Intracellular Calcium Dysregulation by the Alzheimer’s Disease-Linked Protein Presenilin 2


 and
and {kind=link}
{kind=link}
Abstract
:1. APP, PS1, and PS2 Physiopathology: Focus on Alzheimer’s Disease
2. Sporadic Alzheimer’s Disease (SAD)
3. Familial Alzheimer’s Disease (FAD)
3.1. Amyloid Precursor Protein
3.2. Presenilins
4. Ca2+ Molecular Toolkit and Signalling: A General Overview
5. Presenilin 2 and Ca2+ Homeostasis
6. Concluding Remarks
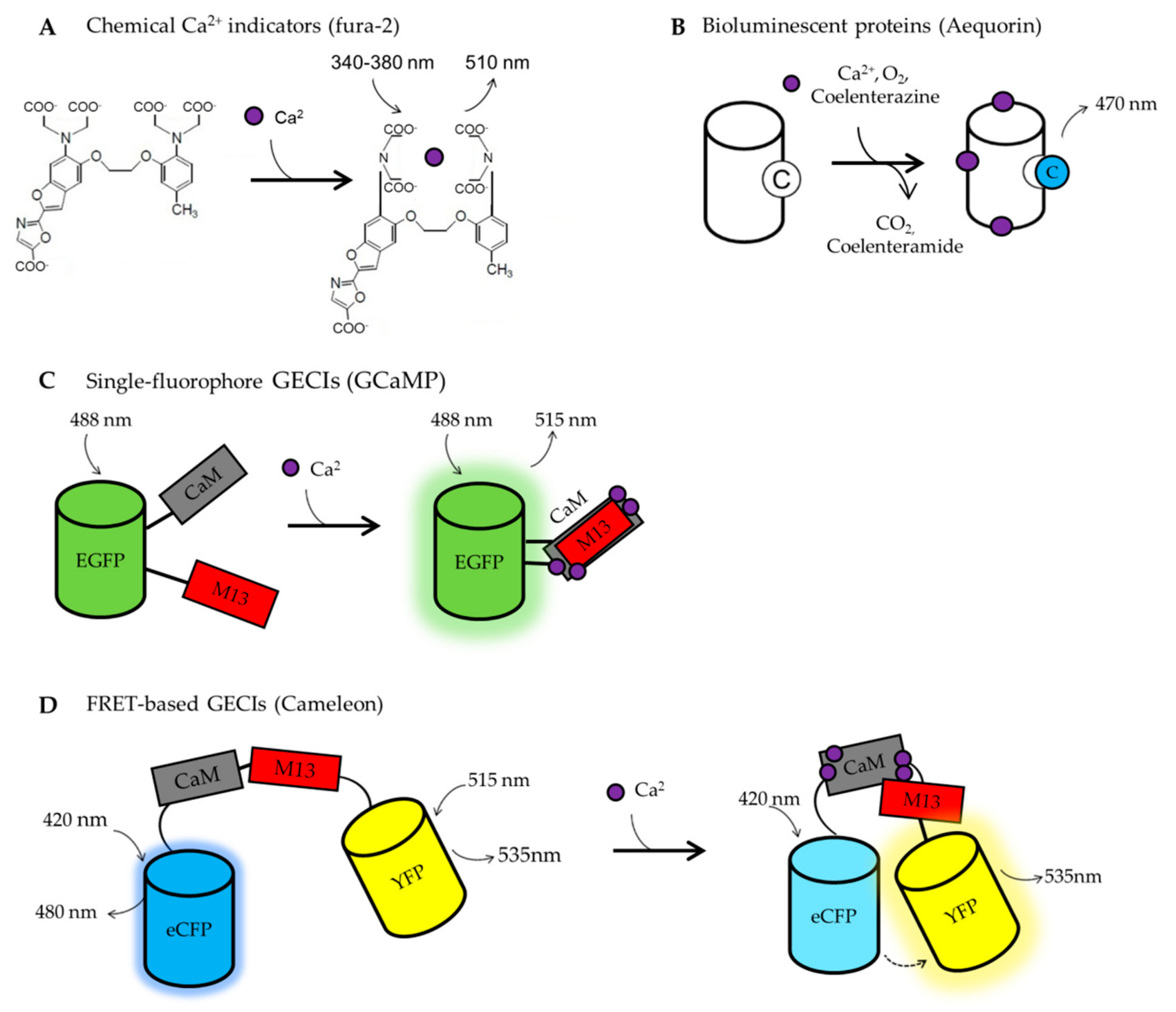
6.1. BOX1. Calcium Imaging Tools
6.2. BOX2. PS2-Based Mouse AD Models
Funding
Conflicts of Interest
References
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossor, M.N.; Fox, N.C.; Freeborough, P.A.; Harvey, R.J. Clinical features of sporadic and familial Alzheimer’s disease. Neurodegeneration 1996, 5, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Schachter, A.S.; Davis, K.L. Alzheimer’s disease. Dialogues Clin. Neurosci. 2000, 2, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Bondi, M.W.; Edmonds, E.C.; Salmon, D.P. Alzheimer’s disease: Past, present, and future. J. Int. Neuropsychol. Soc. 2017, 23, 818–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weller, J.; Budson, A. Current understanding of Alzheimer’s disease diagnosis and treatment. F1000Research 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padovani, A.; Benussi, A.; Cantoni, V.; Dell’Era, V.; Cotelli, M.S.; Caratozzolo, S.; Turrone, R.; Rozzini, L.; Alberici, A.; Altomare, D.; et al. Diagnosis of mild cognitive impairment due to Alzheimer’s disease with transcranial magnetic stimulation. J. Alzheimer’s Dis. 2018, 65, 221–230. [Google Scholar] [CrossRef]
- Murphy, M.P.; Levine, H. Alzheimer’s disease and the amyloid-β peptide. J. Alzheimer’s Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Dorszewska, J.; Prendecki, M.; Oczkowska, A.; Dezor, M.; Kozubski, W. Molecular Basis of Familial and Sporadic Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 952–963. [Google Scholar] [CrossRef]
- Duara, R.; Lopez-Alberola, R.F.; Barker, W.W.; Loewenstein, D.A.; Zatinsky, M.; Eisdorfer, C.E.; Weinberg, G.B. A comparison of familial and sporadic alzheimer’s disease. Neurology 1993, 43, 1377–1384. [Google Scholar] [CrossRef]
- Ray, W.J.; Ashall, F.; Goate, A.M. Molecular pathogenesis of sporadic and familial forms of Alzheimer’s disease. Mol. Med. Today 1998, 4, 151–157. [Google Scholar] [CrossRef]
- Piaceri, I.; Nacmias, B.; Sorbi, S. Genetics of familial and sporadic Alzheimer’s disease. Front. Biosci. - Elit. 2013, 5 E, 167–177. [Google Scholar] [CrossRef] [Green Version]
- Peila, R.; Rodriguez, B.L.; Launer, L.J. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes 2002, 51, 1256–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreses-Werringloer, U.; Lambert, J.C.; Vingtdeux, V.; Zhao, H.; Vais, H.; Siebert, A.; Jain, A.; Koppel, J.; Rovelet-Lecrux, A.; Hannequin, D.; et al. A Polymorphism in CALHM1 Influences Ca2+ Homeostasis, Aβ Levels, and Alzheimer’s Disease Risk. Cell 2008, 133, 1149–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.C.; Sleegers, K.; González-Pérez, A.; Ingelsson, M.; Beecham, G.W.; Hiltunen, M.; Combarros, O.; Bullido, M.J.; Brouwers, N.; Bettens, K.; et al. The CALHM1 P86L polymorphism is a genetic modifier of age at onset in Alzheimer’s disease: A meta-analysis study. J. Alzheimer’s Dis. 2010, 22, 247–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minster, R.L.; Demirci, F.Y.; Dekosky, S.T.; Kamboh, M.I. No association between CALHM1 variation and risk of alzheimer disease. Hum. Mutat. 2009, 30. [Google Scholar] [CrossRef] [Green Version]
- Tan, E.K.; Ho, P.; Cheng, S.Y.; Yih, Y.; Li, H.H.; Fook-Chong, S.; Lee, W.L.; Zhao, Y. CALHM1 variant is not associated with Alzheimer’s disease among Asians. Neurobiol. Aging 2011, 32, 546.e11–546.e12. [Google Scholar] [CrossRef]
- Edwards, G.A.; Gamez, N.; Escobedo, G.; Calderon, O.; Moreno-Gonzalez, I. Modifiable risk factors for Alzheimer’s disease. Front. Aging Neurosci. 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahiri, D.K.; Maloney, B.; Basha, M.R.; Ge, Y.W.; Zawia, N.H. How and When Environmental Agents and Dietary Factors Affect the Course of Alzheimers Disease: The “LEARn” Model (Latent Early-Life Associated Regulation) May Explain the Triggering of AD. Curr. Alzheimer Res. 2007, 4, 219–228. [Google Scholar] [CrossRef]
- Lahiri, D.K.; Zawia, N.H.; Greig, N.H.; Sambamurti, K.; Maloney, B. Early-life events may trigger biochemical pathways for Alzheimer’s disease: The “LEARn” model. Biogerontology 2008, 9, 375–379. [Google Scholar] [CrossRef] [Green Version]
- Lahiri, D.K.; Maloney, B. The “LEARn” (Latent Early-life Associated Regulation) model integrates environmental risk factors and the developmental basis of Alzheimer’s disease, and proposes remedial steps. Exp. Gerontol. 2010, 45, 291–296. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, S.; Khemka, V.K.; Banerjee, A.; Chatterjee, G.; Ganguly, A.; Biswas, A. Metabolic risk factors of sporadic Alzheimer’s disease: Implications in the pathology, pathogenesis and treatment. Aging Dis. 2015, 6, 282–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, R.H.; Khan, S.M. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 2004, 63, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta - Mol. Basis Dis. 2014, 1842, 1219–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridge, P.G.; Kauwe, J.S.K. Mitochondria and Alzheimer’s Disease: the Role of Mitochondrial Genetic Variation. Curr. Genet. Med. Rep. 2018, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Rosa-Neto, P.; Hsiung, G.Y.R.; Sadovnick, A.D.; Masellis, M.; Black, S.E.; Jia, J.; Gauthier, S. Early-onset familial alzheimer’s disease (EOFAD). Can. J. Neurol. Sci. 2012, 39, 436–445. [Google Scholar] [CrossRef] [Green Version]
