Molecular Targets in Precision Chemoprevention of Colorectal Cancer: An Update from Pre-Clinical to Clinical Trials


Abstract
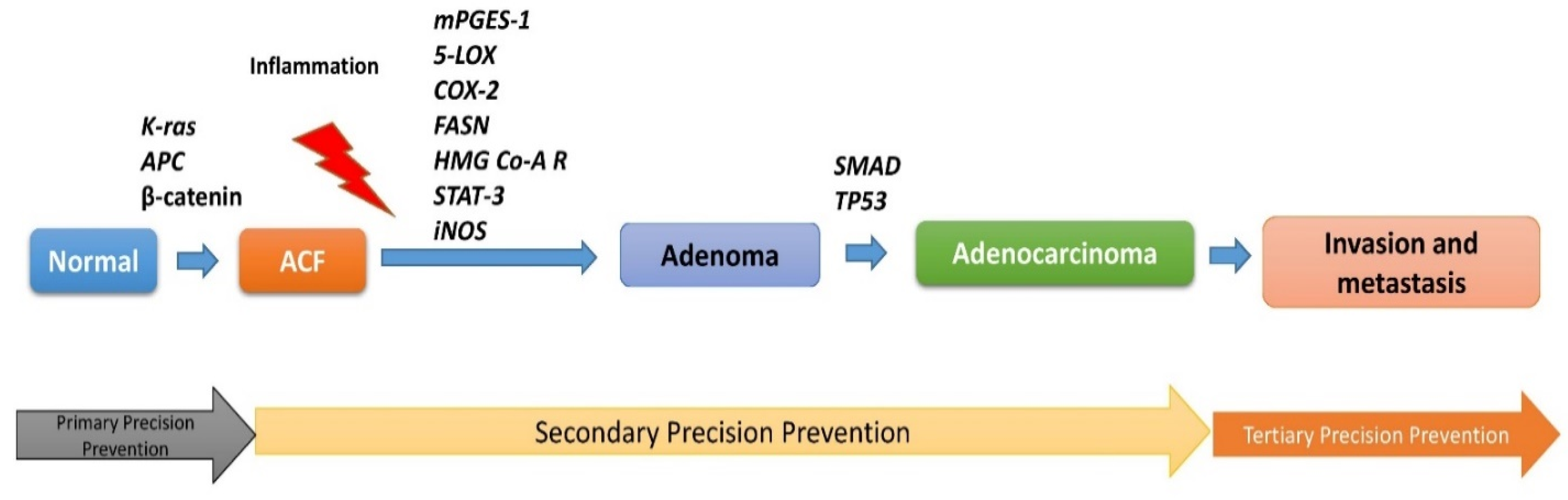
:1. Introduction
2. Lesions-Specific Molecular Targets in CRC
2.1. Mutated and Dysregulated Signaling Precision-Molecular Targets
2.1.1. APC/β-Catenin
2.1.2. KRAS/EGFR
2.1.3. TGF-β1/SMAD
2.1.4. TP53
2.1.5. AKT
2.2. Inflammatory Targets: ACF Progression to Adenoma
2.2.1. Cyclooxygenase-2
2.2.2. Microsomal Prostaglandin E Synthase-1
2.2.3. 5-Lipoxygenase
2.2.4. Prostacyclin I2 Synthase
2.2.5. 15-Hydroxyprostaglandin Dehydrogenase
2.2.6. 15-Lipoxygenase-1
2.2.7. Inducible Nitric Oxide Synthase
2.2.8. STAT-3
2.2.9. Chemokine Receptor 5
2.3. Metabolic and Growth Factor-Related Targets
2.3.1. Fatty Acid Synthase
2.3.2. Ornithine Decarboxylase
2.3.3. HMG Co-A Reductase
3. Molecular Targets and Tertiary Precision Prevention
4. Pre-Clinical Rodent Animal Models for Biomarkers-Based Precision Chemoprevention
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CRC | Colorectal cancer |
| ACF | Aberrant crypt foci |
| APC | Adenomatous polyposis coli |
| KRAS | V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog |
| COX-2 | Cyclooxygenase-2 |
| LOX | Lipoxygenase |
| FASN | Fatty acid synthase |
| ODC | Ornithine decarboxylase |
| iNOS | Inducible nitric oxide synthase |
| EGFR | Epidermal growth factor receptor |
| HMG Co-A-R | 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase |
| FASN | Fatty acid synthase |
| TGF-β1 | Transforming growth factor-β1 |
| 15-PGDH | 15-hydroxyprostaglandin dehydrogenase |
| STAT | Signal transducer and activator of transcription |
| NSAIDs | Nonsteroidal anti-inflammatory drugs |
| AOM | Azoxymethane |
| FAP | Familial adenomatous polyposis |
| CCR5 | Chemokine receptor 5 |
| PGI2 | Prostacyclin |
| PGE2 | Prostaglandin E2 |
| mPGES-1 | Microsomal prostaglandin E synthase-1 |
| MDF | Mucin depleted foci |
| COXibs | COX-2 inhibitors |
| CCR5 | Chemokine receptor 5 |
References
- The American Cancer Society Journal, CA: A Cancer Journal for Clinicians. Cancer Facts & Figures. Available online: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/colorectal-cancer-facts-and-figures/colorectal-cancer-facts-and-figures-2020-2022.pdf (accessed on 10 October 2020).
