Endothelial Response Boosted by Platelet Lysate: The Involvement of Calcium Toolkit




Abstract
:1. Introduction
2. Results
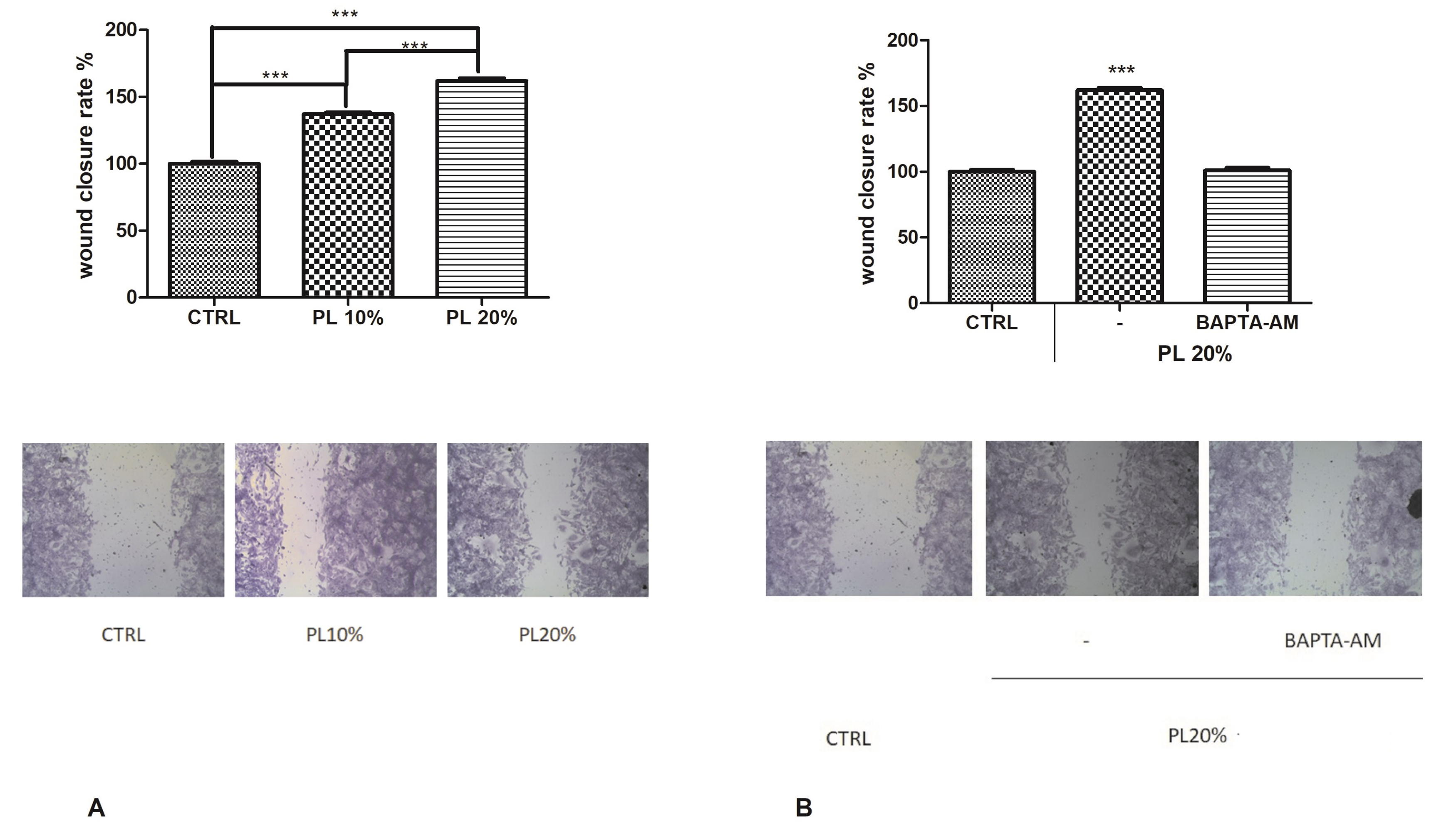
2.1. PL Induces Tissue Regeneration in a Dose-Dependent Manner
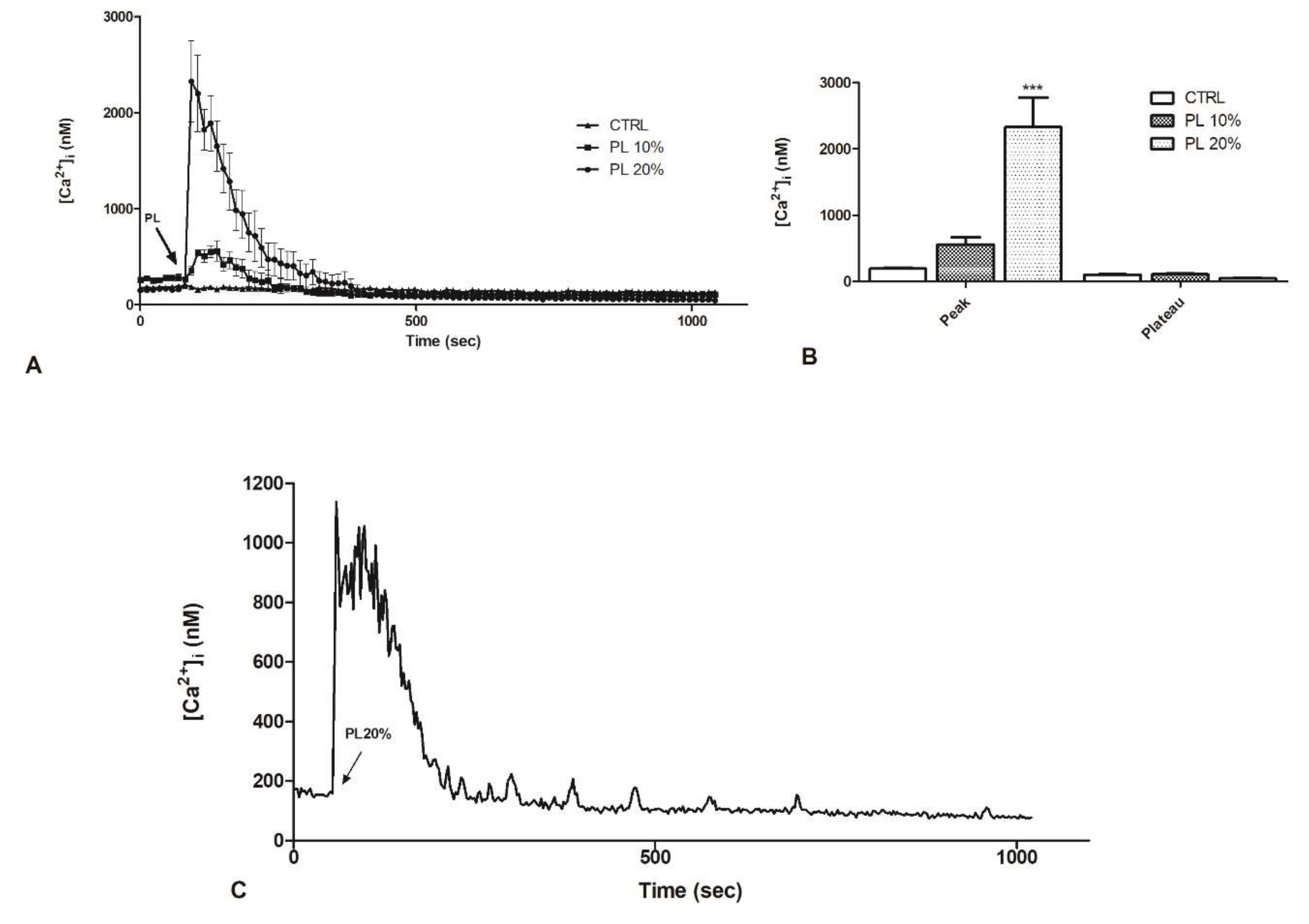
2.2. PL Induces Ca2+ Signals in a Dose-Dependent Manner
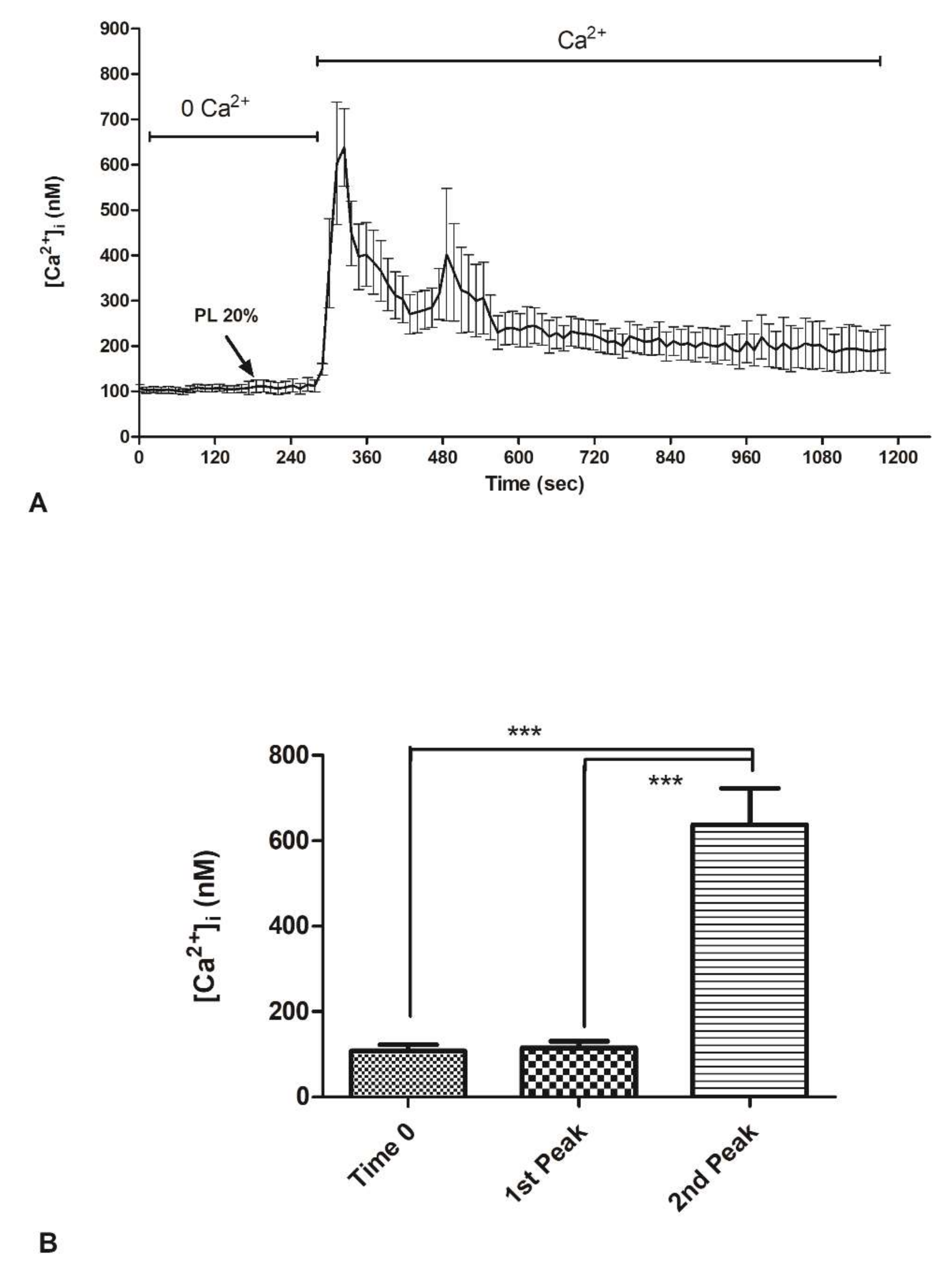
2.3. Contribution of Extracellular Ca2+ to Cytosolic Increase
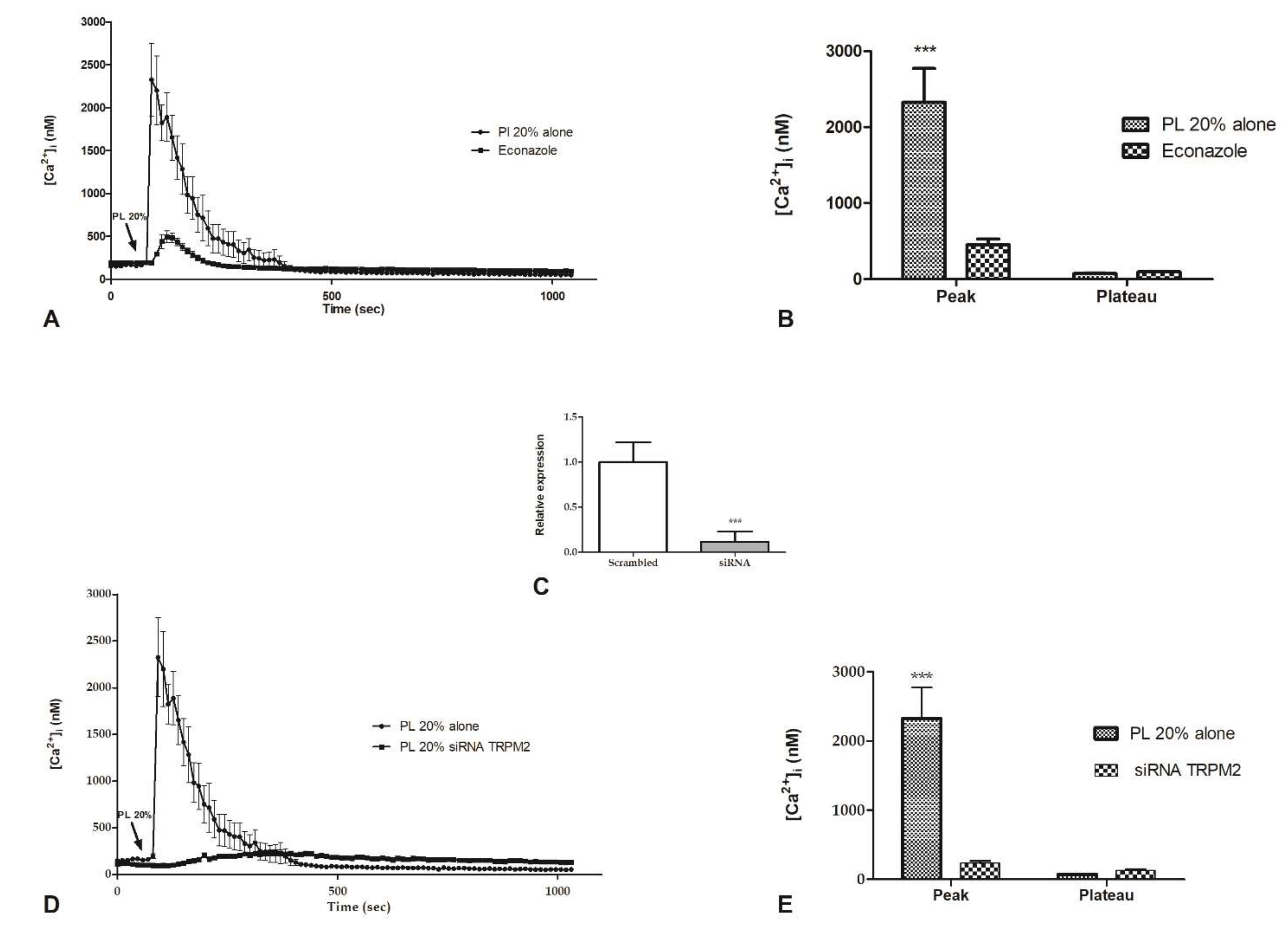
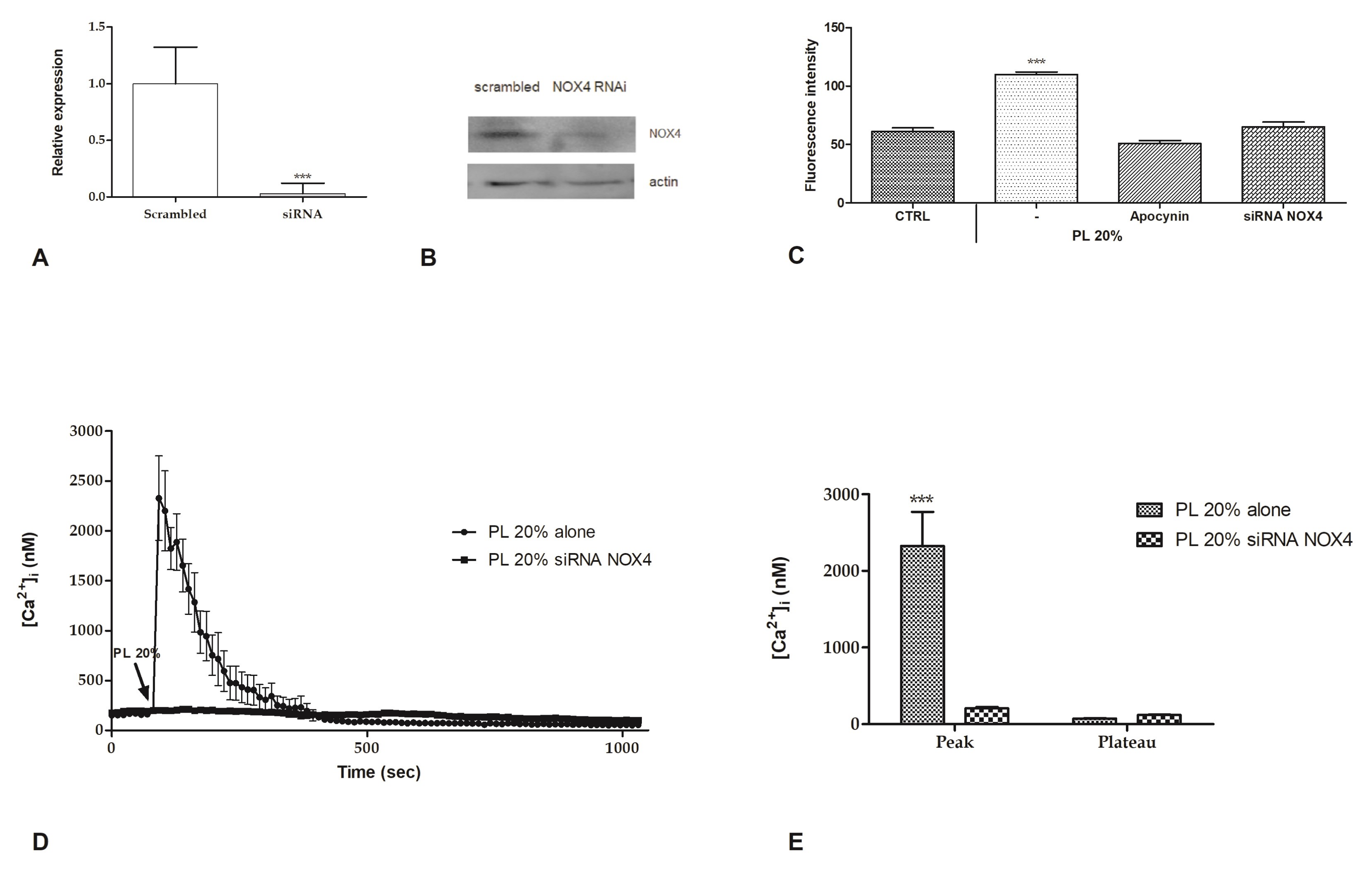
2.4. Intracellular ROS Generation
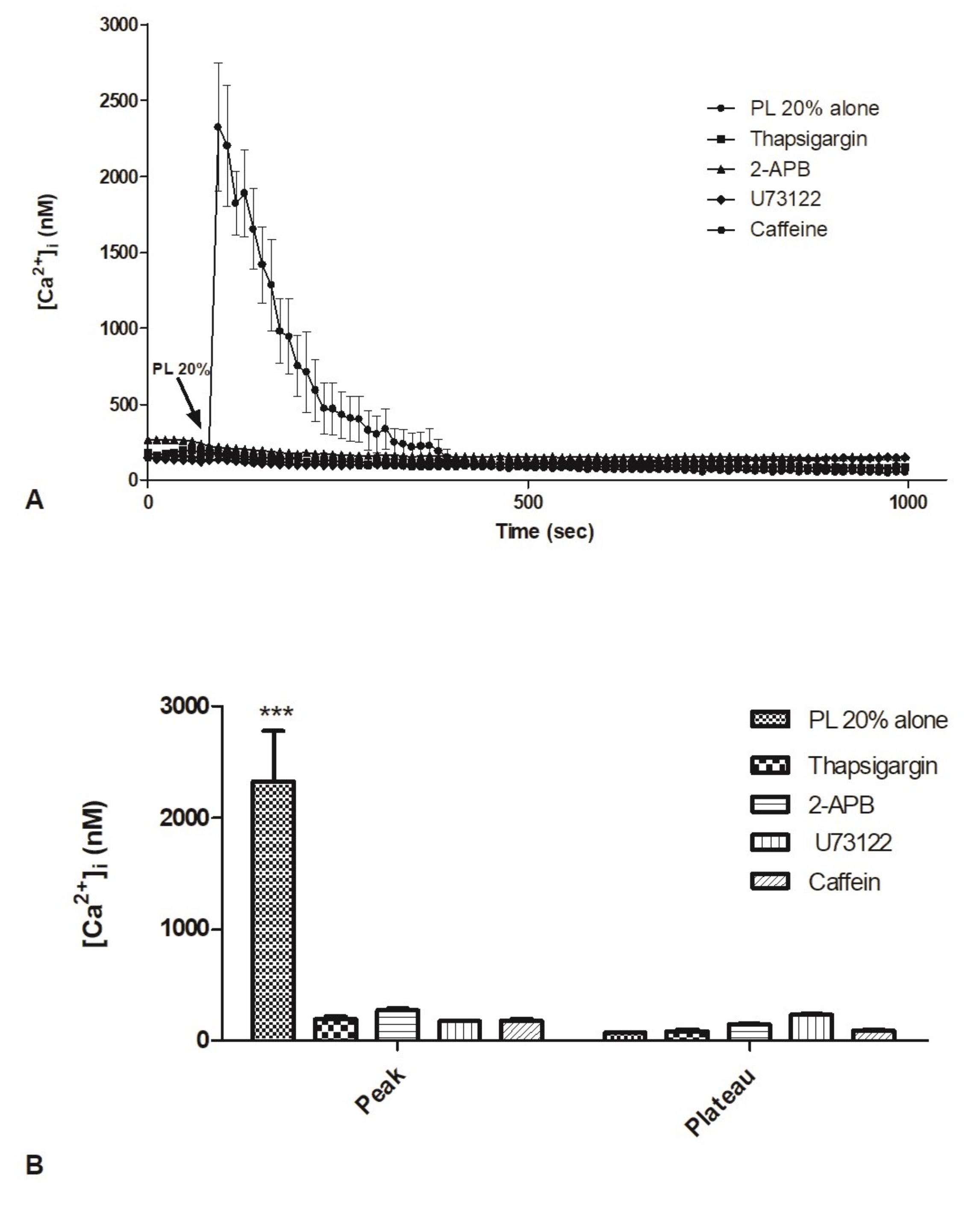
2.5. Contribution of Intracellular Stores to Cytosolic Ca2+ Increase
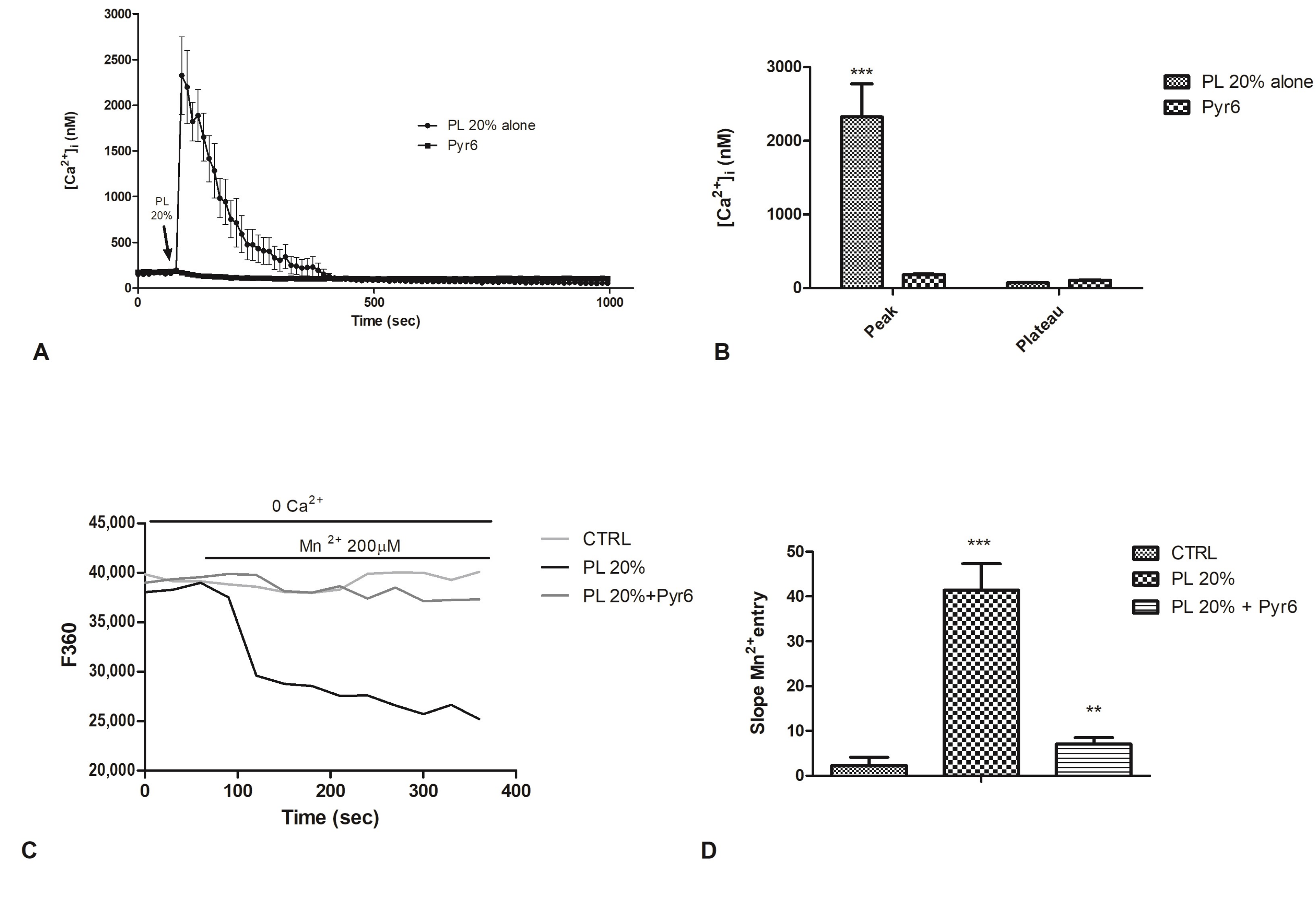
2.6. Involvement of Store-Operated Ca2+ Entry
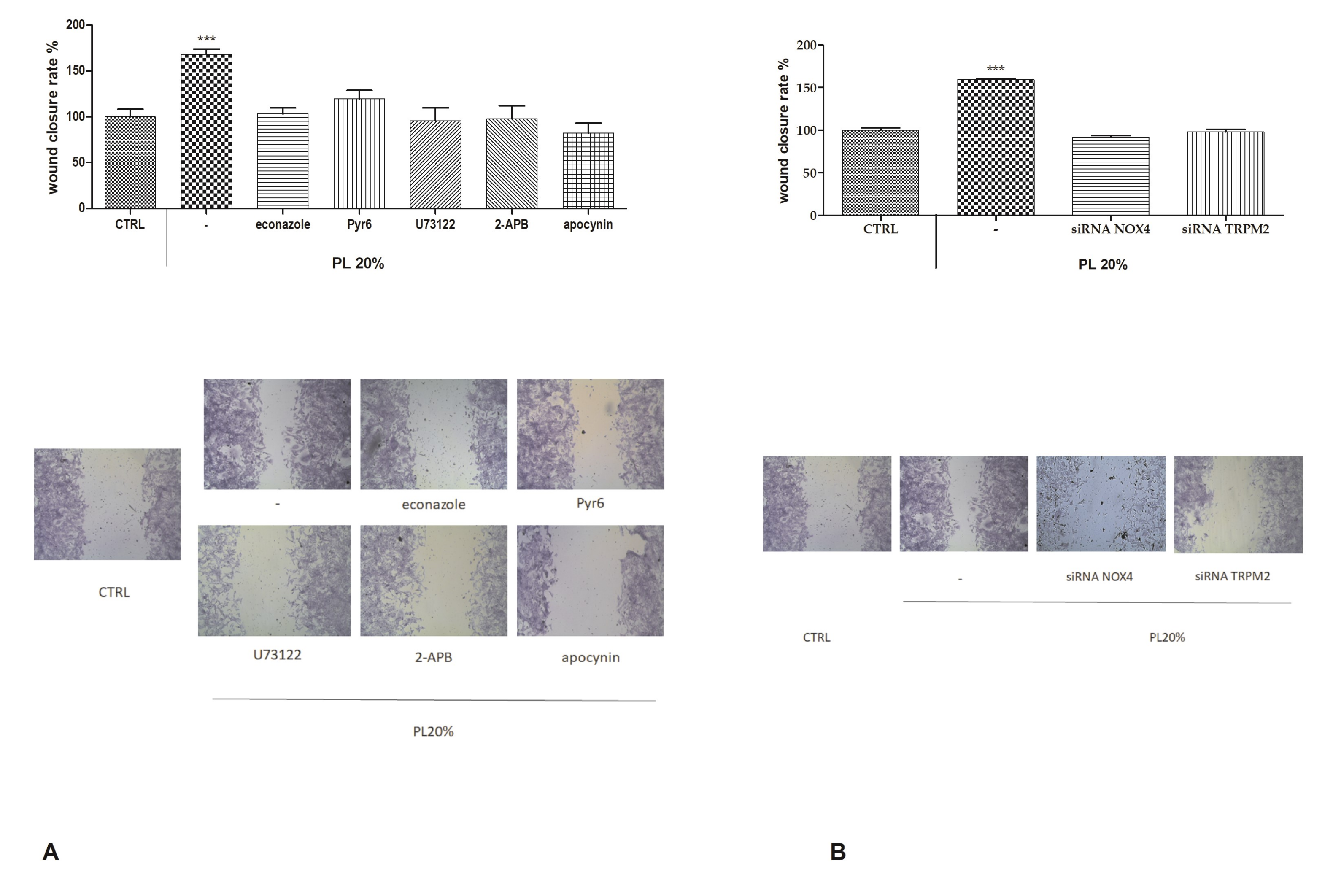
2.7. Functional Role of PL-Induced Tissue Regeneration
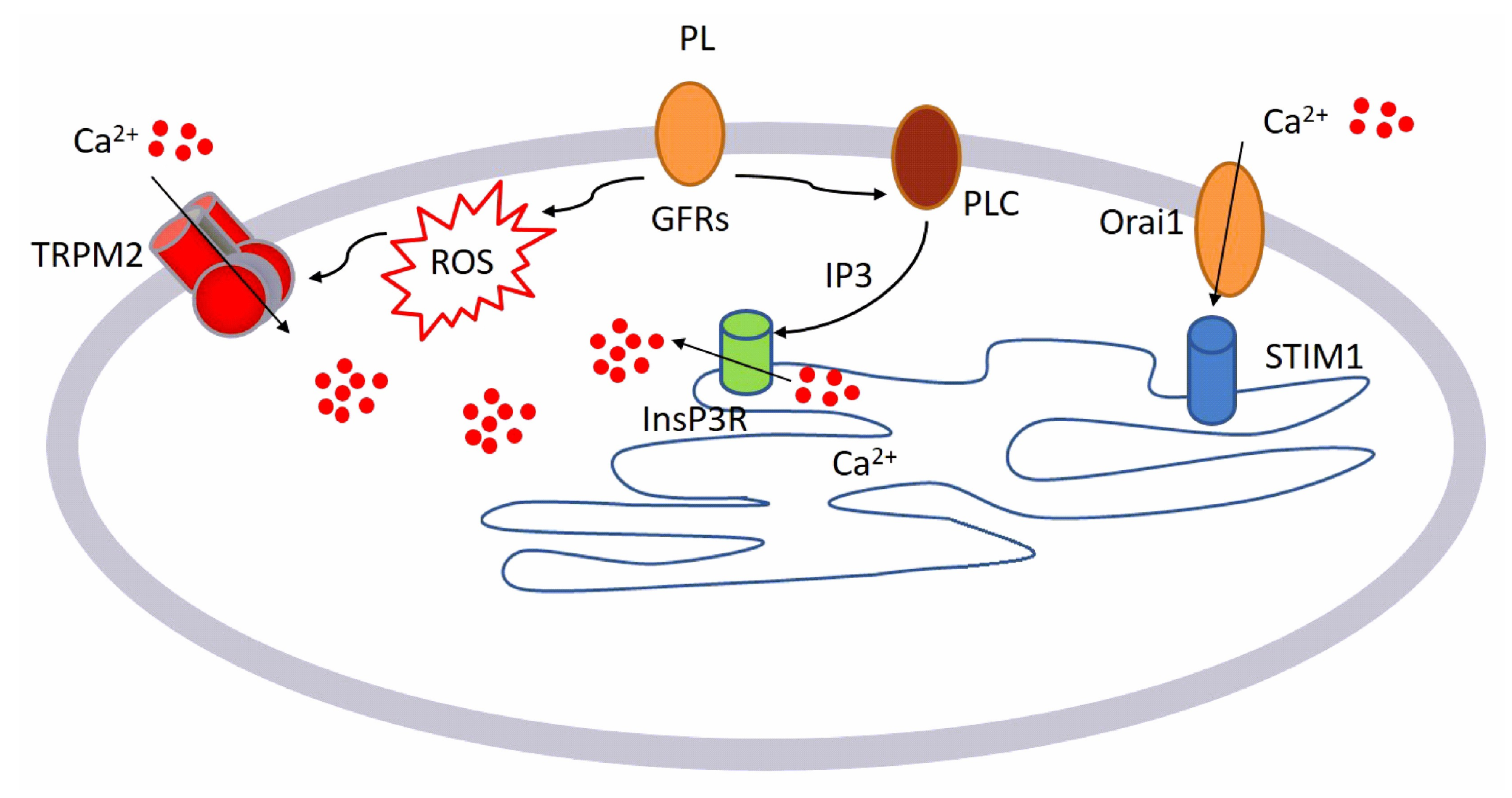
3. Discussion
- GFs contained in PL bind to their specific receptors
- This binding leads to the activation of two different pathways:
- ○
- Generation of intracellular ROS via NOX4
- ○
- Activation of PLCγ, leading to the production of InsP3
- Intracellular ROS activates TRPM2 channel and consequently Ca2+ entry from the extracellular space.
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Platelet Lysate (PL) Preparation
4.3. Scratch Wound Test
4.4. Western Blotting
4.5. Measurements of Intracellular ROS
4.6. Measurements of Free Cytosolic Ca2+ Concentration ([Ca2+]i)
4.7. Quantitative Reverse Transcriptase PCR (qRT-PCR) and RNA Interference (siRNA)
4.8. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Ranzato, E.; Burlando, B. Signaling pathways in wound repair. In Wound Healing: Process, Phases and Promoting; Middleton, J.E., Ed.; Nova Publishers Inc.: Hauppauge, NY, USA, 2011. [Google Scholar]
