossRef] [Green Version]
- Clemetson, K.J.; Capitanio, A.; Lüscher, E.F. High resolution two-dimensional gel electrophoresis of the proteins and glycoproteins of human blood platelets and platelet membranes. Biochim. Biophys. Acta 1979, 553, 11–24. [Google Scholar] [CrossRef]
- O’Neill, E.E.; Brock, C.J.; von Kriegsheim, A.F.; Pearce, A.C.; Dwek, R.A.; Watson, S.P.; Hebestreit, H.F. Towards complete analysis of the platelet proteome. Proteomics 2002, 2, 288–305. [Google Scholar] [CrossRef]
- García, A.; Prabhakar, S.; Hughan, S.; Anderson, T.W.; Brock, C.J.; Pearce, A.C.; Dwek, R.A.; Watson, S.P.; Hebestreit, H.F.; Zitzmann, N. Differential proteome analysis of TRAP- activated platelets: Involvement of DOK-2 and phosphorylation of RGS proteins. Blood 2004, 103, 2088–2095. [Google Scholar] [CrossRef] [Green Version]
- Coppinger, J.A.; Cagney, G.; Toomey, S.; Kislinger, T.; Belton, O.; McRedmond, J.P.; Cahill, D.J.; Emili, A.; Fitzgerald, D.J.; Maguire, P.B. Characterization of the proteins released from activated platelets leads to localization of novel platelet proteins in human atherosclerotic lesions. Blood 2004, 103, 2096–2104. [Google Scholar] [CrossRef] [Green Version]
- Garcia, B.A.; Smalley, D.M.; Cho, H.; Shabanowitz, J.; Ley, K.; Hunt, D.F. The platelet microparticle proteome. J. Proteome Res. 2005, 4, 1516–1521. [Google Scholar] [CrossRef] [Green Version]
- Maynard, D.M.; Heijnen, H.F.; Horne, M.K.; White, J.G.; Gahl, W.A. Proteomic analysis of platelet alpha-granules using mass spectrometry. J. Thromb. Haemost. 2007, 5, 1945–1955. [Google Scholar] [CrossRef]
- Hernandez-Ruiz, L.; Valverde, F.; Jimenez-Nuñez, M.D.; Ocaña, E.; Sáez-Benito, A.; Rodríguez-Martorell, J.; Bohórquez, J.C.; Serrano, A.; Ruiz, F.A. Organellar proteomics of human platelet dense granules reveals that 14-3-3zeta is a granule protein related to atherosclerosis. J. Proteome Res. 2007, 6, 4449–4457. [Google Scholar] [CrossRef] [PubMed]
- Burkhart, J.M.; Vaudel, M.; Gambaryan, S.; Radau, S.; Walter, U.; Martens, L.; Geiger, J.; Sickmann, A.; Zahedi, R.P. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood 2012, 120, e73–e82. [Google Scholar] [CrossRef] [Green Version]
- Wijten, P.; van Holten, T.; Woo, L.L.; Bleijerveld, O.B.; Roest, M.; Heck, A.J.; Scholten, A. High precision platelet releasate definition by quantitative reversed protein profiling--brief report. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1635–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evangelista, V.; Manarini, S.; Di Santo, A.; Capone, M.L.; Ricciotti, E.; Di Francesco, L.; Tacconelli, S.; Sacchetti, A.; D’Angelo, S.; Scilimati, A.; et al. De novo synthesis of cyclooxygenase-1 counteracts the suppression of platelet thromboxane biosynthesis by aspirin. Circ. Res. 2006, 98, 593–595. [Google Scholar] [CrossRef] [PubMed]
- Denis, M.M.; Tolley, N.D.; Bunting, M.; Schwertz, H.; Jiang, H.; Lindemann, S.; Yost, C.C.; Rubner, F.J.; Albertine, K.H.; Swoboda, K.J.; et al. Escaping the nuclear confines: Signal-dependent pre-mRNA splicing in anucleate platelets. Cell 2005, 122, 379–391. [Google Scholar] [CrossRef] [Green Version]
- Ingolia, N.T.; Ghaemmaghami, S.; Newman, J.R.; Weissman, J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 2009, 324, 218–223. [Google Scholar] [CrossRef] [Green Version]
- Mills, E.W.; Green, R.; Ingolia, N.T. Slowed decay of mRNAs enhances platelet specific translation. Blood 2017, 129, e38–e48. [Google Scholar] [CrossRef] [Green Version]
- Kut, C.; Mac Gabhann, F.; Popel, A.S. Where is VEGF in the body? A meta-analysis of VEGF distribution in cancer. Br. J. Cancer 2007, 97, 978–985. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.; Lesyk, G.; Radziwon-Balicka, A.; Jurasz, P. Pharmacological regulation of platelet factors that influence tumor angiogenesis. Semin Oncol. 2014, 41, 370–377. [Google Scholar] [CrossRef]