- Janakiram, N.B.; Rao, C.V. Molecular markers and targets for colorectal cancer prevention. Acta Pharmacol. Sin. 2008, 29, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Müller, M.F.; Ibrahim, A.E.; Arends, M.J. Molecular pathological classification of colorectal cancer. Virchows Arch. 2016, 469, 125–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fodde, R. The APC gene in colorectal cancer. Eur. J. Cancer 2002, 38, 867–871. [Google Scholar] [CrossRef]
- Yuan, P.; Sun, M.H.; Zhang, J.S.; Zhu, X.Z.; Shi, D.R. APC and K-ras gene mutation in aberrant crypt foci of human colon. World J. Gastroenterol. 2001, 7, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Goretsky, T.; Bradford, E.M.; Ye, Q.; Lamping, O.F.; Vanagunas, T.; Moyer, M.P.; Keller, P.C.; Sinh, P.; Llovet, J.M.; Gao, T.; et al. Beta-catenin cleavage enhances transcriptional activation. Sci. Rep. 2018, 8, 671. [Google Scholar] [CrossRef] [PubMed]
- Femia, A.P.; Dolara, P.; Giannini, A.; Salvadori, M.; Biggeri, A.; Caderni, G. Frequent mutation of Apc gene in rat colon tumors and mucin-depleted foci, preneoplastic lesions in experimental colon carcinogenesis. Cancer Res. 2007, 67, 445–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Femia, A.P.; Piero, D.; Maddalena, S.; Giovanna, C. Expression of LGR-5, MSI-1 and DCAMKL-1, putative stem cell markers, in the early phases of 1,2-dimethylhydrazine-induced rat colon carcinogenesis: Correlation with nuclear β-catenin. BMC Cancer 2013, 13, 48. [Google Scholar] [CrossRef] [Green Version]
- Xiaofei, C.; Xiangming, X.; Dong, C.; Feng, Z.; Weilin, W. Therapeutic Potential of Targeting the Wnt/β-catenin Signaling Pathway in Colorectal Cancer. Biomed. Pharmacother. 2019, 110, 473–481. [Google Scholar]
- Markman, B.; Javier, R.F.; Capdevila, J.; Tabernero, J. EGFR and KRAS in colorectal cancer. Adv. Clin. Chem. 2010, 51, 71–119. [Google Scholar]
- Rizzo, S.; Bronte, G.; Fanale, D.; Corsini, L.; Silvestris, N.; Santini, D.; Gulotta, G.; Bazan, V.; Gebbia, N.; Fulfaro, F.; et al. Prognostic vs predictive molecular biomarkers in colorectal cancer: Is KRAS and BRAF wild type status required for anti-EGFR therapy? Cancer Treat. Rev. 2010, 36, S56–S61. [Google Scholar] [CrossRef] [Green Version]
- Pretlow, T.P.; Pretlow, T.G. Mutant KRAS in aberrant crypt foci (ACF): Initiation of colorectal cancer? Biochim. Biophys. Acta 2005, 1756, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Femia, A.P.; Tarquini, E.; Salvadori, M.; Ferri, S.; Giannini, A.; Dolara, P.; Caderni, G. K-ras mutations and mucin profile in preneoplastic lesions and colon tumors induced in rats by 1,2-dimethylhydrazine. Int. J. Cancer 2008, 122, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Mutoh, M.; Kawamori, T.; Sugimura, T.; Wakabayashi, K. Altered expression of beta-catenin, inducible nitric oxide synthase and cyclooxygenase-2 in azoxymethane-induced rat colon carcinogenesis. Carcinogenesis 2000, 21, 1319–1327. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.S.; Cha, J.M.; Cho, Y.H.; Kim, S.H.; Yoon, J.Y.; Jeon, J.W.; Shin, H.P.; Joo, K.R.; Lee, J.I. Clinical Predictors for KRAS Codon 13 Mutations in Patients with Colorectal Cancer. J. Clin. Gastroenterol. 2018, 52, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Valtorta, E.; Misale, S.; Sartore-Bianchi, A.; Nagtegaal, I.D.; Paraf, F.; Lauricella, C.; Dimartino, V.; Hobor, S.; Jacobs, B.; Ercolani, C. KRAS gene amplification in colorectal cancer and impact on response to EGFR-targeted therapy. Int. J. Cancer 2013, 133, 1259–1265. [Google Scholar] [CrossRef]
- Tsushima, H.; Kawata, S.; Tamura, S.; Ito, N.; Shirai, Y.; Kiso, S.; Imai, Y.; Shimomukai, H.; Nomura, Y.; Matsuda, Y.; et al. High levels of transforming growth factor beta 1 in patients with colorectal cancer: Association with disease progression. Gastroenterology 1996, 110, 375–382. [Google Scholar] [CrossRef]
- Szigeti, R.; Pangas, S.A.; Nagy-Szakal, D.; Dowd, S.E.; Shulman, R.J.; Olive, A.P.; Popek, E.J.; Finegold, M.J.; Kellermayer, R. SMAD4 haploinsufficiency associates with augmented colonic inflammation in select humans and mice. Ann. Clin. Lab. Sci. 2012, 42, 401–408. [Google Scholar] [CrossRef] [Green Version]
- Means, A.L.; Freeman, T.J.; Zhu, J.; Woodbury, L.G.; Marincola-Smith, P.; Wu, C.; Meyer, A.R.; Weaver, C.J.; Padmanabhan, C.; An, H.; et al. Epithelial Smad4 Deletion Up-Regulates Inflammation and Promotes Inflammation-Associated Cancer. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 257–276. [Google Scholar] [CrossRef] [Green Version]
- Takayama, T.; Miyanishi, K.; Hayashi, T.; Sato, Y.; Niitsu, Y. Colorectal cancer: Genetics of development and metastasis. J. Gastroenterol. 2006, 41, 185–192. [Google Scholar] [CrossRef]
- Xiao, L.L.; Jianbiao, Z.; Zhi-Rong, C.; Wee-Joo, C. p53 mutations in colorectal cancer- molecular pathogenesis and pharmacological reactivation. World J. Gastroenterol. 2015, 21, 84–93. [Google Scholar]
- Yamashita, N.; Minamoto, T.; Ochiai, A.; Onda, M.; Esumi, H. Frequent and characteristic K-ras activation and absence of p53 protein accumulation in aberrant crypt foci of the colon. Gastroenterology 1995, 108, 434–440. [Google Scholar] [CrossRef]
- Losi, L.; Roncucci, L.; Di Gregorio, C.; Leon, M.P.; Benhattar, J. K-ras and p53 mutations in human colorectal aberrant crypt foci. J. Pathol. 1996, 178, 259–263. [Google Scholar] [CrossRef]
- Chang, W.C.; Coudry, R.A.; Clapper, M.L.; Zhang, X.; Williams, K.L.; Spittle, C.S.; Li, T.; Cooper, H.S. Loss of p53 enhances the induction of colitis-associated neoplasia by dextran sulfate sodium. Carcinogenesis 2007, 28, 2375–2381. [Google Scholar] [CrossRef] [PubMed]