- Martinotti, S.; Ranzato, E. Dynamic interplay between cell types during wound healing. In Keratinocytes: Structure, Molecular Mechanisms and Role in Immunity; Ranzato, E., Ed.; Nova Publishers Inc.: Hauppauge, NY, USA, 2013. [Google Scholar]
- Mao, A.S.; Mooney, D.J. Regenerative medicine: Current therapies and future directions. Proc. Natl. Acad. Sci. USA 2015, 112, 14452–14459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ioannidou, E. Therapeutic modulation of growth factors and cytokines in regenerative medicine. Curr. Pharm. Des. 2006, 12, 2397–2408. [Google Scholar] [CrossRef] [PubMed]
- Ranzato, E.; Mazzucco, L.; Burlando, B. Platelet Derivatives: A New Horizon in Regenerative Medicine. In Advances in Medicine and Biology; Lv, B., Ed.; Nova Publishers Inc.: Hauppauge, NY, USA, 2010. [Google Scholar]
- Burnouf, T.; Goubran, H.A.; Chen, T.M.; Ou, K.L.; El-Ekiaby, M.; Radosevic, M. Blood-derived biomaterials and platelet growth factors in regenerative medicine. Blood Rev. 2013, 27, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Rauch, C.; Feifel, E.; Amann, E.M.; Spotl, H.P.; Schennach, H.; Pfaller, W.; Gstraunthaler, G. Alternatives to the Use of Fetal Bovine Serum: Human Platelet Lysates as a Serum Substitute in Cell Culture Media. Altex-Altern. Anim. Ex. 2011, 28, 305–316. [Google Scholar]
- Ranzato, E.; Boccafoschi, F.; Mazzucco, L.; Patrone, M.; Burlando, B. Role of ERK1/2 in Platelet Lysate-Driven Endothelial Cell Repair. J. Cell Biochem. 2010, 110, 783–793. [Google Scholar] [CrossRef]
- Castelnovo, L.; Dosquet, C.; Gaudric, A.; Sahel, J.; Hicks, D. Human platelet suspension stimulates porcine retinal glial proliferation and migration in vitro. Invest. Ophth. Vis. Sci. 2000, 41, 601–609. [Google Scholar]
- Soffer, E.; Ouhayoun, J.P.; Anagnostou, F. Fibrin sealants and platelet preparations in bone and periodontal healing. Oral Surg. Oral Med. O. 2003, 95, 521–528. [Google Scholar] [CrossRef]