- Salgado, R.; Benoy, I.; Bogers, J.; Weytjens, R.; Vermeulen, P.; Dirix, L.; Van Marck, E. Platelets and vascular endothelial growth factor (VEGF): A morphological and functional study. Angiogenesis 2001, 4, 37–43. [Google Scholar] [CrossRef]
- Peterson, J.E.; Zurakowski, D.; Italiano, J.E., Jr.; Michel, L.V.; Connors, S.; Oenick, M.; D’Amato, R.J.; Klement, G.L.; Folkman, J. VEGF, PF4 and PDGF are elevated in platelets of colorectal cancer patients. Angiogenesis 2012, 15, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Sabrkhany, S.; Kuijpers, M.J.E.; Knol, J.C.; Olde Damink, S.W.M.; Dingemans, A.C.; Verheul, H.M.; Piersma, S.R.; Pham, T.V.; Griffioen, A.W.; Oude Egbrink, M.G.A.; et al. Exploration of the platelet proteome in patients with early-stage cancer. J. Proteom. 2018, 177, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Braza-Boïls, A.; Barwari, T.; Gutmann, C.; Thomas, M.R.; Judge, H.M.; Joshi, A.; Pechlaner, R.; Shankar-Hari, M.; Ajjan, R.A.; Sabroe, I.; et al. Circulating MicroRNA Levels Indicate Platelet and Leukocyte Activation in Endotoxemia Despite Platelet P2Y12 Inhibition. Int. J. Mol. Sci. 2020, 21, 2897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutmann, C.; Joshi, A.; Mayr, M. Platelet “-omics” in health and cardiovascular disease. Atherosclerosis 2020, 307, 87–96. [Google Scholar] [CrossRef]
- McManus, D.D.; Beaulieu, L.M.; Mick, E.; Tanriverdi, K.; Larson, M.G.; Keaney, J.F., Jr.; Benjamin, E.J.; Freedman, J.E. Relationship among circulating inflammatory proteins, platelet gene expression, and cardiovascular risk. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2666–2673. [Google Scholar] [CrossRef] [Green Version]
- Raghavachari, N.; Xu, X.; Harris, A.; Villagra, J.; Logun, C.; Barb, J.; Solomon, M.A.; Suffredini, A.F.; Danner, R.L.; Kato, G.; et al. Amplified expression profiling of platelet transcriptome reveals changes in arginine metabolic pathways in patients with sickle cell disease. Circulation 2007, 115, 1551–1562. [Google Scholar] [CrossRef] [Green Version]
- Best, M.G.; Sol, N.; Kooi, I.; Tannous, J.; Westerman, B.A.; Rustenburg, F.; Schellen, P.; Verschueren, H.; Post, E.; Koster, J.; et al. RNA-Seq of Tumor-Educated Platelets Enables Blood-Based Pan-Cancer, Multiclass, and Molecular Pathway Cancer Diagnostics. Cancer Cell 2015, 28, 666–676. [Google Scholar] [CrossRef] [Green Version]
- Lood, C.; Amisten, S.; Gullstrand, B.; Jönsen, A.; Allhorn, M.; Truedsson, L.; Sturfelt, G.; Erlinge, D.; Bengtsson, A.A. Platelet transcriptional profile and protein expression in patients with systemic lupus erythematosus: Up-regulation of the type I interferon system is strongly associated with vascular disease. Blood 2010, 116, 1951–1957. [Google Scholar] [CrossRef] [Green Version]
- Freedman, J.E.; Larson, M.G.; Tanriverdi, K.; O’Donnell, C.J.; Morin, K.; Hakanson, A.S.; Vasan, R.S.; Johnson, A.D.; Iafrati, M.D.; Benjamin, E.J. Relation of platelet and leukocyte inflammatory transcripts to body mass index in the Framingham heart study. Circulation 2010, 122, 119–129. [Google Scholar] [CrossRef] [Green Version]
- Plé, H.; Maltais, M.; Corduan, A.; Rousseau, G.; Madore, F.; Provost, P. Alteration of the platelet transcriptome in chronic kidney disease. Thromb. Haemost. 2012, 108, 605–615. [Google Scholar] [CrossRef]
- Healy, A.M.; Pickard, M.D.; Pradhan, A.D.; Wang, Y.; Chen, Z.; Croce, K.; Sakuma, M.; Shi, C.; Zago, A.C.; Garasic, J.; et al. Platelet expression profiling and clinical validation of myeloid- related protein-14 as a novel determinant of cardiovascular events. Circulation 2006, 113, 2278–2284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colombo, G.; Gertow, K.; Marenzi, G.; Brambilla, M.; De Metrio, M.; Tremoli, E.; Camera, M. Gene expression profiling reveals multiple differences in platelets from patients with stable angina or non-ST elevation acute coronary syndrome. Thromb. Res. 2011, 128, 161–168. [Google Scholar] [CrossRef]
