Characterization and Identification of Natural Antimicrobial Peptides on Different Organisms

 ,
, 
Abstract
:1. Introduction
2. Results
2.1. Characterization of AMPs
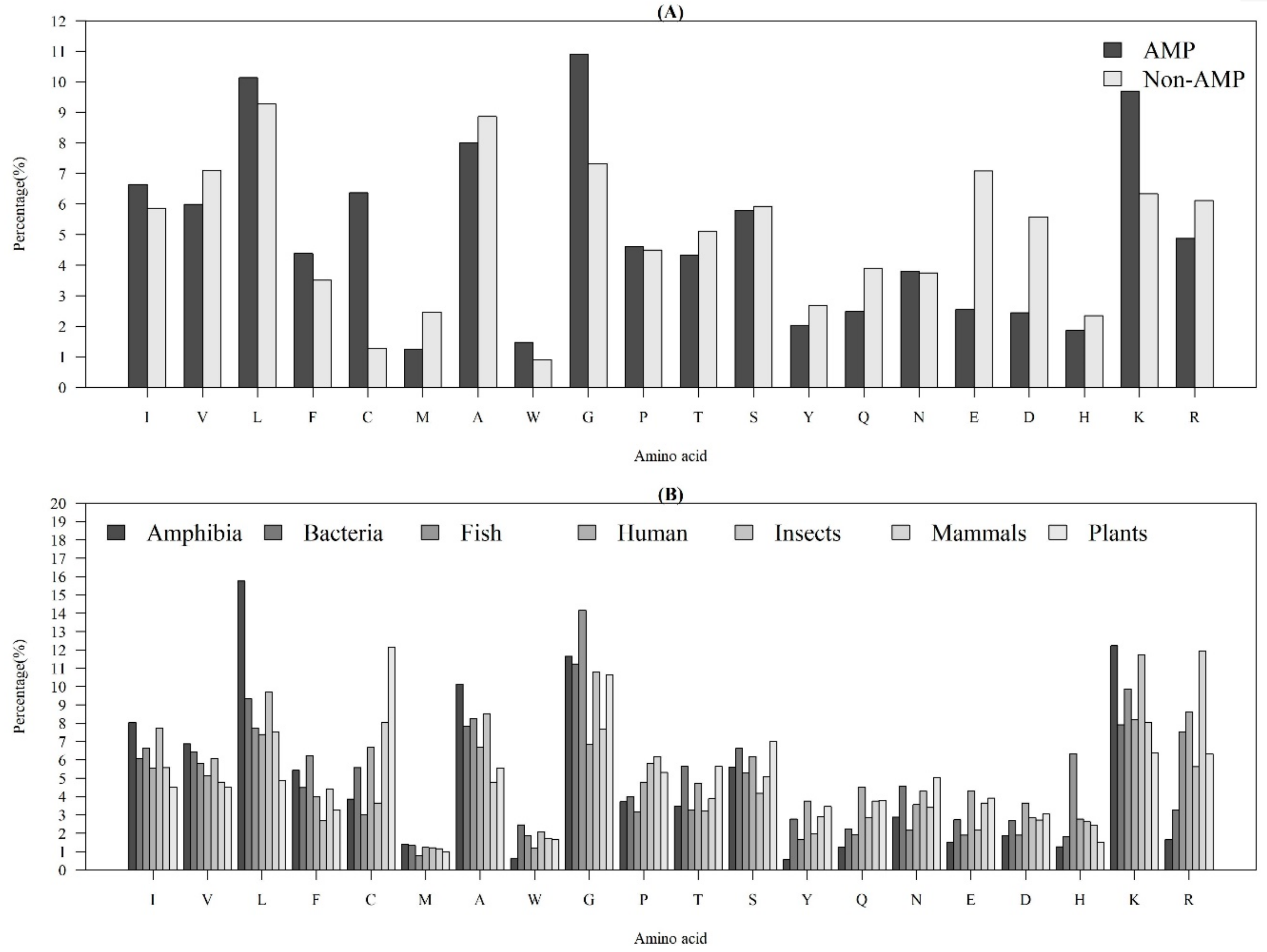
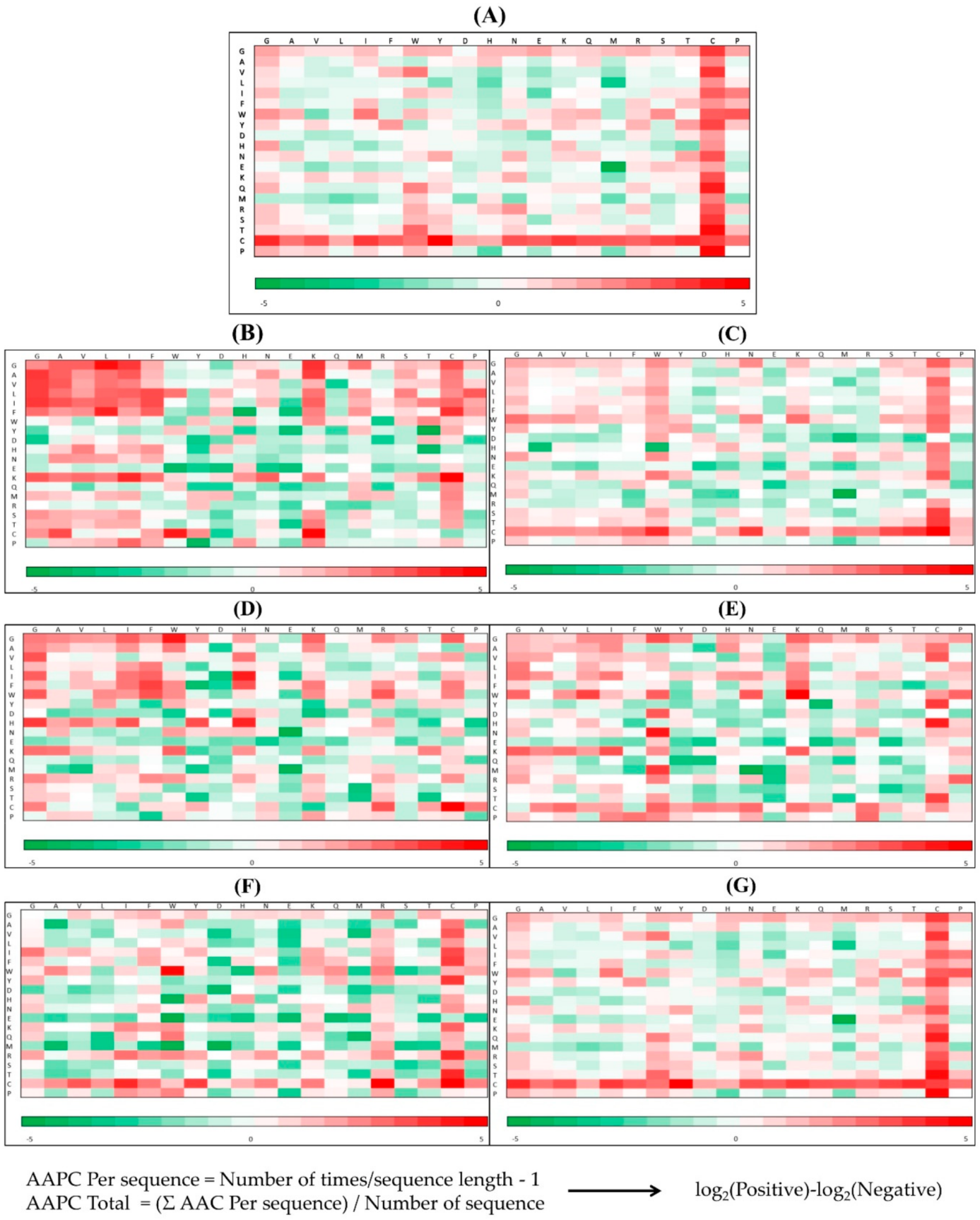