- Martinotti, S.; Mazzucco, L.; Balbo, V.; Patrone, M.; Mozzati, M.; Ranzato, E.; Burlando, B. Platelet-Rich Plasma Induces Mixed Osteogenic/Osteoclastogenic Phenotype in Osteosarcoma SaOS-2 Cells: Role of TGF-Beta. Curr. Pharm. Biotechno. 2014, 15, 120–126. [Google Scholar] [CrossRef]
- Ranzato, E.; Patrone, M.; Mazzucco, L.; Burlando, B. Platelet lysate stimulates wound repair of HaCaT keratinocytes. Br. J. Dermatol. 2008, 159, 537–545. [Google Scholar] [CrossRef]
- Cipriani, V.; Ranzato, E.; Balbo, V.; Mazzucco, L.; Cavaletto, M.; Patrone, M. Long-term effect of platelet lysate on primary fibroblasts highlighted with a proteomic approach. J. Tissue Eng. Regen. M. 2009, 3, 531–538. [Google Scholar] [CrossRef]
- Borowiec, A.S.; Bidaux, G.; Pigat, N.; Goffin, V.; Bernichtein, S.; Capiod, T. Calcium channels, external calcium concentration and cell proliferation. Eur. J. Pharmacol. 2014, 739, 19–25. [Google Scholar] [CrossRef]
- Moe, A.M.; Golding, A.E.; Bement, W.M. Cell healing: Calcium, repair and regeneration. Semin. Cell Dev. Biol. 2015, 45, 10–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Socha, M.J.; Behringer, E.J.; Segal, S.S. Calcium and electrical signalling along endothelium of the resistance vasculature. Basic Clin. Pharmacol. Toxicol. 2012, 110, 80–86. [Google Scholar] [CrossRef]
- Moccia, F.; Tanzi, F.; Munaron, L. Endothelial Remodelling and Intracellular Calcium Machinery. Curr. Mol. Med. 2014, 14, 457–480. [Google Scholar] [CrossRef] [PubMed]
- Martinotti, S.; Patrone, M.; Mazzucco, L.; Ranzato, E. Platelet lysate-driven endothelial cell repair: The mastermind role of intracellular calcium. Vasc. Pharmacol. 2018, 103, 62. [Google Scholar] [CrossRef]