- Eicher, J.D.; Wakabayashi, Y.; Vitseva, O.; Esa, N.; Yang, Y.; Zhu, J.; Freedman, J.E.; McManus, D.D.; Johnson, A.D. Characterization of the platelet transcriptome by RNA sequencing in patients with acute myocardial infarction. Platelets 2016, 27, 230–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reilly, S.J.; Li, N.; Liska, J.; Ekström, M.; Tornvall, P. Coronary artery bypass graft surgery up-regulates genes involved in platelet aggregation. J. Thromb. Haemost. 2012, 10, 557–563. [Google Scholar] [CrossRef]
- Saenz-Pipaon, G.; San Martín, P.; Planell, N.; Maillo, A.; Ravassa, S.; Vilas-Zornoza, A.; Martinez-Aguilar, E.; Rodriguez, J.A.; Alameda, D.; Lara-Astiaso, D.; et al. Functional and transcriptomic analysis of extracellular vesicles identifies calprotectin as a new prognostic marker in peripheral arterial disease (PAD). J. Extracell. Vesicles 2020, 9, 1729646. [Google Scholar] [CrossRef]
- Wysokinski, W.E.; Tafur, A.; Ammash, N.; Asirvatham, S.J.; Wu, Y.; Gosk-Bierska, I.; Grill, D.E.; Slusser, J.P.; Mruk, J.; McBane, R.D. Impact of atrial fibrillation on platelet gene expression. Eur. J. Haematol. 2017, 98, 615–621. [Google Scholar] [CrossRef] [Green Version]
- Heitzer, E.; Haque, I.S.; Roberts, C.E.S.; Speicher, M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet 2019, 2, 71–88. [Google Scholar] [CrossRef]
- Babayan, A.; Pantel, K. Advances in liquid biopsy approaches for early detection and monitoring of cancer. Genome Med. 2018, 10, 21. [Google Scholar] [CrossRef]
- Aravanis, A.M.; Lee, M.; Klausner, R.D. Next-Generation Sequencing of Circulating Tumor DNA for Early Cancer Detection. Cell 2017, 168, 571–574. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Calverley, D.C.; Phang, T.L.; Choudhury, Q.G.; Gao, B.; Oton, A.B.; Weyant, M.J.; Geraci, M.W. Significant downregulation of platelet gene expression in metastatic lung cancer. Clin. Transl. Sci. 2010, 3, 227–232. [Google Scholar] [CrossRef]
- Nilsson, R.J.; Balaj, L.; Hulleman, E.; van Rijn, S.; Pegtel, D.M.; Walraven, M.; Widmark, A.; Gerritsen, W.R.; Verheul, H.M.; Vandertop, W.P.; et al. Blood platelets contain tumor-derived RNA biomarkers. Blood 2011, 118, 3680–3683. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.J.; Karachaliou, N.; Berenguer, J.; Gimenez-Capitan, A.; Schellen, P.; Teixido, C.; Tannous, J.; Kuiper, J.L.; Drees, E.; Grabowska, M.; et al. Rearranged EML4-ALK fusion transcripts sequester in circulating blood platelets and enable blood-based crizotinib response monitoring in non-small-cell lung cancer. Oncotarget 2016, 7, 1066–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tjon-Kon-Fat, L.A.; Lundholm, M.; Schröder, M.; Wurdinger, T.; Thellenberg-Karlsson, C.; Widmark, A.; Wikström, P.; Nilsson, R.J.A. Platelets harbor prostate cancer biomarkers and the ability to predict therapeutic response to abiraterone in castration resistant patients. Prostate 2018, 78, 48–53. [Google Scholar] [CrossRef]
- Klement, G.L.; Yip, T.T.; Cassiola, F.; Kikuchi, L.; Cervi, D.; Podust, V.; Italiano, J.E.; Wheatley, E.; Abou-Slaybi, A.; Bender, E.; et al. Platelets actively sequester angiogenesis regulators. Blood 2009, 113, 2835–2842. [Google Scholar] [CrossRef]
- Best, M.G.; Sol, N.; In ‘t Veld, S.G.J.G.; Vancura, A.; Muller, M.; Niemeijer, A.N.; Fejes, A.V.; Tjon Kon Fat, L.A.; Huis In ‘t Veld, A.E.; Leurs, C.; et al. Swarm Intelligence-Enhanced Detection of Non-Small-Cell Lung Cancer Using Tumor-Educated Platelets. Cancer Cell 2017, 32, 238–252.e9. [Google Scholar] [CrossRef]