- Alpert, L.; Yassan, L.; Poon, R.; Kadri, S.; Niu, N.; Patil, S.A.; Mujacic, I.; Montes, D.; Galbo, F.; Wurst, M.N.; et al. Targeted mutational analysis of inflammatory bowel disease-associated colorectal cancers. Hum. Pathol. 2019, 89, 44–50. [Google Scholar] [CrossRef]
- Du, L.; Kim, J.J.; Shen, J.; Chen, B.; Dai, N. KRAS and TP53 mutations in inflammatory bowel disease-associated colorectal cancer: A meta-analysis. Oncotarget 2017, 8, 22175–22186. [Google Scholar] [CrossRef] [Green Version]
- Rao, C.V.; Swamy, M.V.; Patlolla, J.M.; Kopelovich, L. Suppression of familial adenomatous polyposis by CP-31398, a TP53 modulator, in APCmin/+ mice. Cancer Res. 2008, 68, 7670–7675. [Google Scholar] [CrossRef] [Green Version]
- Narayanankutty, A. PI3K/Akt/mTOR Pathway as a Therapeutic Target for Colorectal Cancer: A Review of Preclinical and Clinical Evidence. Curr. Drug Targets 2019, 20, 1217–1226. [Google Scholar] [CrossRef]
- Saglam, O.; Garrett, C.R.; Boulware, D.; Sayegh, Z.; Shibata, D.; Malafa, M.; Yeatman, T.; Cheng, J.Q.; Sebti, S.; Coppola, D. Activation of the serine/threonine protein kinase AKT during the progression of colorectal neoplasia. Clin. Colorectal Cancer 2007, 6, 652–656. [Google Scholar] [CrossRef]
- Roy, H.K.; Olusola, B.F.; Clemens, D.L.; Karolski, W.J.; Ratashak, A.; Lynch, H.T.; Smyrk, T.C. AKT proto-oncogene overexpression is an early event during sporadic colon carcinogenesis. Carcinogenesis 2002, 23, 201–205. [Google Scholar] [CrossRef] [Green Version]
- Lara, P.N., Jr.; Longmate, J.; Mack, P.C.; Kelly, K.; Socinski, M.A.; Salgia, R.; Gitlitz, B.; Li, T.; Koczywas, M.; Reckamp, K.L.; et al. Phase II Study of the AKT Inhibitor MK-2206 plus Erlotinib in Patients with Advanced Non-Small Cell Lung Cancer Who Previously Progressed on Erlotinib. Clin. Cancer Res. 2015, 21, 4321–4326. [Google Scholar] [CrossRef] [Green Version]
- Sano, H.; Kawahito, Y.; Wilder, R.L.; Hashiramoto, A.; Mukai, S.; Asai, K.; Kimura, S.; Kato, H.; Kondo, M.; Hla, T. Expression of cyclooxygenase-1 and -2 in human colorectal cancer. Cancer Res. 1995, 55, 3785–3789. [Google Scholar] [PubMed]
- Gupta, R.A.; Dubois, R.N. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat. Rev. Cancer 2001, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Femia, A.P.; Dolara, P.; Luceri, C.; Salvadori, M.; Caderni, G. Mucin-depleted foci show strong activation of inflammatory markers in 1,2-dimethylhydrazine-induced carcinogenesis and are promoted by the inflammatory agent sodium dextran sulfate. Int. J. Cancer 2009, 125, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Negi, R.R.; Rana, S.V.; Gupta, V.; Gupta, R.; Chadha, V.D.; Prasad, K.K.; Dhawan, D.K. Over-Expression of Cyclooxygenase-2 in Colorectal Cancer Patients. Asian Pac. J. Cancer Prev. 2019, 20, 1675–1681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roelofs, H.M.; Te Morsche, R.H.; van Heumen, B.W.; Nagengast, F.M.; Peters, W.H. Over-expression of COX-2 mRNA in colorectal cancer. BMC Gastroenterol. 2014, 2, 14. [Google Scholar] [CrossRef] [Green Version]
- Cossiolo, D.C.; Costa, H.C.M.; Fernandes, K.B.P.; Laranjeira, L.L.S.; Fernandes, M.T.P.; Poli-Frederico, R.C. Polymorphism of the COX-2 gene and susceptibility to colon and rectal cancer. Arq. Bras. Cir. Dig. 2017, 30, 114–117. [Google Scholar] [CrossRef] [Green Version]
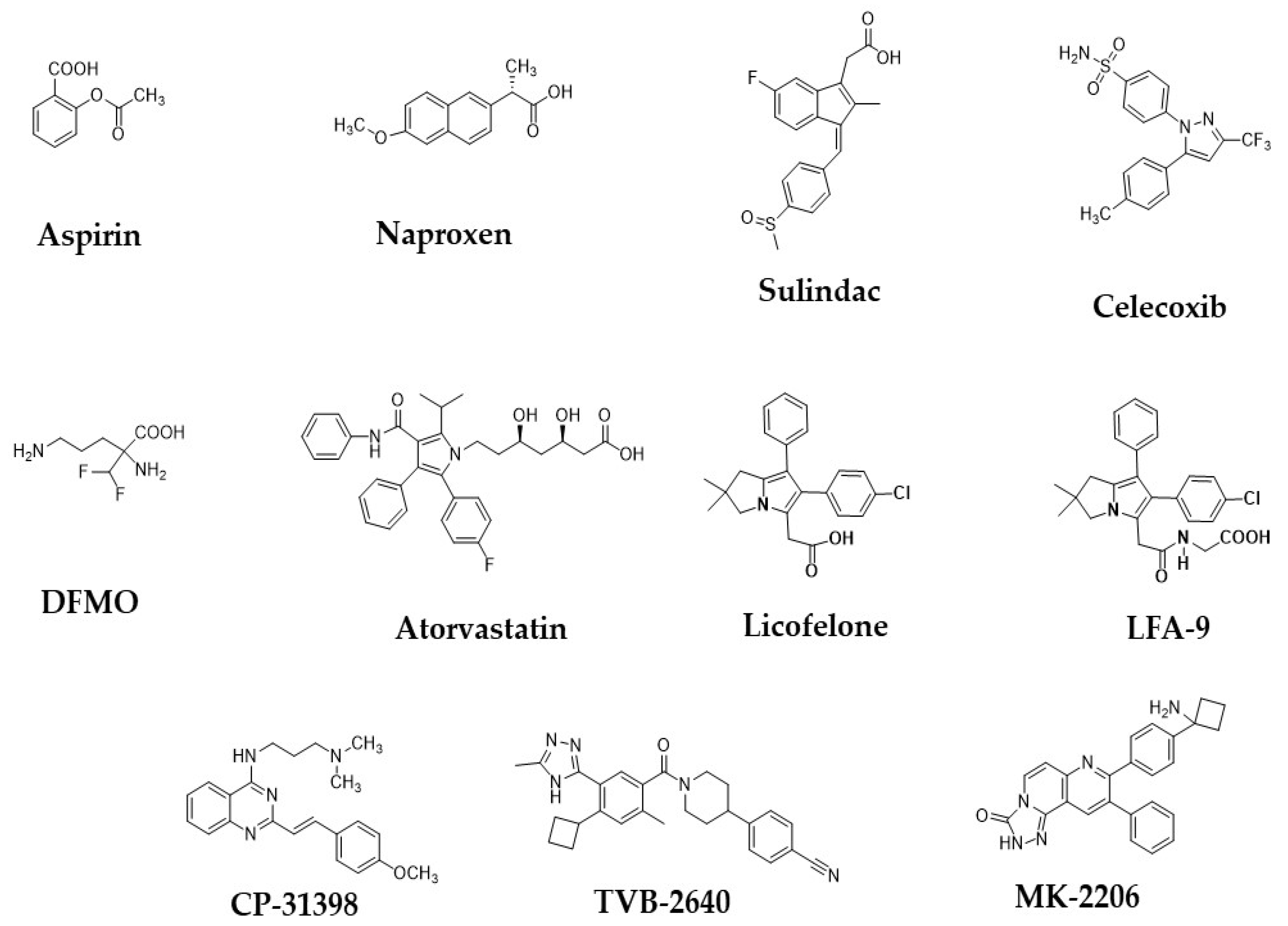
- Katona, B.W.; Weiss, J.M. Chemoprevention of Colorectal Cancer. Gastroenterology 2020, 158, 368–388. [Google Scholar] [CrossRef]
- Mohammed, A.; Yarla, N.S.; Madka, V.; Rao, C.V. Clinically Relevant Anti-Inflammatory Agents for Chemoprevention of Colorectal Cancer: New Perspectives. Int. J. Mol. Sci. 2018, 19, 2332. [Google Scholar] [CrossRef] [Green Version]
- Loomans-Kropp, H.A.; Umar, A. Cancer prevention and screening: The next step in the era of precision medicine. NPJ Precis Oncol. 2019, 28, 3. [Google Scholar] [CrossRef] [Green Version]
- Drew, D.A.; Cao, Y.; Chan, A.T. Aspirin and colorectal cancer: The promise of precision chemoprevention. Nat. Rev. Cancer 2016, 16, 173–186. [Google Scholar] [CrossRef]
- Chan, A.T.; Ogino, S.; Fuchs, C.S. Aspirin and the risk of colorectal cancer in relation to the expression of COX-2. N. Engl. J. Med. 2007, 356, 2131–2142. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.S.; Song, M.; Bezawada, N.; Wu, K.; Garcia-Albeniz, X.; Morikawa, T.; Fuchs, C.S.; Ogino, S.; Giovannucci, E.L.; Chan, A.T. A prospective study of macrophage inhibitory cytokine-1 (MIC-1/GDF15) and risk of colorectal cancer. J. Natl. Cancer Inst. 2014, 106, dju016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- JAMA United States Preventative Services Task Force. Screening for colorectal cancer: US Preventive Services Task Force recommendation statement. JAMA 2016, 315, 2564–2575. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, M.; Montrose, D.C.; Clark, P.; Nambiar, P.R.; Belinsky, G.S.; Claffey, K.P.; Xu, D.; Rosenberg, D.W. Genetic Deletion of mPGES-1 Suppresses Intestinal Tumorigenesis. Cancer Res. 2008, 68, 3251–3259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, Y.; Kamei, D.; Ishikawa, Y.; Ishii, T.; Uematsu, S.; Akira, S.; Murakami, M.; Hara, S. Microsomal prostaglandin E synthase-1 is involved in multiple steps of colon carcinogenesis. Oncogene 2012, 31, 2943–2952. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.H.; Ryu, Y.M.; Lee, S.M.; Jeong, J.Y.; Yoon, S.M.; Ye, B.D.; Byeon, J.S.; Yang, S.K.; Myung, S.J. 15-Hydroxyprostaglandin dehydrogenase as a marker in colon carcinogenesis: Analysis of the prostaglandin pathway in human colonic tissue. Intest. Res. 2017, 15, 75–82. [Google Scholar] [CrossRef] [Green Version]