- Ranzato, E.; Martinotti, S.; Burlando, B. Epithelial mesenchymal transition traits in honey-driven keratinocyte wound healing: Comparison among different honeys. Wound Repair Regen. 2012, 20, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Hecquet, C.M.; Ahmmed, G.U.; Vogel, S.M.; Malik, A.B. Role of TRPM2 channel in mediating H2O2-induced Ca2+ entry and endothelial hyperpermeability. Circ. Res. 2008, 102, 347–355. [Google Scholar] [CrossRef] [Green Version]
- Oda, S.; Uchida, K.; Wang, X.Y.; Lee, J.; Shimada, Y.; Tominaga, M.; Kadowaki, M. TRPM2 contributes to antigen-stimulated Ca2+ influx in mucosal mast cells. Pflug. Arch. Eur. J. Phy. 2013, 465, 1023–1030. [Google Scholar] [CrossRef]
- Sundaresan, M.; Yu, Z.X.; Ferrans, V.J.; Irani, K.; Finkel, T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science 1995, 270, 296–299. [Google Scholar] [CrossRef] [Green Version]
- Mittal, M.; Urao, N.; Hecquet, C.M.; Zhang, M.; Sudhahar, V.; Gao, X.P.; Komarova, Y.; Ushio-Fukai, M.; Malik, A.B. Novel role of reactive oxygen species-activated Trp melastatin channel-2 in mediating angiogenesis and postischemic neovascularization. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 877–887. [Google Scholar] [CrossRef] [Green Version]
- Urao, N.; Inomata, H.; Razvi, M.; Kim, H.W.; Wary, K.; McKinney, R.; Fukai, T.; Ushio-Fukai, M. Role of nox2-based NADPH oxidase in bone marrow and progenitor cell function involved in neovascularization induced by hindlimb ischemia. Circ. Res. 2008, 103, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Kanaji, T.; Nakade, S.; Kanno, T.; Mikoshiba, K. 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. J. Biochem. 1997, 122, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.J.; Kim, K.S.; Kim, S.H.; Kim, D.K.; Park, H.S. Caffeine and 2-Aminoethoxydiphenyl Borate (2-APB) Have Different Ability to Inhibit Intracellular Calcium Mobilization in Pancreatic Acinar Cell. Korean J. Physiol. Pha. 2010, 14, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilsher, N.E.; Court, W.J.; Ruddle, R.; Newbatt, Y.M.; Aherne, W.; Sheldrake, P.W.; Jones, N.P.; Katan, M.; Eccles, S.A.; Raynaud, F.I. The