- Clancy, L.; Beaulieu, L.M.; Tanriverdi, K.; Freedman, J.E. The role of RNA uptake in platelet heterogeneity. Thromb. Haemost. 2017, 117, 948–961. [Google Scholar]
- Navya, P.N.; Kaphle, A.; Srinivas, S.P.; Bhargava, S.K.; Rotello, V.M.; Daima, H.K. Current trends and challenges in cancer management and therapy using designer nanomaterials. Nano Converg. 2019, 6, 23. [Google Scholar] [CrossRef] [Green Version]
- Wei, Q.; Qian, Y.; Yu, J.; Wong, C.C. Metabolic rewiring in the promotion of cancer metastasis: Mechanisms and therapeutic implications. Oncogene 2020, 39, 6139–6156. [Google Scholar] [CrossRef]
- Li, T.; Dong, H.; Zhang, C.; Mo, R. Cell-Based Drug Delivery Systems for Biomedical Applications. Nano Res. 2018, 11, 5240–5257. [Google Scholar] [CrossRef]
- Zhang, C.L.; Huang, T.; Wu, B.L.; He, W.X.; Liu, D. Stem cells in cancer therapy: Opportunities and challenges. Oncotarget 2017, 8, 75756–75766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muzykantov, V.R. Drug delivery by red blood cells: Vascular carriers designed by mother nature. Expert Opin. Drug Deliv. 2010, 7, 403–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.; Kapate, N.; Shields, C.W.; Mitragotri, S. Drug delivery to macrophages: A review of targeting drugs and drug carriers to macrophages for inflammatory diseases. Adv. Drug Deliv. Rev. 2019, 165–166, 15–40. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.; Dong, X.; Shi, X.; Zhang, C.; Wang, Z. Neutrophil-Based Drug Delivery Systems. Adv. Mater. 2018, 30, e1706245. [Google Scholar] [CrossRef]
- Schmid, D.; Park, C.G.; Hartl, C.A.; Subedi, N.; Cartwright, A.N.; Puerto, R.B.; Zheng, Y.; Maiarana, J.; Freeman, G.J.; Wucherpfennig, K.W.; et al. T cell-targeting nanoparticles focus delivery of immunotherapy to improve antitumor immunity. Nat. Commun. 2017, 8, 1747. [Google Scholar] [CrossRef]
- Xu, X.R.; Yousef, G.M.; Ni, H. Cancer and platelet crosstalk: Opportunities and challenges for aspirin and other antiplatelet agents. Blood 2018, 131, 1777–1789. [Google Scholar] [CrossRef] [Green Version]
- Contursi, A.; Grande, R.; Dovizio, M.; Bruno, A.; Fullone, R.; Patrignani, P. Platelets in cancer development and diagnosis. Biochem. Soc. Trans. 2018, 46, 1517–1527. [Google Scholar] [CrossRef]
- Morrell, C.N.; Aggrey, A.A.; Chapman, L.M.; Modjeski, K.L. Emerging roles for platelets as immune and inflammatory cells. Blood 2014, 123, 2759–2767. [Google Scholar] [CrossRef] [Green Version]
- Huong, P.T.; Nguyen, L.T.; Nguyen, X.B.; Lee, S.K.; Bach, D.H. The Role of Platelets in the Tumor-Microenvironment and the Drug Resistance of Cancer Cells. Cancers 2019, 11, 240. [Google Scholar] [CrossRef] [Green Version]
- Qi, C.; Wei, B.; Zhou, W.; Yang, Y.; Li, B.; Guo, S.; Li, J.; Ye, J.; Li, J.; Zhang, Q.; et al. P-selectin-mediated platelet adhesion promotes tumor growth. Oncotarget 2015, 6, 6584–6596. [Google Scholar] [CrossRef] [Green Version]
- Haemmerle, M.; Bottsford-Miller, J.; Pradeep, S.; Taylor, M.L.; Choi, H.J.; Hansen, J.M.; Dalton, H.J.; Stone, R.L.; Cho, M.S.; Nick, A.M.; et al. FAK regulates platelet extravasation and tumor growth after antiangiogenic therapy withdrawal. J. Clin. Investig. 2016, 126, 1885–1896. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, M.; Wang, X.; Lim, B.; Leitner, E.; Klingberg, F.; Ching, V.; Yao, Y.; Huang, D.; Gao, X.M.; Kiriazis, H.; et al. Platelet-Targeted Delivery of Peripheral Blood Mononuclear Cells to the IschemicHeart Restores Cardiac Function after Ischemia-Reperfusion Injury. Theranostics 2017, 7, 3192–3206. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, M.; Huang, Y.; Joshi, S.; Popa, G.J.; Mendenhall, M.D.; Wang, Q.J.; Garvy, B.A.; Myint, T.; Whiteheart, S.W. Platelets Endocytose Viral Particles and Are Activated via TLR (Toll-Like Receptor) Signaling. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1635–1650. [Google Scholar] [CrossRef] [PubMed]