- Lippa, C.F.; Saunders, A.M.; Smith, T.W.; Swearer, J.M.; Drachman, D.A.; Ghetti, B.; Nee, L.; Pulaski-Salo, D.; Dickson, D.; Robitaille, Y.; et al. Familial and sporadic Alzheimer’s disease: Neuropathology cannot exclude a final common pathway. Neurology 1996, 46, 406–412. [Google Scholar] [CrossRef]
- Yoshikai, S.i.; Sasaki, H.; Doh-ura, K.; Furuya, H.; Sakaki, Y. Genomic organization of the human amyloid beta-protein precursor gene. Gene 1990, 87, 257–263. [Google Scholar] [CrossRef]
- Lamb, B.T.; Sisodia, S.S.; Lawler, A.M.; Slunt, H.H.; Kitt, C.A.; Kearns, W.G.; Pearson, P.L.; Price, D.L.; Gearhart, J.D. Introduction and expression of the 400 kilobase precursor amyloid protein gene in transgenic mice. Nat. Genet. 1993, 5, 22–30. [Google Scholar] [CrossRef]
- Matsui, T.; Ingelsson, M.; Fukumoto, H.; Ramasamy, K.; Kowa, H.; Frosch, M.P.; Irizarry, M.C.; Hyman, B.T. Expression of APP pathway mRNAs and proteins in Alzheimer’s disease. Brain Res. 2007, 1161, 116–123. [Google Scholar] [CrossRef]
- Nguyen, K.V. β-Amyloid precursor protein (APP) and the human diseases. AIMS Neurosci. 2019, 6, 273–281. [Google Scholar] [CrossRef]
- Van der Kant, R.; Goldstein, L.S.B. Cellular Functions of the Amyloid Precursor Protein from Development to Dementia. Dev. Cell 2015, 32, 502–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.J.; Moussa, C.E.H.; Lee, Y.; Sung, Y.; Howell, B.W.; Turner, R.S.; Pak, D.T.S.; Hoe, H.S. Beta amyloid-independent role of amyloid precursor protein in generation and maintenance of dendritic spines. Neuroscience 2010, 169, 344–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, U.C.; Deller, T.; Korte, M. Not just amyloid: Physiological functions of the amyloid precursor protein family. Nat. Rev. Neurosci. 2017, 18, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.F.; Xu, T.H.; Yan, Y.; Zhou, Y.R.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, X.; Li, G.; Zhang, Y.; Wu, Y.; Song, W. Modifications and trafficking of APP in the pathogenesis of alzheimer’s disease. Front. Mol. Neurosci. 2017, 10. [Google Scholar] [CrossRef]
- Hooper, N.M.; Walter, J.; Haass, C. Posttranslational Modifications of Amyloid Precursor Protein: Ectodomain Phosphorylation and Sulfation. In Alzheimer’s Disease; Humana Press Inc.: Totowa, NJ, USA, 2003; pp. 149–168. [Google Scholar]
- Zheng, H.; Koo, E.H. Biology and pathophysiology of the amyloid precursor protein. Mol. Neurodegener. 2011, 6. [Google Scholar] [CrossRef] [Green Version]
- Thinakaran, G.; Koo, E.H. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 2008, 283, 29615–29619. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Sun, Y.; Ma, Q.-H.; Liu, Y. Alzheimer’s disease: Amyloid-based pathogenesis and potential therapies. Cell Stress 2018, 2, 150–161. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.R.; Glabe, C.G. Distinct early folding and aggregation properties of Alzheimer amyloid-β peptides Aβ40 and Aβ42: Stable trimer or tetramer formation by Aβ42. J. Biol. Chem. 2006, 281, 24414–24422. [Google Scholar] [CrossRef] [Green Version]
- Parihar, M.S.; Hemnani, T. Alzheimer’s disease pathogenesis and therapeutic interventions. J. Clin. Neurosci. 2004, 11, 456–467. [Google Scholar] [CrossRef] [PubMed]
- Fiala, M.; Liu, P.T.; Espinosa-Jeffrey, A.; Rosenthal, M.J.; Bernard, G.; Ringman, J.M.; Sayre, J.; Zhang, L.; Zaghi, J.; Dejbakhsh, S.; et al. Innate immunity and transcription of MGAT-III and Toll-like receptors in Alzheimer’s disease patients are improved by bisdemethoxycurcumin. Proc. Natl. Acad. Sci. USA 2007, 104, 12849–12854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-β in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Gandy, S. The role of cerebral amyloid β accumulation in common forms of Alzheimer disease. J. Clin. Invest. 2005, 115, 1121–1129. [Google Scholar] [PubMed]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef]
- Rogaev, E.I.; Sherrington, R.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Liang, Y.; Chi, H.; Lin, C.; Holman, K.; Tsuda, T.; et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 1995, 376, 775–778. [Google Scholar] [CrossRef]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375, 754–760. [Google Scholar] [CrossRef]
- De Strooper, B.; Saftig, P.; Craessaerts, K.; Vanderstichele, H.; Guhde, G.; Annaert, W.; Von Figura, K.; Van Leuven, F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 1998, 391, 387–390. [Google Scholar] [CrossRef]
- Wolfe, M.S.; Xia, W.; Ostaszewski, B.L.; Diehl, T.S.; Kimberly, W.T.; Selkoe, D.J. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature 1999, 398, 513–517. [Google Scholar] [CrossRef]