phosphoinositide-specific phospholipase C inhibitor U73122 (1-(6-((17beta-3-methoxyestra-1,3,5(10)-trien-17-yl)amino)hexyl)-1H-pyrrole-2,5-d ione) spontaneously forms conjugates with common components of cell culture medium. Drug Metab. Dispos. 2007, 35, 1017–1022. [Google Scholar] [CrossRef]
- Moccia, F.; Dragoni, S.; Lodola, F.; Bonetti, E.; Bottino, C.; Guerra, G.; Laforenza, U.; Rosti, V.; Tanzi, F. Store-Dependent Ca2+ Entry in Endothelial Progenitor Cells As a Perspective Tool to Enhance Cell-Based Therapy and Adverse Tumour Vascularization. Curr. Med. Chem. 2012, 19, 5802–5818. [Google Scholar] [CrossRef]
- Moccia, F.; Berra-Romani, R.; Tanzi, F. Update on vascular endothelial Ca(2+) signalling: A tale of ion channels, pumps and transporters. World J. Biol. Chem. 2012, 3, 127–158. [Google Scholar] [CrossRef]
- Santiago, E.; Climent, B.; Munoz, M.; Garcia-Sacristan, A.; Rivera, L.; Prieto, D. Hydrogen peroxide activates store-operated Ca(2+) entry in coronary arteries. Br. J. Pharmacol. 2015, 172, 5318–5332. [Google Scholar] [CrossRef] [Green Version]
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [Green Version]
- Zuccolo, E.; Bottino, C.; Diofano, F.; Poletto, V.; Codazzi, A.C.; Mannarino, S.; Campanelli, R.; Fois, G.; Marseglia, G.L.; Guerra, G.; et al. Constitutive Store-Operated Ca(2+) Entry Leads to Enhanced Nitric Oxide Production and Proliferation in Infantile Hemangioma-Derived Endothelial Colony-Forming Cells. Stem Cells Dev. 2016, 25, 301–319. [Google Scholar] [CrossRef]
- Moccia, F.; Berra-Romani, R.; Baruffi, S.; Spaggiari, S.; Adams, D.J.; Taglietti, V.; Tanzi, F. Basal nonselective cation permeability in rat cardiac microvascular endothelial cells. Microvasc. Res. 2002, 64, 187–197. [Google Scholar] [CrossRef]