- Gaertner, F.; Ahmad, Z.; Rosenberger, G.; Fan, S.; Nicolai, L.; Busch, B.; Yavuz, G.; Luckner, M.; Ishikawa-Ankerhold, H.; Hennel, R.; et al. Migrating Platelets Are Mechano- scavengers that Collect and Bundle Bacteria. Cell 2017, 171, 1368–1382.e23. [Google Scholar] [CrossRef]
- White, J.G. Why human platelets fail to kill bacteria. Platelets 2006, 17, 191–200. [Google Scholar] [CrossRef]
- White, J.G. Platelets are covercytes, not phagocytes: Uptake of bacteria involves channels of the open canalicular system. Platelets 2005, 16, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Alam, M.A.; Shaw, J.; Dasgupta, A.K. Drug delivery using platelet cancer cell interaction. Pharm. Res. 2013, 30, 2785–2794. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Zuo, H.; Chen, B.; Wang, R.; Ahmed, A.; Hu, Y.; Ouyang, J. Doxorubicin-loaded platelets as a smart drug delivery system: An improved therapy for lymphoma. Sci. Rep. 2017, 7, 42632. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, C.; Liu, Z. Red Blood Cells as Smart Delivery Systems. Bioconjug. Chem. 2018, 29, 852–860. [Google Scholar] [CrossRef]
- Feins, S.; Kong, W.; Williams, E.F.; Milone, M.C.; Fraietta, J.A. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am. J. Hematol. 2019, 94, S3–S9. [Google Scholar] [CrossRef] [Green Version]
- Lyde, R.; Sabatino, D.; Sullivan, S.K.; Poncz, M. Platelet-delivered therapeutics. J. Thromb. Haemost. 2015, 13, S143–S150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.; Hodivala-Dilke, K.; Johnson, B.D.; DU, L.M.; Hynes, R.O.; White, G.C.; Wilcox, D.A. Therapeutic expression of the platelet-specific integrin, alphaIIbbeta3, in a murine model for Glanzmann thrombasthenia. Blood 2005, 106, 2671–2679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Q.; Wilcox, D.A.; Morateck, P.A.; Fahs, S.A.; Kenny, D.; Montgomery, R.R. Targeting platelet GPIbalpha transgene expression to human megakaryocytes and forming a complete complex with endogenous GPIbeta and GPIX. J. Thromb. Haemost. 2004, 2, 1989–1997. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Kuether, E.L.; Cooley, B.C.; Fahs, S.A.; Schroeder, J.A.; Wilcox, D.A.; Montgomery, R.R. Sustained Phenotypic Correction of Murine Hemophilia A with Pre-Existing Anti-FVIII Immunity Using Lentivirus-Mediated Platelet-Specific FVIII Gene Transfer. ASH Annu. Meet. Abstr. 2009, 114, 29. [Google Scholar] [CrossRef]
- Levine, R.F. Isolation and characterization of normal human megakaryocytes. Br. J. Haematol. 1980, 45, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Sim, X.; Jarocha, D.; Hayes, V.; Hanby, H.A.; Marks, M.S.; Camire, R.M.; French, D.L.; Poncz, M.; Gadue, P. Identifying and enriching platelet-producing human stem cell-derived megakaryocytes using factor V uptake. Blood 2017, 130, 192–204. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. Dissecting the mechanisms of immune checkpoint therapy. Nat. Rev. Immunol. 2020, 20, 75–76. [Google Scholar] [CrossRef]
- Wei, S.C.; Levine, J.H.; Cogdill, A.P.; Zhao, Y.; Anang, N.A.S.; Andrews, M.C.; Sharma, P.; Wang, J.; Wargo, J.A.; Pe’er, D.; et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell 2017, 170, 1120–1133.e17. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- Chitsike, L.; Duerksen-Hughes, P. The Potential of Immune Checkpoint Blockade in Cervical Cancer: Can Combinatorial Regimens Maximize Response? A Review of the Literature. Curr. Treat. Options Oncol. 2020, 21, 95. [Google Scholar] [CrossRef]