- Kamei, D.; Murakami, M.; Nakatani, Y.; Ishikawa, Y.; Ishii, T.; Kudo, I. Potential role of microsomal prostaglandin E synthase-1 in tumorigenesis. J. Biol. Chem. 2003, 278, 19396–19405. [Google Scholar] [CrossRef] [Green Version]
- Melstrom, L.G.; Bentrem, D.J.; Salabat, M.R.; Kennedy, T.J.; Ding, X.Z.; Strouch, M.; Rao, S.M.; Witt, R.C.; Ternent, C.A.; Talamonti, M.S.; et al. Overexpression of 5-lipoxygenase in colon polyps and cancer and the effect of 5-LOX inhibitors in vitro and in a murine model. Clin. Cancer Res. 2008, 14, 6525–6530. [Google Scholar] [CrossRef] [Green Version]
- Wasilewicz, M.P.; Kołodziej, B.; Bojułko, T.; Kaczmarczyk, M.; Sulzyc-Bielicka, V.; Bielicki, D.; Ciepiela, K. Overexpression of 5-lipoxygenase in sporadic colonic adenomas and a possible new aspect of colon carcinogenesis. Int. J. Colorectal Dis. 2010, 25, 1079–1085. [Google Scholar] [CrossRef] [Green Version]
- Shureiqi, I.; Lippman, S.M. Lipoxygenase modulation to reverse carcinogenesis. Cancer Res. 2001, 61, 6307–6312. [Google Scholar]
- Takahashi, M.; Wakabayashi, K. Gene mutations and altered gene expression in azoxymethane-induced colon carcinogenesis in rodents. Cancer Sci. 2004, 95, 475–480. [Google Scholar] [CrossRef]
- Cianchi, F.; Cortesini, C.; Fantappiè, O.; Messerini, L.; Schiavone, N.; Vannacci, A.; Nistri, S.; Sardi, I.; Baroni, G.; Marzocca, C.; et al. Inducible nitric oxide synthase expression in human colorectal cancer: Correlation with tumor angiogenesis. Am. J. Pathol. 2003, 162, 793–801. [Google Scholar] [CrossRef]
- Zafirellis, K.; Zachaki, A.; Agrogiannis, G.; Gravani, K. Inducible nitric oxide synthase expression and its prognostic significance in colorectal cancer. APMIS 2010, 118, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Kangwan, N.; Kim, Y.; Han, Y.M.; Jeong, M.; Park, J.M.; Hahm, K.B. Concerted actions of ameliorated colitis, aberrant crypt foci inhibition and 15-hydroxyprostaglandin dehydrogenase induction by sonic hedgehog inhibitor led to prevention of colitis-associated cancer. Int. J. Cancer 2016, 138, 1482–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, P.S.; Kim, J.H.; Moon, O.I.; Lim, S.C.; Kim, K.J. Prognostic implication of 15-hydroxyprostaglandin dehydrogenase down-regulation in patients with colorectal cancer. J. Korean Soc. Coloproctol. 2012, 28, 253–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, Y.; Jiao, H.; Teng, G.; Wang, W.; Zhang, R.; Wang, Y.; Hebbard, L.; George, J.; Qiao, L. Embelin reduces colitis-associated tumorigenesis through limiting IL-6/STAT3 signaling. Mol. Cancer Ther. 2014, 13, 1206–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.X.; Bi, X.Y.; Huang, Z.; Zhao, J.J.; Han, Y.; Li, Z.Y.; Zhang, Y.F.; Li, Y.; Chen, X.; Hu, X.H.; et al. Prognostic role of phospho-STAT3 in patients with cancers of the digestive system: A systematic review and meta-analysis. PLoS ONE 2015, 10, e0127356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, Y.; Kamiyama, S.; Kamiyama, A.; Matsumoto, K.; Akatsu, M.; Nakatani, Y.; Kuwata, H.; Ishikawa, Y.; Ishii, T.; Yokoyama, C.; et al. Genetic-deletion of Cyclooxygenase-2 Downstream Prostacyclin Synthase Suppresses Inflammatory Reactions but Facilitates Carcinogenesis, unlike Deletion of Microsomal Prostaglandin E Synthase-1. Sci. Rep. 2015, 5, 17376. [Google Scholar] [CrossRef] [Green Version]
- Poole, E.M.; Bigler, J.; Whitton, J.; Sibert, J.G.; Potter, J.D.; Ulrich, C.M. Prostacyclin synthase and arachidonate 5-lipoxygenase polymorphisms and risk of colorectal polyps. Cancer Epidemiol. Biomark. Prev. 2006, 15, 502–508. [Google Scholar] [CrossRef] [Green Version]
- Rigas, B.; Goldman, I.S.; Levine, L. Altered eicosanoid levels in human colon cancer. J. Lab. Clin. Med. 1993, 122, 518–523. [Google Scholar]
- Zuo, X.; Peng, Z.; Wu, Y.; Moussalli, M.J.; Yang, X.L.; Wang, Y.; Parker-Thornburg, J.; Morris, J.S.; Broaddus, R.R.; Fischer, S.M.; et al. Effects of gut-targeted 15-LOX-1 transgene expression on colonic tumorigenesis in mice. J. Natl. Cancer Inst. 2012, 104, 709–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, F.; Xu, M.; Zuo, X.; Yu, J.; Xu, W.; Moussalli, M.J.; Elias, E.; Li, H.S.; Watowich, S.S.; Shureiqi, I. 15-Lipoxygenase-1 suppression of colitis-associated colon cancer through inhibition of the IL-6/STAT3 signaling pathway. FASEB J. 2015, 29, 2359–2370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, R.; Zuo, X.; Jaoude, J.; Mao, F.; Colby, J.; Shureiqi, I. ALOX15 as a suppressor of inflammation and cancer: Lost in the link. Prostaglandins Other Lipid Mediat. 2017, 132, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Erdman, S.H.; Ignatenko, N.A.; Powell, M.B.; Blohm-Mangone, K.A.; Holubec, H.; Guillén-Rodriguez, J.M.; Gerner, E.W. APC-dependent changes in expression of genes influencing polyamine metabolism, and consequences for gastrointestinal carcinogenesis, in the Min mouse. Carcinogenesis 1999, 20, 1709–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshino, Y.; Terashima, S.; Teranishi, Y.; Terashima, M.; Kogure, M.; Saitoh, T.; Osuka, F.; Kashimura, S.; Saze, Z.; Gotoh, M. Ornithine decarboxylase activity as a prognostic marker for colorectal cancer. Fukushima J. Med. Sci. 2007, 53, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zell, J.A.; Ziogas, A.; Ignatenko, N.; Honda, J.; Qu, N.; Bobbs, A.S.; Neuhausen, S.L.; Gerner, E.W.; Anton-Culver, H. Associations of a polymorphism in the ornithine decarboxylase gene with colorectal cancer survival. Clin. Cancer Res. 2009, 15, 6208–6216. [Google Scholar] [CrossRef] [Green Version]