- Zuccolo, E.; Lim, D.; Kheder, D.A.; Perna, A.; Catarsi, P.; Botta, L.; Rosti, V.; Riboni, L.; Sancini, G.; Tanzi, F.; et al. Acetylcholine induces intracellular Ca(2+) oscillations and nitric oxide release in mouse brain endothelial cells. Cell Calcium 2017, 66, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Anitua, E.; Sanchez, M.; Nurden, A.T.; Nurden, P.; Orive, G.; Andia, I. New insights into and novel applications for platelet-rich fibrin therapies. Trends Biotechnol. 2006, 24, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Doucet, C.; Ernou, I.; Zhang, Y.Z.; Llense, J.R.; Begot, L.; Holy, X.; Lataillade, J.J. Platelet lysates promote mesenchymal stem cell expansion: A safety substitute for animal serum in cell-based therapy applications. J. Cell Physiol. 2005, 205, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Mazzucco, L.; Balbo, V.; Cattana, E.; Borzini, P. Platelet-rich plasma and platelet gel preparation using Plateltex (R). Vox. Sang. 2008, 94, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Ranzato, E.; Mazzucco, L.; Patrone, M.; Burlando, B. Platelet lysate promotes in vitro wound scratch closure of human dermal fibroblasts: Different roles of cell calcium, P38, ERK and PI3K/AKT. J. Cell Mol. Med. 2009, 13, 2030–2038. [Google Scholar] [CrossRef] [PubMed]
- Ranzato, E.; Martinotti, S.; Balbo, V.; Boccafoschi, F.; Patrone, M.; Mazzucco, L.; Burlando, B. Platelet Lysate-Driven in Vitro Wound Healing. Wound Repair. Regen. 2009, 17, A78. [Google Scholar]
- Abdullaev, I.F.; Bisaillon, J.M.; Potier, M.; Gonzalez, J.C.; Motiani, R.K.; Trebak, M. Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ. Res. 2008, 103, 1289–1299. [Google Scholar] [CrossRef]
- Rossello, R.A.; David, H. Cell communication and tissue engineering. Commun. Integr. Biol. 2010, 3, 53–56. [Google Scholar] [CrossRef]