- Levitan, D.; Greenwald, I. Facilitation of lin-12-mediated signalling by sel-12, a Caenorhabditis elegans S182 Alzheimer’s disease gene. Nature 1995, 377, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Boulianne, G.L.; Livne-Bar, I.; Humphreys, J.M.; Liang, Y.; Lin, C.; Rogaev, E.; George-Hyslop, P.S. Cloning and characterization of the Drosophila presenilin homologue. Neuroreport 1997, 8, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Mir, A.; Cañestro, C.; Gonzàlez-Duarte, R.; Albalat, R. Characterization of the amphioxus presenilin gene in a high gene-density genomic region illustrates duplication during the vertebrate lineage. Gene 2001, 279, 157–164. [Google Scholar] [CrossRef]
- Lee, M.K.; Slunt, H.H.; Martin, L.J.; Thinakaran, G.; Kim, G.; Gandy, S.E.; Seeger, M.; Koo, E.; Price, D.L.; Sisodia, S.S. Expression of presenilin 1 and 2 (PS1 and PS2) in human and murine tissues. J. Neurosci. 1996, 16, 7513–7525. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, D.M.; Fausett, H.J.; Page, K.J.; Kim, T.W.; Moir, R.D.; Merriam, D.E.; Hollister, R.D.; Hallmark’, O.G.; Mancini, R.; Felsenstein, K.M.; et al. Alzheimer-associated presenilins 1 and 2: Neuronal expression in brain and localization to intracellular membranes in mammalian cells. Nat. Med. 1996, 2, 224–229. [Google Scholar] [CrossRef]
- Suzuki, T.; Nishiyama, K.; Murayama, S.; Yamamoto, A.; Sato, S.; Kanazawa, I.; Sakaki, Y. Regional and cellular Presenilin 1 gene expression in human and rat tissues. Biochem. Biophys. Res. Commun. 1996, 219, 708–713. [Google Scholar] [CrossRef]
- Brunkan, A.L.; Goate, A.M. Presenilin function and γ-secretase activity. J. Neurochem. 2005, 93, 769–792. [Google Scholar] [CrossRef]
- Area-Gomez, E.; De Groof, A.J.C.; Boldogh, I.; Bird, T.D.; Gibson, G.E.; Koehler, C.M.; Yu, W.H.; Duff, K.E.; Yaffe, M.P.; Pon, L.A.; et al. Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am. J. Pathol. 2009, 175, 1810–1816. [Google Scholar] [CrossRef] [Green Version]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Presenilin 2 Modulates Endoplasmic Reticulum-Mitochondria Coupling by Tuning the Antagonistic Effect of Mitofusin 2. Cell Rep. 2016, 15, 2226–2238. [Google Scholar] [CrossRef] [Green Version]
- Leal, N.S.; Schreiner, B.; Pinho, C.M.; Filadi, R.; Wiehager, B.; Karlström, H.; Pizzo, P.; Ankarcrona, M. Mitofusin-2 knockdown increases ER–mitochondria contact and decreases amyloid β-peptide production. J. Cell. Mol. Med. 2016, 20, 1686–1695. [Google Scholar] [CrossRef]
- Doan, A.; Thinakaran, G.; Borchelt, D.R.; Slunt, H.H.; Ratovitsky, T.; Podlisny, M.; Selkoe, D.J.; Seeger, M.; Gandy, S.E.; Price, D.L.; et al. Protein topology of presenilin 1. Neuron 1996, 17, 1023–1030. [Google Scholar] [CrossRef] [Green Version]
- Laudon, H.; Hansson, E.M.; Melén, K.; Bergman, A.; Farmery, M.R.; Winblad, B.; Lendahl, U.; Von Heijne, G.; Näslund, J. A nine-transmembrane domain topology for presenilin 1. J. Biol. Chem. 2005, 280, 35352–35360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfe, M.S. Toward the structure of presenilin/γ-secretase and presenilin homologs. Biochim. Biophys. Acta - Biomembr. 2013, 1828, 2886–2897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.M.; Lai, M.T.; Xu, M.; Huang, Q.; DiMuzio-Mower, J.; Sardana, M.K.; Shi, X.P.; Yin, K.C.; Shafer, J.A.; Gardell, S.J. Presenilin 1 is linked with γ-secretase activity in the detergent solubilized state. Proc. Natl. Acad. Sci. USA 2000, 97, 6138–6143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, F.; Hasegawa, H.; Schmitt-Ulms, G.; Kawarai, T.; Bohm, C.; Katayama, T.; Gu, Y.; Sanjo, N.; Glista, M.; Rogaeva, E.; et al. TMP21 is a presenilin complex component that modulates γ-secretase but not ε-secretase activity. Nature 2006, 440, 1208–1212. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.; Shelton, C.C.; Tian, Y.; Zhang, X.; Gilchrist, M.L.; Sisodia, S.S.; Li, Y.M. Activation and intrinsic γ-secretase activity of presenilin 1. Proc. Natl. Acad. Sci. USA 2010, 107, 21435–21440. [Google Scholar] [CrossRef] [Green Version]
- Edbauer, D.; Winkler, E.; Regula, J.T.; Pesold, B.; Steiner, H.; Haass, C. Reconstitution of gamma-secretase activity. Nat. Cell Biol. 2003. [Google Scholar] [CrossRef]
- Iwatsubo, T. The γ-secretase complex: Machinery for intramembrane proteolysis. Curr. Opin. Neurobiol. 2004, 14, 379–383. [Google Scholar] [CrossRef]
- McCarthy, J.V.; Twomey, C.; Wujek, P. Presenilin-dependent regulated intramembrane proteolysis and γ-secretase activity. Cell. Mol. Life Sci. 2009, 66, 1534–1555. [Google Scholar] [CrossRef]
- Thinakaran, G.; Borchelt, D.R.; Lee, M.K.; Slunt, H.H.; Spitzer, L.; Kim, G.; Ratovitsky, T.; Davenport, F.; Nordstedt, C.; Seeger, M.; et al. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron 1996, 17, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Steiner, H.; Fluhrer, R.; Haass, C. Intramembrane proteolysis by γ-secretase. J. Biol. Chem. 2008, 283, 29627–29631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Strooper, B.; Annaert, W. Novel Research Horizons for Presenilins and γ-Secretases in Cell Biology and Disease. Annu. Rev. Cell Dev. Biol. 2010, 26, 235–260. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Pettingell, W.H.; Hallmark, O.G.; Moir, R.D.; Wasco, W.; Tanzi, R.E. Endoproteolytic cleavage and proteasomal degradation of presenilin 2 in transfected cells. J. Biol. Chem. 1997, 272, 11006–11010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Center for Molecular Neurology. Available online: https://uantwerpen.vib.be/ (accessed on 17 December 2019).