- Takami, H.; Koudaira, H.; Kodaira, S. Relationship of ornithinedecarboxylase activity and human colon tumorigenesis. Jpn. J. Clin. Oncol. 1994, 24, 141–143. [Google Scholar]
- Wei, T.T.; Lin, Y.T.; Chen, W.S.; Luo, P.; Lin, Y.C.; Shun, C.T.; Lin, Y.H.; Chen, J.B.; Chen, N.W.; Fang, J.M.; et al. Dual Targeting of 3-Hydroxy-3-methylglutaryl Coenzyme A Reductase and Histone Deacetylase as a Therapy for Colorectal Cancer. EBioMedicine 2016, 10, 124–136. [Google Scholar] [CrossRef] [Green Version]
- Barone, M.; Notarnicola, M.; Caruso, M.G.; Scavo, M.P.; Viggiani, M.T.; Tutino, V.; Polimeno, L.; Pesetti, B.; Di Leo, A.; Francavilla, A. Olive oil and omega-3 polyunsaturated fatty acids suppress intestinal polyp growth by modulating the apoptotic process in ApcMin/+ mice. Carcinogenesis 2014, 35, 1613–1619. [Google Scholar] [CrossRef]
- Bengtsson, E.; Nerjovaj, P.; Wangefjord, S.; Nodin, B.; Eberhard, J.; Uhlén, M.; Borgquist, S.; Jirström, K. HMG-CoA reductase expression in primary colorectal cancer correlates with favourable clinicopathological characteristics and an improved clinical outcome. Diagn. Pathol. 2014, 9, 78. [Google Scholar] [CrossRef] [Green Version]
- Cruz, M.D.; Wali, R.K.; Bianchi, L.K.; Radosevich, A.J.; Crawford, S.E.; Jepeal, L.; Goldberg, M.J.; Weinstein, J.; Momi, N.; Roy, P.; et al. Colonic mucosal fatty acid synthase as an early biomarker for colorectal neoplasia: Modulation by obesity and gender. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2413–2421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearney, K.E.; Pretlow, T.G.; Pretlow, T.P. Increased expression of fatty acid synthase in human aberrant crypt foci: Possible target for colorectal cancer prevention. Int. J. Cancer 2009, 125, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, M.; Menoret, A.; Tanaka, T.; Miyamoto, S.; Montrose, D.C.; Vella, A.T.; Rosenberg, D.W. Selective PGE(2) suppression inhibits colon carcinogenesis and modifies local mucosal immunity. Cancer Prev. Res. 2011, 4, 1198–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.C.; Wang, D.X.; Li, C.S.; Ye, Z.P.; Wu, Z.M.; Chen, J.H. Association of 5-lipoxygenase expression and clinicopathological factors in colorectal carcinoma. Zhonghua Wei Chang. Wai Ke Za Zhi 2013, 16, 895–897. [Google Scholar]
- Gounaris, E.; Heiferman, M.J.; Heiferman, J.R.; Shrivastav, M.; Vitello, D.; Blatner, N.R.; Knab, L.M.; Phillips, J.D.; Cheon, E.C.; Grippo, P.J.; et al. Zileuton, 5-lipoxygenase inhibitor, acts as a chemopreventive agent in intestinal polyposis, by modulating polyp and systemic inflammation. PLoS ONE 2015, 10, e0121402. [Google Scholar] [CrossRef]
- Cutler, N.S.; Graves-Deal, R.; LaFleur, B.J.; Gao, Z.; Boman, B.M.; Whitehead, R.H.; Terry, E.; Morrow, J.D.; Coffey, R.J. Stromal production of prostacyclin confers an antiapoptotic effect to colonic epithelial cells. Cancer Res. 2003, 63, 1748–1751. [Google Scholar]
- Frigola, J.; Muñoz, M.; Clark, S.J.; Moreno, V.; Capellà, G.; Peinado, M.A. Hypermethylation of the prostacyclin synthase (PTGIS) promoter is a frequent event in colorectal cancer and associated with aneuploidy. Oncogene 2005, 24, 7320–7326. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Cho, N.L.; Zauber, A.G.; Hsu, M.; Dawson, D.; Srivastava, A.; Mitchell-Richards, K.A.; Markowitz, S.D.; Bertagnolli, M.M. The expression of Cox-2 and 15-PGDH in pre-treatment adenomas provides predictive information in patients treated with celecoxib for prevention of colorectal adenomas. Cancer Epidemiol. Biomark. Prev. 2018, 27, 728–736. [Google Scholar] [CrossRef] [Green Version]
- Backlund, M.G.; Mann, J.R.; Holla, V.R.; Shi, Q.; Daikoku, T.; Dey, S.K.; DuBois, R.N. Repression of 15-hydroxyprostaglandin dehydrogenase involves histone deacetylase 2 and snail in colorectal cancer. Cancer Res. 2008, 68, 9331–9337. [Google Scholar] [CrossRef] [Green Version]
- Gilligan, M.M.; Gartung, A.; Sulciner, M.L.; Norris, P.C.; Sukhatme, V.P.; Bielenberg, D.R.; Huang, S.; Kieran, M.W.; Serhan, C.N.; Panigrahy, D. Aspirin-triggered proresolving mediators stimulate resolution in cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 6292–6297. [Google Scholar] [CrossRef] [Green Version]
- Cimen, I.; Astarci, E.; Banerjee, S. 15-lipoxygenase-1 exerts its tumor suppressive role by inhibiting nuclear factor-kappa B via activation of PPAR gamma. J. Cell Biochem. 2011, 112, 2490–2501. [Google Scholar] [CrossRef] [PubMed]
- Cimen, I.; Tunçay, S.; Banerjee, S. 15-Lipoxygenase-1 expression suppresses the invasive properties of colorectal carcinoma cell lines HCT-116 and HT-29. Cancer Sci. 2009, 100, 2283–2291. [Google Scholar] [CrossRef] [PubMed]
- Tunçer, S.; Tunçay, Ç.S.; Keşküş, A.G.; Çolakoğlu, M.; Konu, Ö.; Banerjee, S. Interplay between 15-lipoxygenase-1 and metastasis-associated antigen 1 in the metastatic potential of colorectal cancer. Cell Prolif. 2016, 49, 448–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tunçer, S.; Keşküş, A.G.; Çolakoğlu, M.; Çimen, I.; Yener, C.; Konu, Ö.; Banerjee, S. 15-Lipoxygenase-1 re-expression in colorectal cancer alters endothelial cell features through enhanced expression of TSP-1 and ICAM-1. Cell. Signal. 2017, 39, 44–54. [Google Scholar] [CrossRef]