- Berra-Romani, R.; Faris, P.; Negri, S.; Botta, L.; Genova, T.; Moccia, F. Arachidonic Acid Evokes an Increase in Intracellular Ca(2+) Concentration and Nitric Oxide Production in Endothelial Cells from Human Brain Microcirculation. Cells 2019, 8, 689. [Google Scholar] [CrossRef] [Green Version]
- Ostrovskaya, O.; Goyal, R.; Osman, N.; McAllister, C.E.; Pessah, I.N.; Hume, J.R.; Wilson, S.M. Inhibition of ryanodine receptors by 4-(2-aminopropyl)-3,5-dichloro-N,N-dimethylaniline (FLA 365) in canine pulmonary arterial smooth muscle cells. J. Pharmacol. Exp. Ther 2007, 323, 381–390. [Google Scholar] [CrossRef] [Green Version]
- Anguita, E.; Villalobo, A. Ca(2+) signaling and Src-kinases-controlled cellular functions. Arch. Biochem. Biophys. 2018, 650, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Berra Romani, R.; Raqeeb, A.; Laforenza, U.; Scaffino, M.F.; Moccia, F.; Avelino-Cruz, J.E.; Oldani, A.; Coltrini, D.; Milesi, V.; Taglietti, V.; et al. Cardiac microvascular endothelial cells express a functional Ca+ -sensing receptor. J. Vasc. Res. 2009, 46, 73–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sammak, B.M.; Yousef, B.A.; Gali, M.H.; al Karawi, M.A.; Mohamed, A.E. Case report: Radiological and endoscopic management of bile leak following laparoscopic cholecystectomy. J. Gastroenterol. Hepatol. 1997, 12, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Ranzato, E.; Martinotti, S.; Volante, A.; Mazzucco, L.; Burlando, B. Platelet lysate modulates MMP-2 and -9 expression, matrix deposition and cell-to-matrix adhesion in keratinocytes and fibroblasts. Exp. Dermatol. 2011, 20, 308–313. [Google Scholar] [CrossRef]
- Ranzato, E.; Martinotti, S.; Magnelli, V.; Murer, B.; Biffo, S.; Mutti, L.; Burlando, B. Epigallocatechin-3-gallate induces mesothelioma cell death via H2O2 -dependent T-type Ca2+ channel opening. J. Cell Mol. Med. 2012, 16, 2667–2678. [Google Scholar] [CrossRef]