- Scheuner, D.; Eckman, C.; Jensen, M.; Song, X.; Citron, M.; Suzuki, N.; Bird, T.D.; Hardy, J.; Hutton, M.; Kukull, W.; et al. Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat. Med. 1996, 2, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Citron, M.; Westaway, D.; Xia, W.; Carlson, G.; Diehl, T.; Levesque, G.; Johnson-Wood, K.; Lee, M.; Seubert, P.; Davis, A.; et al. Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid β-protein in both transfected cells and transgenic mice. Nat. Med. 1997, 3, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Chévez-Gutiérrez, L.; Bammens, L.; Benilova, I.; Vandersteen, A.; Benurwar, M.; Borgers, M.; Lismont, S.; Zhou, L.; Van Cleynenbreugel, S.; Esselmann, H.; et al. The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. EMBO J. 2012, 31, 2261–2274. [Google Scholar] [CrossRef]
- Florean, C.; Zampese, E.; Zanese, M.; Brunello, L.; Ichas, F.; De Giorgi, F.; Pizzo, P. High content analysis of γ-secretase activity reveals variable dominance of presenilin mutations linked to familial Alzheimer’s disease. Biochim. Biophys. Acta - Mol. Cell Res. 2008, 1783, 1551–1560. [Google Scholar] [CrossRef]
- Shimojo, M.; Sahara, N.; Murayama, M.; Ichinose, H.; Takashima, A. Decreased Aβ secretion by cells expressing familial Alzheimer’s disease-linked mutant presenilin 1. Neurosci. Res. 2007, 57, 446–453. [Google Scholar] [CrossRef]
- Walker, E.S.; Martinez, M.; Brunkan, A.L.; Goate, A. Presenilin 2 familial Alzheimer’s disease mutations result in partial loss of function and dramatic changes in Aβ 42/40 ratios. J. Neurochem. 2005, 92, 294–301. [Google Scholar] [CrossRef]
- Quintero-Monzon, O.; Martin, M.M.; Fernandez, M.A.; Cappello, C.A.; Krzysiak, A.J.; Osenkowski, P.; Wolfe, M.S. Dissociation between the processivity and total activity of γ-secretase: Implications for the mechanism of Alzheimer’s disease-causing presenilin mutations. Biochemistry 2011, 50, 9023–9035. [Google Scholar] [CrossRef] [Green Version]
- De Strooper, B.; Iwatsubo, T.; Wolfe, M.S. Presenilins and γ-secretase: Structure, function, and role in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Asahi, M.; Yamaguchi, O.; Hikoso, S.; Nakayama, H.; Kusakari, Y.; Kawai, M.; Hongo, K.; Higuchi, Y.; Kashiwase, K.; et al. Presenilin 2 regulates the systolic function of heart by modulating Ca2+ signaling. FASEB J. 2005, 19, 2069–2071. [Google Scholar] [CrossRef] [PubMed]
- Donoviel, D.B.; Hadjantonakis, A.-K.; Ikeda, M.; Zheng, H.; Hyslop, P.S.G.; Bernstein, A. Mice lacking both presenilin genes exhibitearlyembryonic patterningdefects. Genes Dev. 1999, 13, 2801–2810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Parks, S.B.; Kushner, J.D.; Nauman, D.; Burgess, D.; Ludwigsen, S.; Partain, J.; Nixon, R.R.; Allen, C.N.; Irwin, R.P.; et al. Mutations of Presenilin Genes in Dilated Cardiomyopathy and Heart Failure. Am. J. Hum. Genet. 2006, 79, 1030–1039. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Li, C.; Zhang, Y.; Ren, J. Interrelationship between Alzheimer’s disease and cardiac dysfunction: the brain–heart continuum? Acta Biochim. Biophys. Sin. 2020. [Google Scholar] [CrossRef] [Green Version]
- An, S.S.; Cai, Y.; Kim, S. Mutations in presenilin 2 and its implications in Alzheimer’s disease and other dementia-associated disorders. Clin. Interv. Aging 2015, 1163. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, K.; Gianni, D.; Balla, C.; Assenza, G.E.; Joshi, M.; Semigran, M.J.; Macgillivray, T.E.; Van Eyk, J.E.; Agnetti, G.; Paolocci, N.; et al. Cofilin-2 Phosphorylation and Sequestration in Myocardial Aggregates. J. Am. Coll. Cardiol. 2015, 65, 1199–1214. [Google Scholar] [CrossRef] [Green Version]
- Gianni, D.; Li, A.; Tesco, G.; McKay, K.M.; Moore, J.; Raygor, K.; Rota, M.; Gwathmey, J.K.; Dec, G.W.; Aretz, T.; et al. Protein Aggregates and Novel Presenilin Gene Variants in Idiopathic Dilated Cardiomyopathy. Circulation 2010, 121, 1216–1226. [Google Scholar] [CrossRef] [Green Version]
- Tublin, J.M.; Adelstein, J.M.; del Monte, F.; Combs, C.K.; Wold, L.E. Getting to the Heart of Alzheimer Disease. Circ. Res. 2019, 124, 142–149. [Google Scholar] [CrossRef]
- Berridge, M.J. Neuronal calcium signaling. Neuron 1998, 21, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Zampese, E.; Pizzo, P. Intracellular organelles in the saga of Ca2+ homeostasis: Different molecules for different purposes? Cell. Mol. Life Sci. 2012, 69, 1077–1104. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. Calcium Signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santulli, G.; Nakashima, R.; Yuan, Q.; Marks, A.R. Intracellular calcium release channels: An update. J. Physiol. 2017, 595, 3041–3051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prole, D.L.; Taylor, C.W. Structure and function of ip3 receptors. Cold Spring Harb. Perspect. Biol. 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Pinton, P.; Pozzan, T.; Rizzuto, R. The Golgi apparatus is an inositol 1,4,5-trisphosphate-sensitive Ca2+ store, with functional properties distinct from those of the endoplasmic reticulum. EMBO J. 1998, 17, 5298–5308. [Google Scholar] [CrossRef] [Green Version]
- Pizzo, P.; Lissandron, V.; Capitanio, P.; Pozzan, T. Ca2+ signalling in the Golgi apparatus. Cell Calcium 2011, 50, 184–192. [Google Scholar] [CrossRef]
- Morgan, A.J. Ca2+ dialogue between acidic vesicles and ER. Biochem. Soc. Trans. 2016, 44, 546–553. [Google Scholar] [CrossRef]
- Putney, J.W.; Steinckwich-Besançon, N.; Numaga-Tomita, T.; Davis, F.M.; Desai, P.N.; D’Agostin, D.M.; Wu, S.; Bird, G.S. The functions of store-operated calcium channels. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 900–906. [Google Scholar] [CrossRef]
- Kar, P.; Bakowski, D.; Di Capite, J.; Nelson, C.; Parekh, A.B. Different agonists recruit different stromal interaction molecule proteins to support cytoplasmic Ca2+ oscillations and gene expression. Proc. Natl. Acad. Sci. USA 2012, 109, 6969–6974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chemaly, E.R.; Troncone, L.; Lebeche, D. SERCA control of cell death and survival. Cell Calcium 2018, 69, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Baba-Aissa, F.; Raeymaekers, L.; Wuytack, F.; Dode, L.; Casteels, R. Distribution and isoform diversity of the organellar Ca2+ pumps in the brain. Mol. Chem. Neuropathol. 1998, 33, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Lissandron, V.; Podini, P.; Pizzo, P.; Pozzan, T. Unique characteristics of Ca2+ homeostasis of the trans-Golgi compartment. Proc. Natl. Acad. Sci. USA 2010, 107, 9198–9203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, A.K.C.; Capitanio, P.; Lissandron, V.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Heterogeneity of Ca2+ handling among and within Golgi compartments. J. Mol. Cell Biol. 2013, 5, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.C.; Zheng, Q.; Tan, H.; Zhang, B.; Li, X.; Yang, Y.; Yu, J.; Liu, Y.; Chai, H.; Wang, X.; et al. TMCO1 is an ER Ca2+ load-activated Ca2+ channel. Cell 2016, 165, 1454–1466. [Google Scholar] [CrossRef] [Green Version]