- Yoshinaga, M.; Murao, H.; Kitamura, Y.; Koga, K.; Tsuruta, S.; Igarashi, H.; Nakamura, K.; Takayanagi, R. The 15-lipoxygenase-1 expression may enhance the sensitivity to non-steroidal anti-inflammatory drug-induced apoptosis in colorectal cancers from patients who are treated with the compounds. J. Gastroenterol. Hepatol. 2007, 22, 2324–2329. [Google Scholar] [CrossRef]
- Xu, M.H.; Deng, C.S.; Zhu, Y.Q.; Lin, J. Role of inducible nitric oxide synthase expression in aberrant crypt foci-adenoma-carcinoma sequence. World J. Gastroenterol. 2003, 9, 1246–1250. [Google Scholar] [CrossRef]
- Aoi, W.; Naito, Y.; Takagi, T.; Kokura, S.; Mizushima, K.; Takanami, Y.; Kawai, Y.; Tanimura, Y.; Hung, L.P.; Koyama, R.; et al. Regular exercise reduces colon tumorigenesis associated with suppression of iNOS. Biochem. Biophys. Res. Commun. 2010, 399, 14–19. [Google Scholar] [CrossRef]
- Hofseth, L.J.; Hussain, S.P.; Wogan, G.N.; Harris, C.C. Nitric oxide in cancer and chemoprevention. Free Radic. Biol. Med. 2003, 34, 955–968. [Google Scholar] [CrossRef]
- Crowell, J.A.; Steele, V.E.; Sigman, C.C.; Fay, J.R. Is inducible nitric oxide synthase a target for chemoprevention? Mol. Cancer Ther. 2003, 2, 815–823. [Google Scholar]
- Ahn, B.; Ohshima, H. Suppression of intestinal polyposis in ApcMin/+ mice by inhibiting nitric oxide production. Cancer Res. 2001, 61, 8357–8360. [Google Scholar] [PubMed]
- Gao, Y.; Zhou, S.; Xu, Y.; Sheng, S.; Qian, S.Y.; Huo, X. Nitric oxide synthase inhibitors 1400W and L-NIO inhibit angiogenesis pathway of colorectal cancer. Nitric Oxide. 2019, 83, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Atreya, R.; Neurath, M.F. Signaling molecules: The pathogenic role of the IL-6/STAT-3 trans signaling pathway in intestinal inflammation and in colonic cancer. Curr. Drug Targets 2008, 9, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Wang, G.; Li, W.; Hu, X.; Huang, Q.; Xu, K.; Lou, W.; Wu, J.; Liang, C.; Lou, Q.; et al. Activation of the JAK-STAT3 pathway is associated with the growth of colorectal carcinoma cells. Oncol. Rep. 2014, 31, 335–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janakiram, N.B.; Mohammed, A.; Bryant, T.; Brewer, M.; Biddick, L.; Lightfoot, S.; Lang, M.L.; Rao, C.V. Adoptive transfer of regulatory T cells promotes intestinal tumorigenesis and is associated with decreased NK cells and IL-22 binding protein. Mol. Carcinog. 2015, 54, 986–998. [Google Scholar] [CrossRef]
- Chen, L.; Jiang, B.; Zhong, C.; Guo, J.; Zhang, L.; Mu, T.; Zhang, Q.; Bi, X. Chemoprevention of colorectal cancer by black raspberry anthocyanins involved the modulation of gut microbiota and SFRP2 demethylation. Carcinogenesis 2018, 39, 471–481. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, S.; Baba, T.; Shinagawa, K.; Matsushima, K.; Mukaida, N. Crucial involvement of the CCL3-CCR5 axis-mediated fibroblast accumulation in colitis-associated carcinogenesis in mice. Int. J. Cancer 2014, 135, 1297–1306. [Google Scholar] [CrossRef] [Green Version]
- Löfroos, A.B.; Kadivar, M.; Resic, L.S.; Marsal, J. Colorectal cancer-infiltrating T lymphocytes display a distinct chemokine receptor expression profile. Eur. J. Med. Res. 2017, 22, 40. [Google Scholar] [CrossRef]
- Xing, J.; Li, X.; Sui, J.; Cao, G.; Fu, C. C-X-C chemokine receptor type 5 gene polymorphism affects gene expression in CD4+ T cells and is associated with increased risk of colorectal cancer. Tumour. Biol. 2014, 35, 7929–7934. [Google Scholar] [CrossRef]
- Tanabe, Y.; Sasaki, S.; Mukaida, N.; Baba, T. Blockade of the chemokine receptor, CCR5, reduces the growth of orthotopically injected colon cancer cells via limiting cancer-associated fibroblast accumulation. Oncotarget 2016, 7, 48335–48345. [Google Scholar] [CrossRef] [Green Version]
- Halama, N.; Zoernig, I.; Berthel, A.; Kahlert, C.; Klupp, F.; Suarez-Carmona, M.; Suetterlin, T.; Brand, K.; Krauss, J.; Lasitschka, F.; et al. Tumoral Immune Cell Exploitation in Colorectal Cancer Metastases Can Be Targeted Effectively by Anti-CCR5 Therapy in Cancer Patients. Cancer Cell 2016, 29, 587–601. [Google Scholar] [CrossRef] [Green Version]
- Jafari, N.; Drury, J.; Morris, A.J.; Onono, F.O.; Stevens, P.D.; Gao, T.; Liu, J.; Wang, C.; Lee, E.Y.; Weiss, H.L.; et al. De Novo Fatty Acid Synthesis-Driven Sphingolipid Metabolism Promotes Metastatic Potential of Colorectal Cancer. Mol. Cancer Res. 2019, 17, 140–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yekaterina, Y.; Piotr, G.R.; Tianyan, G.; Eun, Y.L.; Heidi, L.W.; Timothy, S.H.; George, K.; Mark, B.E. Evaluation of small-molecule FASN inhibitors in preclinical models of colorectal cancer. In Proceedings of the 107th Annual Meeting of the American Association for Cancer Research, New Orleans, LA, USA, 16–20 April 2016; AACR: Philadelphia, PA, USA, 2016. [Google Scholar]
- Zaytseva, Y.Y.; Rychahou, P.G.; Le, A.T.; Scott, T.L.; Flight, R.M.; Kim, J.T.; Harris, J.; Liu, J.; Wang, C.; Morris, A.J.; et al. Preclinical evaluation of novel fatty acid synthase inhibitors in primary colorectal cancer cells and a patient-derived xenograft model of colorectal cancer. Oncotarget 2018, 9, 24787–24800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, Q.Q.; Yi, Y.X.; Qiu, J.; Xu, C.J.; Huang, P.L. Fatty acid synthase (FASN) levels in serum of colorectal cancer patients: Correlation with clinical outcomes. Tumor. Biol. 2014, 35, 3855–3859. [Google Scholar] [CrossRef] [PubMed]
- Clinical Trial Identification Number. Available online: https://clinicaltrials.gov/ct2/show/NCT02980029 (accessed on 10 January 2020).