- Martinotti, S.; Laforenza, U.; Patrone, M.; Moccia, F.; Ranzato, E. Honey-Mediated Wound Healing: H2O2 Entry through AQP3 Determines Extracellular Ca2+ Influx. Int. J. Mol. Sci. 2019, 20, 764. [Google Scholar] [CrossRef] [Green Version]
- Ranzato, E.; Magnelli, V.; Martinotti, S.; Waheed, Z.; Cain, S.M.; Snutch, T.P.; Marchetti, C.; Burlando, B. Epigallocatechin-3-gallate elicits Ca2+ spike in MCF-7 breast cancer cells: Essential role of Cav3.2 channels. Cell Calcium 2014, 56, 285–295. [Google Scholar] [CrossRef]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TARGET GENE | FORWARD SEQUENCE | REVERSE SEQUENCE |
|---|---|---|
| β-actin | 5’-TCCCTGGAGAAGAGCTACGA-3′ | 5’-AGCACTGTGTTGGCGTACAG-3′ |
| GADPH | 5’-AATCCCATCACCATCTTCCA-3′ | 5’-TGGACTCCACGACGTACTCA-3′ |
| NOX4 | 5’-GGTATTGTTCCTCATGGTTAC-3’ | 5’-TGGGATGATGTCTGGTTAAG-3’ |
| TRPM2 | 5’-GTGAAGTCATCACTATTGGC-3′ | 5’-GAATCTCCACACCATATTGC-3′ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinotti, S.; Patrone, M.; Balbo, V.; Mazzucco, L.; Ranzato, E. Endothelial Response Boosted by Platelet Lysate: The Involvement of Calcium Toolkit. Int. J. Mol. Sci. 2020, 21, 808. https://doi.org/10.3390/ijms21030808
Martinotti S, Patrone M, Balbo V, Mazzucco L, Ranzato E. Endothelial Response Boosted by Platelet Lysate: The Involvement of Calcium Toolkit. International Journal of Molecular Sciences. 2020; 21(3):808. https://doi.org/10.3390/ijms21030808
Chicago/Turabian StyleMartinotti, Simona, Mauro Patrone, Valeria Balbo, Laura Mazzucco, and Elia Ranzato. 2020. "Endothelial Response Boosted by Platelet Lysate: The Involvement of Calcium Toolkit" International Journal of Molecular Sciences 21, no. 3: 808. https://doi.org/10.3390/ijms21030808
APA StyleMartinotti, S., Patrone, M., Balbo, V., Mazzucco, L., & Ranzato, E. (2020). Endothelial Response Boosted by Platelet Lysate: The Involvement of Calcium Toolkit. International Journal of Molecular Sciences, 21(3), 808. https://doi.org/10.3390/ijms21030808