- Kwapisz, D. Pembrolizumab and atezolizumab in triple-negative breast cancer. Cancer Immunol. Immunother. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hallqvist, A.; Rohlin, A.; Raghavan, S. Immune checkpoint blockade and biomarkers of clinical response in Non-small cell lung cancer (NSCLC). Scand. J. Immunol. 2020, 92, e12980. [Google Scholar] [CrossRef] [PubMed]
- De Melo Gagliato, D.; Buzaid, A.C.; Perez-Garcia, J.; Cortes, J. Immunotherapy in Breast Cancer: Current Practice and Clinical Challenges. BioDrugs 2020, 34, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, J.; Chen, Z.; Hu, Q.; Wang, C.; Yan, J.; Dotti, G.; Huang, P.; Gu, Z. Engineering PD-1-Presenting Platelets for Cancer Immunotherapy. Nano Lett. 2018, 18, 5716–5725. [Google Scholar] [CrossRef]
- Scurr, M.; Pembroke, T.; Bloom, A.; Roberts, D.; Thomson, A.; Smart, K.; Bridgeman, H.; Adams, R.; Brewster, A.; Jones, R.; et al. Low-Dose Cyclophosphamide Induces Antitumor T-Cell Responses, which Associate with Survival in Metastatic Colorectal Cancer. Clin. Cancer Res. 2017, 23, 6771–6780. [Google Scholar] [CrossRef] [Green Version]
- Deng, D.; Shah, K. TRAIL of Hope Meeting Resistance in Cancer. Trends Cancer 2020, 6, 989–1001. [Google Scholar] [CrossRef]
- Li, J.; Sharkey, C.C.; Wun, B.; Liesveld, J.L.; King, M.R. Genetic engineering of platelets to neutralize circulating tumor cells. J. Control Release 2016, 228, 38–47. [Google Scholar] [CrossRef] [Green Version]
- Chan, V.; Novakowski, S.K.; Law, S.; Klein-Bosgoed, C.; Kastrup, C.J. Controlled Transcription of Exogenous mRNA in Platelets Using Protocells. Angew. Chem. Int. Ed. Engl. 2015, 54, 13590–13593. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Sun, W.; Ye, Y.; Hu, Q.; Bomba, H.N.; Gu, Z. In situ activation of platelets with checkpoint inhibitors for post-surgical cancer immunotherapy. Nat. Biomed. Eng. 2017, 1, 0011. [Google Scholar] [CrossRef]
- Hu, Q.; Sun, W.; Wang, J.; Ruan, H.; Zhang, X.; Ye, Y.; Shen, S.; Wang, C.; Lu, W.; Cheng, K.; et al. Conjugation of haematopoietic stem cells andplatelets decorated with anti-PD-1 antibodies augments anti-leukaemia efficacy. Nat. Biomed. Eng. 2018, 2, 831–840. [Google Scholar] [CrossRef]
- Kolb, H.C.; Sharpless, K.B. The growing impact of click chemistry on drug discovery. Drug Discov. Today 2003, 8, 1128–1137. [Google Scholar] [CrossRef]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Bose, R.J.; Paulmurugan, R.; Moon, J.; Lee, S.H.; Park, H. Cell membrane-coated nanocarriers: The emerging targeted delivery system for cancer theranostics. Drug Discov. Today 2018, 23, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Le, W.; Mei, T.; Wang, Y.; Chen, B.; Liu, Z.; Xue, C. Cell membrane camouflaged nanoparticles: A new biomimetic platform for cancer photothermal therapy. Int. J. Nanomed. 2019, 14, 4431–4448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Q.; Sun, W.; Qian, C.; Wang, C.; Bomba, H.N.; Gu, Z. Anticancer Platelet-Mimicking Nanovehicles. Adv. Mater. 2015, 27, 7043–7050. [Google Scholar] [CrossRef] [PubMed]
- Jing, L.; Qu, H.; Wu, D.; Zhu, C.; Yang, Y.; Jin, X.; Zheng, J.; Shi, X.; Yan, X.; Wang, Y. Platelet-camouflaged nanococktail: Simultaneous inhibition of drug-resistant tumor growth and metastasis via a cancer cells and tumor vasculature dual- targeting strategy. Theranostics 2018, 8, 2683–2695. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, G.; Wang, S.; He, Y.; Han, S.; Du, C.; Li, S.; Fan, Z.; Wang, C.; Wang, J. Platelet-membrane-camouflaged bismuth sulfide nanorods for synergistic radio- photothermal therapy against cancer. Biomater. Sci. 2019, 7, 3450–3459. [Google Scholar] [CrossRef]
- Jiang, Q.; Wang, K.; Zhang, X.; Ouyang, B.; Liu, H.; Pang, Z.; Yang, W. Platelet Membrane- Camouflaged Magnetic Nanoparticles for Ferroptosis-Enhanced Cancer Immunotherapy. Small 2020, 16, e2001704. [Google Scholar] [CrossRef]