- Wang, H.; Xi, Q.; Wu, G. Fatty acid synthase regulates invasion and metastasis of colorectal cancer via Wnt signaling pathway. Cancer Med. 2016, 5, 1599–1606. [Google Scholar] [CrossRef] [Green Version]
- Zaytseva, Y.Y.; Rychahou, P.G.; Gulhati, P.; Elliott, V.A.; Mustain, W.C.; O’Connor, K.; Morris, A.J.; Sunkara, M.; Weiss, H.L.; Lee, E.Y.; et al. Inhibition of fatty acid synthase attenuates CD44-associated signaling and reduces metastasis in colorectal cancer. Cancer Res. 2012, 72, 1504–1517. [Google Scholar] [CrossRef] [Green Version]
- Kuchiba, A.; Morikawa, T.; Yamauchi, M.; Imamura, Y.; Liao, X.; Chan, A.T.; Meyerhardt, J.A.; Giovannucci, E.; Fuchs, C.S.; Ogino, S. Body mass index and risk of colorectal cancer according to fatty acid synthase expression in the nurses’ health study. J. Natl. Cancer Inst. 2012, 104, 415–420. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.; Wu, P.; Senthilkumar, R.; Tian, X.; Liu, H.; Shen, X.; Tao, Z.; Huang, P. Loss of fatty acid synthase suppresses the malignant phenotype of colorectal cancer cells by down-regulating energy metabolism and mTOR signaling pathway. J. Cancer Res. Clin. Oncol. 2016, 142, 59–72. [Google Scholar] [CrossRef]
- Meyskens, F.L.; Emerson, S.S.; Pelot, D.; Meshkinpour, H.; Shassetz, L.R.; Einspahr, J.; Alberts, D.S.; Gerner, E.W. Dose de-escalation chemoprevention trial of α-difluoromethylornithine in patients with colon polyps. J. Natl. Cancer Inst. 1994, 86, 1122–1130. [Google Scholar] [CrossRef] [Green Version]
- Meyskens, F.L.; McLaren, C.E.; Pelot, D.; Fujikawa-Brooks, S.; Carpenter, P.M.; Hawk, E.; Kelloff, G.; Lawson, M.J.; Kidao, J.; McCracken, J.; et al. Difluoromethylornithine plus sulindac for the prevention of sporadic colorectal adenomas: A randomized placebo-controlled, double-blind trial. Cancer Prev. Res. 2008, 1, 32–38. [Google Scholar] [CrossRef] [Green Version]
- Danzin, C.; Mamont, P.S. Polyamine inhibition in vivo and in organ growth and repair. In Inhibition of Polyamine Metabolism; McCann, P.P., Pegg, A.E., Sjoerdsma, A., Eds.; Academic Press: New York, NY, USA, 1987; pp. 141–164. [Google Scholar]
- Janakiram, N.B.; Mohammed, A.; Bryant, T.; Zhang, Y.; Brewer, M.; Duff, A.; Biddick, L.; Singh, A.; Lightfoot, S.; Steele, V.E.; et al. Potentiating NK cell activity by combination of Rosuvastatin and Difluoromethylornithine for effective chemopreventive efficacy against Colon Cancer. Sci. Rep. 2016, 6, 37046. [Google Scholar] [CrossRef] [Green Version]
- Rao, C.V.; Tokumo, K.; Rigotty, J.; Zang, E.; Kelloff, G.; Reddy, B.S. Chemoprevention of colon carcinogenesis by dietary administration of piroxicam, alpha-difluoromethylornithine, 16 alpha-fluoro-5-androsten-17-one, and ellagic acid individually and in combination. Cancer Res. 1991, 51, 4528–4534. [Google Scholar] [PubMed]
- Kumar, K.N.; Raja, S.B.; Vidhya, N.; Devaraj, S.N. Ellagic acid modulates antioxidant status, ornithine decarboxylase expression, and aberrant crypt foci progression in 1,2-dimethylhydrazine-instigated colon preneoplastic lesions in rats. J. Agric. Food Chem. 2012, 60, 3665–3672. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; He, X.; Ding, Y.; Chen, H.; Sun, L. Statin uses and mortality in colorectal cancer patients: An updated systematic review and meta-analysis. Cancer Med. 2019, 8, 3305–3313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karagkounis, G.; DeVecchio, J.; Ferrandon, S.; Kalady, M.F. Simvastatin enhances radiation sensitivity of colorectal cancer cells. Surg. Endosc. 2018, 32, 1533–1539. [Google Scholar] [CrossRef]
- Wei, T.T.; Lin, Y.T.; Tseng, R.Y.; Shun, C.T.; Lin, Y.C.; Wu, M.S.; Fang, J.M.; Chen, C.C. Prevention of Colitis and Colitis-Associated Colorectal Cancer by a Novel Polypharmacological Histone Deacetylase Inhibitor. Clin. Cancer Res. 2018, 24, 499. [Google Scholar] [CrossRef] [Green Version]
- Suh, N.; Reddy, B.S.; DeCastro, A.; Paul, S.; Lee, H.J.; Smolarek, A.K.; So, J.Y.; Simi, B.; Wang, C.X.; Janakiram, N.B.; et al. Combination of atorvastatin with sulindac or naproxen profoundly inhibits colonic adenocarcinomas by suppressing the p65/β-catenin/cyclin D1 signaling pathway in rats. Cancer Prev. Res. 2011, 4, 1895–1902. [Google Scholar] [CrossRef] [Green Version]
- Swamy, M.V.; Patlolla, J.M.; Steele, V.E.; Kopelovich, L.; Reddy, B.S.; Rao, C.V. Chemoprevention of familial adenomatous polyposis by low doses of atorvastatin and celecoxib given individually and in combination to APCMin mice. Cancer Res. 2006, 66, 7370–7377. [Google Scholar] [CrossRef] [Green Version]
- Lipkin, S.M.; Chao, E.C.; Moreno, V.; Rozek, L.S.; Rennert, H.; Pinchev, M.; Dizon, D.; Rennert, G.; Kopelovich, L.; Gruber, S.B. Genetic variation in 3-hydroxy-3-methylglutaryl CoA reductase modifies the chemopreventive activity of statins for colorectal cancer. Cancer Prev. Res. 2010, 3, 597–603. [Google Scholar] [CrossRef] [Green Version]
- Ricciardiello, L.; Ahnen, D.J.; Lynch, P.M. Chemoprevention of hereditary colon cancers: Time for new strategies. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 352–361. [Google Scholar] [CrossRef]
- Clinical Trial Identification Number. Available online: https://clinicaltrials.gov/ct2/show/NCT02301286 (accessed on 10 January 2020).