- Stafford, N.; Wilson, C.; Oceandy, D.; Neyses, L.; Cartwright, E.J. The plasma membrane calcium ATPases and their role as major new players in human disease. Physiol. Rev. 2017, 97, 1089–1125. [Google Scholar] [CrossRef] [Green Version]
- DiPolo, R.; Beaugé, L. Sodium/calcium exchanger: Influence of metabolic regulation on ion carrier interactions. Physiol. Rev. 2006, 86, 155–203. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; Pozzan, T. Microdomains of intracellular Ca2+: Molecular determinants and functional consequences. Physiol. Rev. 2006, 86, 369–408. [Google Scholar] [CrossRef]
- Schwaller, B. The continuing disappearance of “pure” Ca2+ buffers. Cell. Mol. Life Sci. 2009, 66, 275–300. [Google Scholar] [CrossRef]
- Giorgi, C.; Danese, A.; Missiroli, S.; Patergnani, S.; Pinton, P. Calcium Dynamics as a Machine for Decoding Signals. Trends Cell Biol. 2018, 28, 258–273. [Google Scholar] [CrossRef] [PubMed]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabó, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Zhao, L.; Clapham, D.E. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 2009, 326, 144–147. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R. Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca2+ Responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef]
- Filadi, R.; Greotti, E.; Pizzo, P. Highlighting the endoplasmic reticulum-mitochondria connection: Focus on Mitofusin 2. Pharmacol. Res. 2018, 128, 42–51. [Google Scholar] [CrossRef]
- Pinton, P. Mitochondria-associated membranes (MAMs) and pathologies editorial. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Khachaturian, Z.S.; Radebaugh, T.S. Alzheimer disease: Where are we now? Where are we going? Alzheimer Dis Assoc Disord 1988, 12 (Suppl. 3), S24–S28. [Google Scholar]
- Peterson, C.; Ratan, R.R.; Shelanski, M.L.; Goldman, J.E. Altered response of fibroblasts from aged and Alzheimer donors to drugs that elevate cytosolic free calcium. Neurobiol. Aging 1988, 9, 261–266. [Google Scholar] [CrossRef]
- Khachaturian, Z.S. Calcium Hypothesis of Alzheimer’s Disease and Brain Aging. Ann. N. Y. Acad. Sci. 2006, 747, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Agostini, M.; Fasolato, C. When, where and how? Focus on neuronal calcium dysfunctions in Alzheimer’s Disease. Cell Calcium 2016, 60, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A. Calcium Signaling During Brain Aging and Its Influence on the Hippocampal Synaptic Plasticity. In Advances in Experimental Medicine and Biology; Springer: Basel, Switzerland, 2020; Volume 1131, pp. 985–1012. [Google Scholar]
- Emilsson, L.; Saetre, P.; Jazin, E. Alzheimer’s disease: MRNA expression profiles of multiple patients show alterations of genes involved with calcium signaling. Neurobiol. Dis. 2006, 21, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Ito, E.; Oka, K.; Etcheberrigaray, R.; Nelson, T.J.; McPhie, D.L.; Tofel-Grehl, B.; Gibson, G.E.; Alkon, D.L. Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 534–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etcheberrigaray, R.; Hirashima, N.; Nee, L.; Prince, J.; Govoni, S.; Racchi, M.; Tanzi, R.E.; Alkon, D.L. Calcium responses in fibroblasts from asymptomatic members of Alzheimer’s disease families. Neurobiol. Dis. 1998, 5, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Giacomello, M.; Barbiero, L.; Zatti, G.; Squitti, R.; Binetti, G.; Pozzan, T.; Fasolato, C.; Ghidoni, R.; Pizzo, P. Reduction of Ca2+ stores and capacitative Ca2+ entry is associated with the familial Alzheimer’s disease presenilin-2 T122R mutation and anticipates the onset of dementia. Neurobiol. Dis. 2005, 18, 638–648. [Google Scholar] [CrossRef]
- LaFerla, F.M. Calcium dyshomeostasis and intracellular signalling in alzheimer’s disease. Nat. Rev. Neurosci. 2002, 3, 862–872. [Google Scholar] [CrossRef]
- Leissring, M.A.; Paul, B.A.; Parker, I.; Cotman, C.W.; Laferla, F.M. Alzheimer’s presenilin-1 mutation potentiates inositol 1,4,5- trisphosphate-mediated calcium signaling in Xenopus oocytes. J. Neurochem. 1999, 72, 1061–1068. [Google Scholar] [CrossRef]
- Chan, S.L.; Mayne, M.; Holden, C.P.; Geiger, J.D.; Mattson, M.P. Presenilin-1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons. J. Biol. Chem. 2000, 275, 18195–18200. [Google Scholar] [CrossRef] [Green Version]
- Leissring, M.A.; Akbari, Y.; Fanger, C.M.; Cahalan, M.D.; Mattson, M.P.; LaFerla, F.M. Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. J. Cell Biol. 2000, 149, 793–797. [Google Scholar] [CrossRef] [Green Version]
- Smith, I.F.; Boyle, J.P.; Vaughan, P.F.T.; Pearson, H.A.; Peers, C. Effects of chronic hypoxia on Ca2+ stores and capacitative Ca2+ entry in human neuroblastoma (SH-SY5Y) cells. J. Neurochem. 2001, 79, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Stutzmann, G.E.; Caccamo, A.; LaFerla, F.M.; Parker, I. Dysregulated IP3 Signaling in Cortical Neurons of Knock-In Mice Expressing an Alzheimer’s-Linked Mutation in Presenilin1 Results in Exaggerated Ca2+ Signals and Altered Membrane Excitability. J. Neurosci. 2004, 24, 508–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stutzmann, G.E.; Smith, I.; Caccamo, A.; Oddo, S.; LaFerla, F.M.; Parker, I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer’s disease mice. J. Neurosci. 2006, 26, 5180–5189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayrapetyan, V.; Rybalchenko, V.; Rybalchenko, N.; Koulen, P. The N-terminus of presenilin-2 increases single channel activity of brain ryanodine receptors through direct protein-protein interaction. Cell Calcium 2008, 44, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Rybalchenko, V.; Hwang, S.Y.; Rybalchenko, N.; Koulen, P. The cytosolic N-terminus of presenilin-1 potentiates mouse ryanodine receptor single channel activity. Int. J. Biochem. Cell Biol. 2008, 40, 84–97. [Google Scholar] [CrossRef]
- Chakroborty, S.; Goussakov, I.; Miller, M.B.; Stutzmann, G.E. Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J. Neurosci. 2009, 29, 9458–9470. [Google Scholar] [CrossRef]
- Leissring, M.A.; Parker, I.; LaFerla, F.M. Presenilin-2 mutations modulate amplitude and kinetics of inositol 1,4,5-trisphosphate-mediated calcium signals. J. Biol. Chem. 1999, 274, 32535–32538. [Google Scholar] [CrossRef] [Green Version]
- Schneider, I.; Reversé, D.; Dewachter, I.; Ris, L.; Caluwaerts, N.; Kuipér, C.; Gilis, M.; Geerts, H.; Kretzschmar, H.; Godaux, E.; et al. Mutant Presenilins Disturb Neuronal Calcium Homeostasis in the Brain of Transgenic Mice, Decreasing the Threshold for Excitotoxicity and Facilitating Long-term Potentiation. J. Biol. Chem. 2001, 276, 11539–11544. [Google Scholar] [CrossRef] [Green Version]