- Choi, J.; Fenando, A. Sulfasalazine. 2020 Jun 28. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and Guillem function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Gasic, G.J.; Gasic, T.B.; Stewart, C.C. Antimetastatic effects associated with platelet reduction. Proc. Natl. Acad. Sci. USA 1968, 61, 46–52. [Google Scholar] [CrossRef] [Green Version]
- Palmblad, J. To give and take—Life of a platelet. Blood 2009, 113, 2617. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wang, S.; Liu, P.; Dai, L.; Chen, B.; Luan, J.; Zhou, J. Platelet-inspired medicine for tumor therapy. Oncotarget 2017, 8, 115748–115753. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Platelet Constituent | Cancer Cell Constituent | Cells Responses | Pharmacological Tools | References |
|---|---|---|---|---|
| Integrin αIIbβ3 (or GPIIb/IIIa) | Integrin αvβ3 | Cancer cell capacity to adhere to the endothelium was increased by platelet activation under flow | Abciximab | Lonsdorf et al., 2012; Felding-Habermann et al., 1996 [60,61] |
| Integrin αIIbβ3 (or GPIIb/IIIa) | ? | Platelet receptor FcɣRIIa activation and ADP release induced platelet aggregation | Abciximab | Mitrugno et al., 2014 [62] |
| Integrin αIIbβ3 (or GPIIb/IIIa) | GPIIb/IIa-like complex | ADP released from cancer cells induces platelet aggregation, degranulation, and the formation of platelet–tumor cell aggregates | mAb LYP18 against GPIIb/IIIa | Boukerche et al., 1995 [63] |
| Integrin α6β1 | ADAM9 | Induction of platelet activation, granule secretion, and subsequent endothelial transmigration of tumor cells | GoH3 (Integrin α6-blocking antibody) | Mammadova-Bach et al., 2016 [64] |
| P-selectin | Mucin-type glycoprotein | Direct interaction between cancer cells and activated platelets | Crizanlizumab | Mannori et al., 1995; Man et al., 2020 [65,66] |
| P-selectin | PSGL-1 | Induction of platelet activation and platelet/cancer cell aggregates | Crizanlizumab | Man et al., 2020; Gong et al., 2012 [66,67] |
| P-selectin | CDC44 | Induction of platelet/cancer cell aggregates under shear stress | Crizanlizumab | Man et al., 2020; Alves et al., 2008 [66,68] |
| P-selectin | PCLP1 | Induction of platelet activation and formation of platelet/cancer cell aggregates | Crizanlizumab | Man et al., 2020; Larrucea et al., 2007 [66,69] |
| GPVI | Galectin-3 | COX-2 overexpression and EMT induction in cancer cells | Revacept | Dovizio et al., 2013 [17] |
| CLEC-2 | Podoplanin | Platelet aggregation and induction of platelet/cancer cell aggregates | mAb NZ-1 against CLEC-2 | Suzuki-Inoue et al., 2007; Chang et al., 2015; Kato et al., 2006; [70,71,72] |
| Treatment | Patients | Primary End-Point | Results (95%CI) | References |
|---|---|---|---|---|
| Placebo or 325 mg of aspirin each day for 5 years | Male physicians aged between 40 and 84 years | Incidence of total cancer | RR: 1.15 (0.80–1.65) for colorectal cancer | Gann et al., 1993. [91] |
| Placebo or aspirin 100 mg of aspirin every other day for an average of 10.1 years | Healthy women aged at least 45 years | Confirmed newly diagnosed invasive cancer at any site | RR: 1.01 (0.94–1.08) for total cancer; RR: 0.97 (0.77–1.24) for colorectal cancer | Cook et al., 2005 [92] |
| Placebo or 81 mg or 325 mg of aspirin daily for 2.8 years | Patients with a recent history of histologically documented (removed) adenomas | Proportion of patients in whom one or more colorectal adenomas were detected | Any adenoma; RR: 0.81 (0.69–0.96) for ASA 81 mg, p = 0.04; RR: 0.96 (0.81–1.13) for ASA 325 mg Advanced lesion; RR: 0.59 (0.38–0.92) for ASA 81 mg; RR: 0.83 (0.55–1.23) for ASA 325 mg | Baron et al., 2003. [93] |
| Placebo or 325 mg daily of enteric-coated aspirin for 2.6 years | Patients who had histologically documented colon or rectal cancer with a low risk of disease recurrence | Detection of adenomas in the large bowel by either colonoscopy or sigmoidoscopy after randomization | RR: 0.65 (0.46–0.91) p = 0.004 | Sandler et al., 2003. [94] |