- Seo, T.; Tatsuguchi, A.; Shinji, S.; Yonezawa, M.; Mitsui, K.; Tanaka, S.; Fujimori, S.; Gudis, K.; Fukuda, Y.; Sakamoto, C. Microsomal prostaglandin E synthase protein levels correlate with prognosis in colorectal cancer patients. Virchows Arch. 2009, 454, 667–676. [Google Scholar] [CrossRef]
- Reddy, B.S.; Hirose, Y.; Lubet, R.; Steele, V.; Kelloff, G.; Paulson, S.; Seibert, K.; Rao, C.V. Chemoprevention of colon cancer by specific cyclooxygenase-2 inhibitor, celecoxib, administered during different stages of carcinogenesis. Cancer Res. 2000, 60, 293–297. [Google Scholar] [PubMed]
- Li, P.; Wu, H.; Zhang, H.; Shi, Y.; Xu, J.; Ye, Y.; Xia, D.; Yang, J.; Cai, J.; Wu, Y. Aspirin use after diagnosis but not prediagnosis improves established colorectal cancer survival: A meta-analysis. Gut 2015, 64, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- De Robertis, M.; Massi, E.; Poeta, M.L.; Carotti, S.; Morini, S.; Cecchetelli, L.; Signori, E.; Fazio, V.M. The AOM/DSS murine model for the study of colon carcinogenesis: From pathways to diagnosis and therapy studies. J. Carcinog. 2011, 10, 9. [Google Scholar] [PubMed]
- Zeineldin, M.; Neufeld, K.L. More than two decades of Apc modeling in rodents. Biochim. Biophys. Acta 2013, 1836, 80–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tetteh, P.W.; Kretzschmar, K.; Begthel, H.; van den Born, M.; Korving, J.; Morsink, F.; Farin, H.; van Es, J.H.; Offerhaus, G.J.; Clevers, H. Generation of an inducible colon-specific Cre enzyme mouse line for colon cancer research. Proc. Natl. Acad. Sci. USA 2016, 113, 11859–11864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Femia, A.P.; Luceri, C.; Soares, P.V.; Lodovici, M.; Caderni, G. Multiple mucin depleted foci, high proliferation and low apoptotic response in the onset of colon carcinogenesis of the PIRC rat, mutated in Apc. Int. J. Cancer 2015, 136, E488–E495. [Google Scholar] [CrossRef]
- Navarra, M.; Femia, A.P.; Romagnoli, A.; Tortora, K.; Luceri, C.; Cirmi, S.; Ferlazzo, N.; Caderni, G. A flavonoid-rich extract from bergamot juice prevents carcinogenesis in a genetic model of colorectal cancer, the Pirc rat (F344/NTac-Apcam1137). Eur. J. Nutr. 2020, 59, 885–894. [Google Scholar] [CrossRef]
- Tong, Y.; Yang, W.; Koeffler, H.P. Mouse models of colorectal cancer. Chin. J. Cancer 2011, 30, 450–462. [Google Scholar] [CrossRef] [Green Version]
- Taketo, M.M.; Edelmann, W. Mouse models of colon cancer. Gastroenterology 2009, 136, 780–798. [Google Scholar] [CrossRef]
- Young, M.; Ordonez, L.; Clarke, A.R. What are the best routes to effectively model human colorectal cancer? Mol. Oncol. 2013, 7, 178–189. [Google Scholar] [CrossRef]
- Oliveira, R.C.; Abrantes, A.M.; Tralhao, J.G.; Botelho, M.F. The role of mouse models in colorectal cancer research-The need and the importance of the orthotopic models. Anim. Models Exp. Med. 2020, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Deem, A.K.; Kopetz, S.; Heffernan, T.P.; Draetta, G.F.; Carugo, A. Current and Future Horizons of Patient-Derived Xenograft Models in Colorectal Cancer Translational Research. Cancers 2019, 11, 1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarla, N.S.; Pathuri, G.; Gali, H.; Panneerselvam, J.; Chandrakesan, P.; Madka, V.; Rao, C.V. LFA-9, a Selective Inhibitor of Microsomal Prostaglandin Synthase-1 and 5-Lipoxygenase. Prev. Inflamm. Oncol. Dis. 2020, 34, S1. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Normal | ACF | Adenoma | Adenocarcinoma | References | |
|---|---|---|---|---|---|
| Mutant/Dysregulated Signaling | |||||
| APC | |||||
| Pre-clinical | + | − | − | − | [6] |
| Clinical | + | − | − | − | [3,4,5] |
| β-catenin | |||||
| Pre-clinical | + | ++ | ++++ | +++++ | [7,8] |
| Clinical | + | ++ | ++++ | +++++ | [6] |
| TP53 | |||||
| Pre-clinical | + | + | − | − | [24] |
| Clinical | + | + | − | − | [20,21,22,23] |
| KRAS | |||||
| Pre-clinical | − | ? | +++ | ++++ | [12,13] |
| Clinical | − | + | +++ | ++++ | [12] |
| AKT | |||||
| Pre-clinical | − | ? | +++ | +++++ | [30] |
| Clinical | − | ? | +++ | +++++ | [29] |
| SMAD4 | |||||
| Pre-clinical | + | ? | + | − | [19] |
| Clinical | + | ? | + | − | [19] |
| Inflammatory | |||||
| COX-2 | |||||
| Pre-clinical | − | + * | ++++ | +++++ | [34] |
| Clinical | ? | ++++ | +++++ | [32,35] | |
| mPGES-1 | |||||
| Pre-clinical | − | ? | ++++ | +++++ | [45,46] |
| Clinical | − | ? | ++++ | +++++ | [47,48] |
| 5-LOX | |||||
| Pre-clinical | − | ? | +++ | ++++ | [49] |
| Clinical | − | ? | +++ | ++++ | [50,51] |
| iNOS | |||||
| Pre-clinical | − | + * | +++ | ++++ | [52] |
| Clinical | − | ? | +++ | ++++ | [53,54] |
| 15-PGDH | |||||
| Pre-clinical | +++ | ? | − | − | [55] |
| Clinical | +++ | ? | − | − | [56] |
| STAT-3 | |||||
| Pre-clinical | + | ? | +++ | +++++ | [57] |
| Clinical | + | ? | +++ | +++++ | [58] |
| Prostaglandin I2 Synthase | ++ | − | − | − | |
| Pre-clinical | ++ | − | − | − | [59,60] |
| Clinical | ++ | − | − | − | [61] |
| 15-LOX | |||||
| Pre-clinical | ++ | ? | − | − | [62,63] |
| Clinical | ++ | ? | − | − | [64] |
| Growth and metabolism | |||||
| Ornithine decarboxylase | |||||
| Pre-clinical | + | ? | ++++ | +++++ | [65] |
| Clinical | + | ? | ++++ | +++++ | [66,67,68] |
| HMG Co-A-Reductase | |||||
| Pre-clinical | + | ? | ++++ | +++++ | [69,70] |
| Clinical | + | ? | ++++ | +++++ | [71] |
| Fatty acid synthase | |||||
| Pre-clinical | + | + | +++ | ++++ | [72] |
| Clinical | + | + | +++ | ++++ | [72,73] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yarla, N.S.; Madka, V.; Pathuri, G.; Rao, C.V. Molecular Targets in Precision Chemoprevention of Colorectal Cancer: An Update from Pre-Clinical to Clinical Trials. Int. J. Mol. Sci. 2020, 21, 9609. https://doi.org/10.3390/ijms21249609
Yarla NS, Madka V, Pathuri G, Rao CV. Molecular Targets in Precision Chemoprevention of Colorectal Cancer: An Update from Pre-Clinical to Clinical Trials. International Journal of Molecular Sciences. 2020; 21(24):9609. https://doi.org/10.3390/ijms21249609
Chicago/Turabian StyleYarla, Nagendra S., Venkateshwar Madka, Gopal Pathuri, and Chinthalapally V. Rao. 2020. "Molecular Targets in Precision Chemoprevention of Colorectal Cancer: An Update from Pre-Clinical to Clinical Trials" International Journal of Molecular Sciences 21, no. 24: 9609. https://doi.org/10.3390/ijms21249609
APA StyleYarla, N. S., Madka, V., Pathuri, G., & Rao, C. V. (2020). Molecular Targets in Precision Chemoprevention of Colorectal Cancer: An Update from Pre-Clinical to Clinical Trials. International Journal of Molecular Sciences, 21(24), 9609. https://doi.org/10.3390/ijms21249609