- Cai, D.; Netzer, W.J.; Zhong, M.; Lin, Y.; Du, G.; Frohman, M.; Foster, D.A.; Sisodia, S.S.; Xu, H.; Gorelick, F.S.; et al. Presenilin-1 uses phospholipase D1 as a negative regulator of β-amyloid formation. Proc. Natl. Acad. Sci. USA 2006, 103, 1941–1946. [Google Scholar] [CrossRef] [Green Version]
- Tu, H.; Nelson, O.; Bezprozvanny, A.; Wang, Z.; Lee, S.F.; Hao, Y.H.; Serneels, L.; De Strooper, B.; Yu, G.; Bezprozvanny, I. Presenilins Form ER Ca2+ Leak Channels, a Function Disrupted by Familial Alzheimer’s Disease-Linked Mutations. Cell 2006, 126, 981–993. [Google Scholar] [CrossRef] [Green Version]
- Nelson, O.; Supnet, C.; Liu, H.; Bezprozvanny, I. Familial Alzheimer’s disease mutations in presenilins: Effects on endoplasmic reticulum calcium homeostasis and correlation with clinical phenotypes. J. Alzheimer’s Dis. 2010, 21, 781–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, O.; Supnet, C.; Tolia, A.; Horre, K.; De Strooper, B.; Bezprozvanny, I. Mutagenesis mapping of the presenilin 1 calcium leak conductance pore. J. Biol. Chem. 2011, 286, 22339–22347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, K.H.; Shineman, D.; Müller, M.; Cárdenas, C.; Mei, L.; Yang, J.; Tomita, T.; Iwatsubo, T.; Lee, V.M.Y.; Foskett, J.K. Mechanism of Ca2+ Disruption in Alzheimer’s Disease by Presenilin Regulation of InsP3 Receptor Channel Gating. Neuron 2008, 58, 871–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, K.H.; Mei, L.; Mak, D.O.D.; Hayashi, I.; Iwatsubo, T.; Kang, D.E.; Foskett, J.K. Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer’s disease-linked presenilin mutants in human cells and mouse neurons. Sci. Signal. 2010, 3. [Google Scholar] [CrossRef] [Green Version]
- Zatti, G.; Burgo, A.; Giacomello, M.; Barbiero, L.; Ghidoni, R.; Sinigaglia, G.; Florean, C.; Bagnoli, S.; Binetti, G.; Sorbi, S.; et al. Presenilin mutations linked to familial Alzheimer’s disease reduce endoplasmic reticulum and Golgi apparatus calcium levels. Cell Calcium 2006, 39, 539–550. [Google Scholar] [CrossRef] [PubMed]
- McCombs, J.E.; Gibson, E.A.; Palmer, A.E. Using a genetically targeted sensor to investigate the role of presenilin-1 in ER Ca2+ levels and dynamics. Mol. Biosyst. 2010, 6, 1640–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shilling, D.; Mak, D.O.D.; Kang, D.E.; Foskett, J.K. Lack of evidence for presenilins as endoplasmic reticulum Ca2+ leak channels. J. Biol. Chem. 2012, 287, 10933–10944. [Google Scholar] [CrossRef] [Green Version]
- Zatti, G.; Ghidoni, R.; Barbiero, L.; Binetti, G.; Pozzan, T.; Fasolato, C.; Pizzo, P. The presenilin 2 M239I mutation associated with familial Alzheimer’s disease reduces Ca2+ release from intracellular stores. Neurobiol. Dis. 2004, 15, 269–278. [Google Scholar] [CrossRef]
- Zampese, E.; Fasolato, C.; Kipanyula, M.J.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. USA 2011, 108, 2777–2782. [Google Scholar] [CrossRef] [Green Version]
- Greotti, E.; Capitanio, P.; Wong, A.; Pozzan, T.; Pizzo, P.; Pendin, D. Familial Alzheimer’s disease-linked presenilin mutants and intracellular Ca2+ handling: A single-organelle, FRET-based analysis. Cell Calcium 2019, 79, 44–56. [Google Scholar] [CrossRef]
- Pack-Chung, E.; Meyers, M.B.; Pettingell, W.P.; Moir, R.D.; Brownawell, A.M.; Cheng, I.; Tanzi, R.E.; Kim, T.W. Presenilin 2 interacts with sorcin, a modulator of the ryanodine receptor. J. Biol. Chem. 2000, 275, 14440–14445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buxbaum, J.D.; Choi, E.K.; Luo, Y.; Lilliehook, C.; Crowley, A.C.; Merriam, D.E.; Wasco, W. Calsenilin: A calcium-binding protein that interacts with the presenilins and regulates the levels of a presenilin fragment. Nat. Med. 1998, 4, 1177–1181. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Christakos, S.; Robinson, N.; Mattson, M.P. Calbindin D28K blocks the proapoptotic actions of mutant presenilin 1: Reduced oxidative stress and preserved mitochondrial function. Proc. Natl. Acad. Sci. USA 1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, K.N.; Demuro, A.; Akbari, Y.; Hitt, B.D.; Smith, I.F.; Parker, I.; LaFerla, F.M. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid β production. J. Cell Biol. 2008, 181, 1107–1116. [Google Scholar] [CrossRef] [Green Version]
- Filadi, R.; Theurey, P.; Pizzo, P. The endoplasmic reticulum-mitochondria coupling in health and disease: Molecules, functions and significance. Cell Calcium 2017, 62, 1–15. [Google Scholar] [CrossRef]
- Kipanyula, M.J.; Contreras, L.; Zampese, E.; Lazzari, C.; Wong, A.K.C.; Pizzo, P.; Fasolato, C.; Pozzan, T. Ca2+ dysregulation in neurons from transgenic mice expressing mutant presenilin 2. Aging Cell 2012, 11, 885–893. [Google Scholar] [CrossRef] [Green Version]
- Hedskog, L.; Pinho, C.M.; Filadi, R.; Rönnbäck, A.; Hertwig, L.; Wiehager, B.; Larssen, P.; Gellhaar, S.; Sandebring, A.; Westerlund, M.; et al. Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc. Natl. Acad. Sci. USA 2013, 110, 7916–7921. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhu, X. Endoplasmic reticulum-mitochondria tethering in neurodegenerative diseases. Transl. Neurodegener. 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Brunello, L.; Zampese, E.; Florean, C.; Pozzan, T.; Pizzo, P.; Fasolato, C. Presenilin-2 dampens intracellular Ca2+ stores by increasing Ca2+ leakage and reducing Ca2+ uptake. J. Cell. Mol. Med. 2009, 13, 3358–3369. [Google Scholar] [CrossRef] [Green Version]
- Pendin, D.; Fasolato, C.; Basso, E.; Filadi, R.; Greotti, E.; Galla, L.; Gomiero, C.; Leparulo, A.; Redolfi, N.; Scremin, E.; et al. Familial Alzheimer’s disease presenilin-2 mutants affect Ca2+ homeostasis and brain network excitability. Aging Clin. Exp. Res. 2019. [Google Scholar] [CrossRef]
- Lee, S.Y.; Hwang, D.Y.; Kim, Y.K.; Lee, J.W.; Shin, I.C.; Oh, K.W.; Lee, M.K.; Lim, J.S.; Yoon, D.Y.; Hwang, S.J.; et al. PS2 mutation increases neuronal cell vulnerability to neurotoxicants through activation of caspase-3 by enhancing of ryanodine receptor-mediated calcium release. FASEB J. 2006, 20, 151–153. [Google Scholar] [CrossRef]
- Stutzmann, G.E.; Smith, I.; Caccamo, A.; Oddo, S.; Laferla, F.M.; Parker, I. Enhanced Ryanodine-Mediated Calcium Release in Mutant PS1-Expressing Alzheimer’s Mouse Models. Ann. N. Y. Acad. Sci. 2007, 1097, 265–277. [Google Scholar] [CrossRef]
- Tong, B.C.K.; Lee, C.S.K.; Cheng, W.H.; Lai, K.O.; Foskett, J.K.; Cheung, K.H. Familial Alzheimer’s disease-associated presenilin 1 mutants promote γ-secretase cleavage of STIM1 to impair store-operated Ca2+ entry. Sci. Signal. 2016, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, I.F.; Boyle, J.P.; Vaughan, P.F.T.; Pearson, H.A.; Cowburn, R.F.; Peers, C.S. Ca2+ stores and capacitative Ca2+ entry in human neuroblastoma (SH-SY5Y) cells expressing a familial Alzheimer’s disease presenilin-1 mutation. Brain Res. 2002, 949, 105–111. [Google Scholar] [CrossRef]