| Placebo or soluble aspirin (160 or 300 mg daily) for 1 year | Patients with a history of colorectal adenomas | Adenoma recurrence after 1 year | RR: 0.73 (0.52–1.04) p = 0.04 for both doses | Benamouzig et al., 2003 (APACC trial) [95] |
| Placebo or soluble aspirin (160 or 300 mg daily) for 4 years | Patients with a history of colorectal adenomas | Adenoma recurrence after 4 years | RR: 0.96 (0.75–1.22) for both doses | Benamouzig et al., 2012 (APACC trial) [96] |
| Aspirin (300 mg daily) versus folate supplements (0.5 mg/d) for about 2.6 years | Patients with an adenoma removed in the 6 months before recruitment | A colorectal adenoma diagnosed after baseline | RR: 0.79 (0.63–0.99) p = 0.04 | Logan et al., 2008. [97] |
| Aspirin (600 mg/d) and/or resistant starch (30 g/d) for 17 years | FAP young patients | Polyp number in the rectum and sigmoid colon | RR: 0.77 (0.54–1.10) | Burn et al., 2011 (CAPP1 trial) [98] |
| Aspirin (600 mg/d) and/or resistant starch (30 g/d) or placebo for a mean of 10 years | Lynch syndrome (hereditary nonpolyposis colon cancer or HNPCC) | Development of colorectal cancer | HR: 0.65 (0.43–0.97; p = 0.035) for aspirin vs. placebo | Burn et al., 2020 (CAPP2 trial). [100] |
| 81 or 100 mg of aspirin daily for 10.7 years | Type 2 diabetic patients | Time to first cancer incidence | HR: 0.92 (0.73–1.14) p = 0.4; adjusted HR, 0.66 (0.43–0.99) p = 0.04 | Okada et al., 2018. [101] |
| 100 mg of aspirin or placebo for 7.4 years | Subjects with a diagnosis of diabetes mellitus (any type) aged >40 years | First serious vascular event | RR: 0.98 (0.84–1.15) for cancer-related mortality | Bowman et al., 2018 (ASCEND study) [102] |
| 100 mg of aspirin or placebo for 4.7 years | Healthy Elderly (>70 years) | Composite of death, dementia, or persistent physical disability | HR: 1.35 (1.13 to 1.61) for cancer mortality; HR: 1.04 (0.95–1.14) for cancer incidence | McNeil et al., 2018; McNeil et al., 2020. [103,104] |
| 100 mg of aspirin or 300 mg or placebo daily for at least five years | Patients who have undergone potentially curative treatment for breast, colorectal, gastroesophageal, or prostate cancer | Invasive disease-free survival for the breast cohort, disease-free survival for the colorectal cohort, overall survival for the gastroesophageal cohort | Ongoing | Coyle et al., 2016 (ADD-ASPIRIN trial) [105] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dovizio, M.; Ballerini, P.; Fullone, R.; Tacconelli, S.; Contursi, A.; Patrignani, P. Multifaceted Functions of Platelets in Cancer: From Tumorigenesis to Liquid Biopsy Tool and Drug Delivery System. Int. J. Mol. Sci. 2020, 21, 9585. https://doi.org/10.3390/ijms21249585
Dovizio M, Ballerini P, Fullone R, Tacconelli S, Contursi A, Patrignani P. Multifaceted Functions of Platelets in Cancer: From Tumorigenesis to Liquid Biopsy Tool and Drug Delivery System. International Journal of Molecular Sciences. 2020; 21(24):9585. https://doi.org/10.3390/ijms21249585
Chicago/Turabian StyleDovizio, Melania, Patrizia Ballerini, Rosa Fullone, Stefania Tacconelli, Annalisa Contursi, and Paola Patrignani. 2020. "Multifaceted Functions of Platelets in Cancer: From Tumorigenesis to Liquid Biopsy Tool and Drug Delivery System" International Journal of Molecular Sciences 21, no. 24: 9585. https://doi.org/10.3390/ijms21249585
APA StyleDovizio, M., Ballerini, P., Fullone, R., Tacconelli, S., Contursi, A., & Patrignani, P. (2020). Multifaceted Functions of Platelets in Cancer: From Tumorigenesis to Liquid Biopsy Tool and Drug Delivery System. International Journal of Molecular Sciences, 21(24), 9585. https://doi.org/10.3390/ijms21249585