- Pascual-Caro, C.; Berrocal, M.; Lopez-Guerrero, A.M.; Alvarez-Barrientos, A.; Pozo-Guisado, E.; Gutierrez-Merino, C.; Mata, A.M.; Martin-Romero, F.J. STIM1 deficiency is linked to Alzheimer’s disease and triggers cell death in SH-SY5Y cells by upregulation of L-type voltage-operated Ca2+ entry. J. Mol. Med. 2018, 96, 1061–1079. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Chauhan, A.; Sukumaran, P.; Sharma, J.; Singh, B.B.; Mishra, B.B. Inhibition of store-operated calcium entry in microglia by helminth factors: Implications for immune suppression in neurocysticercosis. J. Neuroinflammation 2014, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Wu, L.; Pchitskaya, E.; Zakharova, O.; Saito, T.; Saido, T.; Bezprozvanny, I. Neuronal store-operated calcium entry and mushroom spine loss in amyloid precursor protein knock-in mouse model of Alzheimer’s disease. J. Neurosci. 2015, 35, 13275–13286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbari, Y.; Hitt, B.D.; Murphy, M.P.; Dagher, N.N.; Tseng, B.P.; Green, K.N.; Golde, T.E.; LaFerla, F.M. Presenilin regulates capacitative calcium entry dependently and independently of γ-secretase activity. Biochem. Biophys. Res. Commun. 2004, 322, 1145–1152. [Google Scholar] [CrossRef]
- Shideman, C.R.; Reinardy, J.L.; Thayer, S.A. γ-Secretase activity modulates store-operated Ca2+ entry into rat sensory neurons. Neurosci. Lett. 2009, 451, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Greotti, E.; Wong, A.; Pozzan, T.; Pendin, D.; Pizzo, P. Characterization of the ER-targeted low affinity Ca2+ probe D4ER. Sensors 2016, 16, 1419. [Google Scholar] [CrossRef] [Green Version]
- Fedeli, C.; Filadi, R.; Rossi, A.; Mammucari, C.; Pizzo, P. PSEN2 (presenilin 2) mutants linked to familial Alzheimer disease impair autophagy by altering Ca2+ homeostasis. Autophagy 2019, 15, 2044–2062. [Google Scholar] [CrossRef] [PubMed]
- Filadi, R.; Pizzo, P. Defective autophagy and Alzheimer’s disease: Is calcium the key? Neural Regen. Res. 2019, 14, 2081–2082. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Biophys. Acta - Mol. Cell Res. 2019, 1866, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Rigotto, G.; Valente, G.; Giorgio, V.; Basso, E.; Filadi, R.; Pizzo, P. Defective mitochondrial pyruvate flux affects cell bioenergetics in Alzheimer’s disease related models. Cell Rep. 2020. In press. [Google Scholar]
- Theurey, P.; Connolly, N.M.C.; Fortunati, I.; Basso, E.; Lauwen, S.; Ferrante, C.; Moreira Pinho, C.; Joselin, A.; Gioran, A.; Bano, D.; et al. Systems biology identifies preserved integrity but impaired metabolism of mitochondria due to a glycolytic defect in Alzheimer’s disease neurons. Aging Cell 2019, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, R.; Agostini, M.; Murana, E.; Mahmud, M.; Scremin, E.; Rubega, M.; Sparacino, G.; Vassanelli, S.; Fasolato, C. Early hippocampal hyperexcitability in PS2APP mice: Role of mutant PS2 and APP. Neurobiol. Aging 2017, 50, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Leparulo, A.; Mahmud, M.; Scremin, E.; Pozzan, T.; Vassanelli, S.; Fasolato, C. Dampened Slow Oscillation Connectivity Anticipates Amyloid Deposition in the PS2APP Mouse Model of Alzheimer’s Disease. Cells 2019, 9, 54. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Aron, L.; Zullo, J.; Pan, Y.; Kim, H.; Chen, Y.; Yang, T.H.; Kim, H.M.; Drake, D.; Liu, X.S.; et al. REST and stress resistance in ageing and Alzheimer’s disease. Nature 2014, 507, 448–454. [Google Scholar] [CrossRef] [Green Version]
- Pinton, P.; Romagnoli, A.; Rizzuto, R.; Giorgi, C. Ca2+ Signaling, Mitochondria and Cell Death. Curr. Mol. Med. 2008, 8, 119–130. [Google Scholar] [CrossRef]
- Tsien, R.Y. New Calcium Indicators and Buffers with High Selectivity Against Magnesium and Protons: Design, Synthesis, and Properties of Prototype Structures. Biochemistry 1980, 19, 2396–2404. [Google Scholar] [CrossRef]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [PubMed]
- Tsien, R.Y. A non-disruptive technique for loading calcium buffers and indicators into cells. Nature 1981, 290, 527–528. [Google Scholar] [CrossRef] [PubMed]
- Pendin, D.; Norante, R.; De Nadai, A.; Gherardi, G.; Vajente, N.; Basso, E.; Kaludercic, N.; Mammucari, C.; Paradisi, C.; Pozzan, T.; et al. A Synthetic Fluorescent Mitochondria-Targeted Sensor for Ratiometric Imaging of Calcium in Live Cells. Angew. Chemie Int. Ed. 2019, 58, 9917–9922. [Google Scholar] [CrossRef] [PubMed]
- Shimomura, O.; Johnson, F.H.; Saiga, Y. Further data on the bioluminescent protein, aequorin. J. Cell. Comp. Physiol. 1963, 62, 1–8. [Google Scholar] [CrossRef]
- Tsien, R.Y. The Green Fluorescent Protein. Annu. Rev. Biochem. 1998, 67, 509–544. [Google Scholar] [CrossRef]
- Shimomura, O.; Johnson, F.H.; Saiga, Y. Extraction, purification and properties of aequorin, a bioluminescent. J. Cell. Comp. Physiol. 1962, 59, 223–239. [Google Scholar] [CrossRef]
- Pendin, D.; Greotti, E.; Filadi, R.; Pozzan, T. Spying on organelle Ca2+ in living cells: The mitochondrial point of view. J. Endocrinol. Invest. 2015, 38, 39–45. [Google Scholar] [CrossRef]
- Bonora, M.; Giorgi, C.; Bononi, A.; Marchi, S.; Patergnani, S.; Rimessi, A.; Rizzuto, R.; Pinton, P. Subcellular calcium measurements in mammalian cells using jellyfish photoprotein aequorin-based probes. Nat. Protoc. 2013, 8, 2105–2118. [Google Scholar] [CrossRef]
- Kendall, J.M.; Sala-Newby, G.; Ghalaut, V.; Dormer, R.L.; Cambell, A.K. Engineering the Ca(2+)-activated photoprotein aequorin with reduced affinity for calcium. Biochem. Biophys. Res. Commun. 1992, 187, 1091–1097. [Google Scholar] [CrossRef]
- de la Fuente, S.; Fonteriz, R.I.; de la Cruz, P.J.; Montero, M.; Alvarez, J. Mitochondrial free [Ca2+] dynamics measured with a novel low-Ca2+ affinity aequorin probe. Biochem. J. 2012, 445, 371–376. [Google Scholar] [CrossRef] [Green Version]
- De la Fuente, S.; Fonteriz, R.I.; Montero, M.; Alvarez, J. Ca2+ homeostasis in the endoplasmic reticulum measured with a new low-Ca2+-affinity targeted aequorin. Cell Calcium 2013, 54, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Nakai, J.; Ohkura, M.; Imoto, K. A high signal-to-noise Ca2+ probe composed of a single green fluorescent protein. Nat. Biotechnol. 2001, 19, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.J.; Luo, A.F.; Hu, L.Y.; Wang, D.C.; Shao, F. Structural basis of the ultrasensitive calcium indicator GCaMP6. Sci. China Life Sci. 2014, 57, 269–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Jackson Laboratory. Available online: https://www.jax.org (accessed on 17 December 2019).
- Dana, H.; Chen, T.W.; Hu, A.; Shields, B.C.; Guo, C.; Looger, L.L.; Kim, D.S.; Svoboda, K. Thy1-GCaMP6 transgenic mice for neuronal population imaging in vivo. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Dana, H.; Sun, Y.; Mohar, B.; Hulse, B.K.; Kerlin, A.M.; Hasseman, J.P.; Tsegaye, G.; Tsang, A.; Wong, A.; Patel, R.; et al. High-performance calcium sensors for imaging activity in neuronal populations and microcompartments. Nat. Methods 2019, 16, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Steinmetz, N.A.; Buetfering, C.; Lecoq, J.; Lee, C.R.; Peters, A.J.; Jacobs, E.A.K.; Coen, P.; Ollerenshaw, D.R.; Valley, M.T.; De Vries, S.E.J.; et al. Aberrant cortical activity in multiple GCaMP6-expressing transgenic mouse lines. eNeuro 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Liu, N.; He, Y.; Liu, Y.; Ge, L.; Zou, L.; Song, S.; Xiong, W.; Liu, X. Improved calcium sensor GCaMP-X overcomes the calcium channel perturbations induced by the calmodulin in GCaMP. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Miyawaki, A.; Llopis, J.; Heim, R.; Michael McCaffery, J.; Adams, J.A.; Ikura, M.; Tsien, R.Y. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 1997, 388, 882–887. [Google Scholar] [CrossRef]
- Persechini, A.; Lynch, J.A.; Romoser, V.A. Novel fluorescent indicator proteins for monitoring free intracellular Ca2+. Cell Calcium 1997, 22, 209–216. [Google Scholar] [CrossRef]
- Heim, N.; Griesbeck, O. Genetically Encoded Indicators of Cellular Calcium Dynamics Based on Troponin C and Green Fluorescent Protein. J. Biol. Chem. 2004, 279, 14280–14286. [Google Scholar] [CrossRef] [Green Version]
- Palmer, A.E.; Tsien, R.Y. Measuring calcium signaling using genetically targetable fluorescent indicators. Nat. Protoc. 2006, 1, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Galla, L.; Pizzo, P.; Greotti, E. Exploiting Cameleon Probes to Investigate Organelles Ca2+ Handling. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2019; pp. 15–30. [Google Scholar]
- Huang, X.G.; Yee, B.K.; Nag, S.; Chan, S.T.H.; Tang, F. Behavioral and neurochemical characterization of transgenic mice carrying the human presenilin-1 gene with or without the leucine-to-proline mutation at codon 235. Exp. Neurol. 2003, 183, 673–681. [Google Scholar] [CrossRef]
- Sun, X.; Beglopoulos, V.; Mattson, M.P.; Shen, J. Hippocampal spatial memory impairments caused by the familial Alzheimer’s disease-linked presenilin 1 M146V mutation. Neurodegener. Dis. 2005, 2, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.H.; Hof, P.R.; Chen, X.; Gluck, K.; Austin, G.; Younkin, S.G.; Younkin, L.H.; DeGasperi, R.; Gama Sosa, M.A.; Robakis, N.K.; et al. The presenilin-1 familial Alzheimer disease mutant P117L impairs neurogenesis in the hippocampus of adult mice. Exp. Neurol. 2004, 188, 224–237. [Google Scholar] [CrossRef]
- Alzforum Networking for a Cure. Available online: www.alzforum.org/research-models/alzheimers-disease (accessed on 17 December 2019).
- Sasaguri, H.; Nilsson, P.; Hashimoto, S.; Nagata, K.; Saito, T.; De Strooper, B.; Hardy, J.; Vassar, R.; Winblad, B.; Saido, T.C. APP mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017, 36, 2473–2487. [Google Scholar] [CrossRef]
- Webster, S.J.; Bachstetter, A.D.; Nelson, P.T.; Schmitt, F.A.; Van Eldik, L.J. Using mice to model Alzheimer’s dementia: An overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front. Genet. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Richards, J.G.; Higgins, G.A.; Ouagazzal, A.M.; Ozmen, L.; Kew, J.N.C.; Bohrmann, B.; Malherbe, P.; Brockhaus, M.; Loetscher, H.; Czech, C.; et al. PS2APP transgenic mice, coexpressing hPS2mut and hAPPswe, show age-related cognitive deficits associated with discrete brain amyloid deposition and inflammation. J. Neurosci. 2003, 23, 8989–9003. [Google Scholar] [CrossRef] [Green Version]
- Ozmen, L.; Albientz, A.; Czech, C.; Jacobsen, H. Expression of transgenic APP mRNA is the key determinant for beta-amyloid deposition in PS2APP transgenic mice. Neurodegener. Dis. 2008, 6, 29–36. [Google Scholar] [CrossRef]
- Weidensteiner, C.; Metzger, F.; Bruns, A.; Bohrmann, B.; Kuennecke, B.; Von Kienlin, M. Cortical hypoperfusion in the B6.PS2APP mouse model for Alzheimer’s disease: Comprehensive phenotyping of vascular and tissular parameters by MRI. Magn. Reson. Med. 2009, 62, 35–45. [Google Scholar] [CrossRef]
- Poirier, R.; Veltman, I.; Pflimlin, M.C.; Knoflach, F.; Metzger, F. Enhanced dentate gyrus synaptic plasticity but reduced neurogenesis in a mouse model of amyloidosis. Neurobiol. Dis. 2010, 40, 386–393. [Google Scholar] [CrossRef]
- Grueninger, F.; Bohrmann, B.; Czech, C.; Ballard, T.M.; Frey, J.R.; Weidensteiner, C.; von Kienlin, M.; Ozmen, L. Phosphorylation of Tau at S422 is enhanced by Aβ in TauPS2APP triple transgenic mice. Neurobiol. Dis. 2010, 37, 294–306. [Google Scholar] [CrossRef] [PubMed]
- Esquerda-Canals, G.; Montoliu-Gaya, L.; Güell-Bosch, J.; Villegas, S. Mouse Models of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1171–1183. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galla, L.; Redolfi, N.; Pozzan, T.; Pizzo, P.; Greotti, E. Intracellular Calcium Dysregulation by the Alzheimer’s Disease-Linked Protein Presenilin 2. Int. J. Mol. Sci. 2020, 21, 770. https://doi.org/10.3390/ijms21030770
Galla L, Redolfi N, Pozzan T, Pizzo P, Greotti E. Intracellular Calcium Dysregulation by the Alzheimer’s Disease-Linked Protein Presenilin 2. International Journal of Molecular Sciences. 2020; 21(3):770. https://doi.org/10.3390/ijms21030770
Chicago/Turabian StyleGalla, Luisa, Nelly Redolfi, Tullio Pozzan, Paola Pizzo, and Elisa Greotti. 2020. "Intracellular Calcium Dysregulation by the Alzheimer’s Disease-Linked Protein Presenilin 2" International Journal of Molecular Sciences 21, no. 3: 770. https://doi.org/10.3390/ijms21030770
APA StyleGalla, L., Redolfi, N., Pozzan, T., Pizzo, P., & Greotti, E. (2020). Intracellular Calcium Dysregulation by the Alzheimer’s Disease-Linked Protein Presenilin 2. International Journal of Molecular Sciences, 21(3), 770. https://doi.org/10.3390/ijms21030770