2.1.1. Compositional Characteristics of AMPs
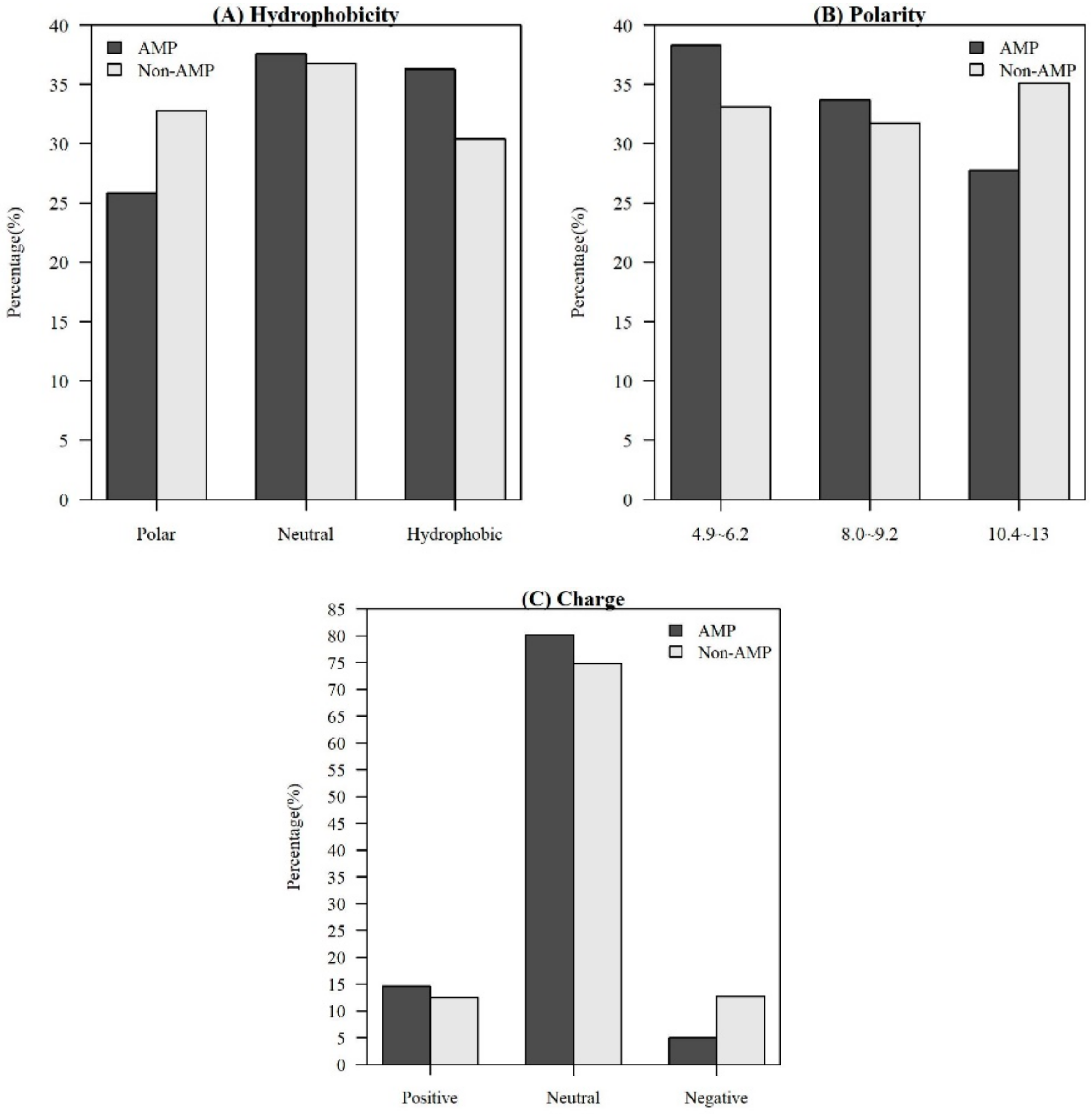
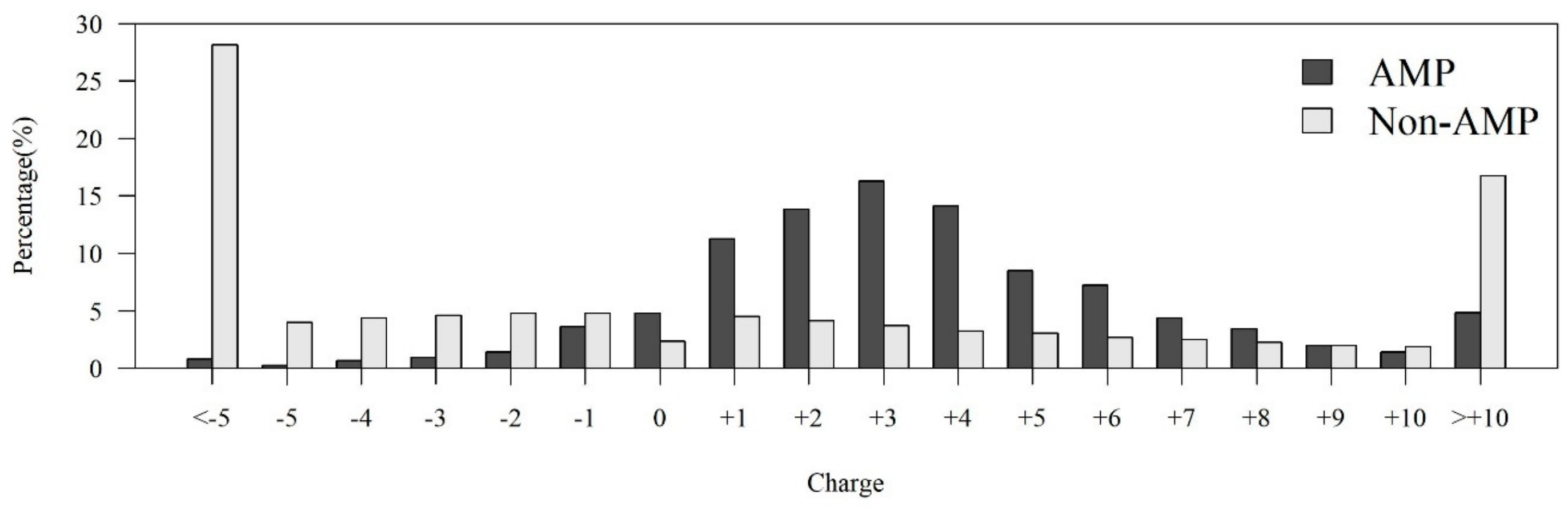
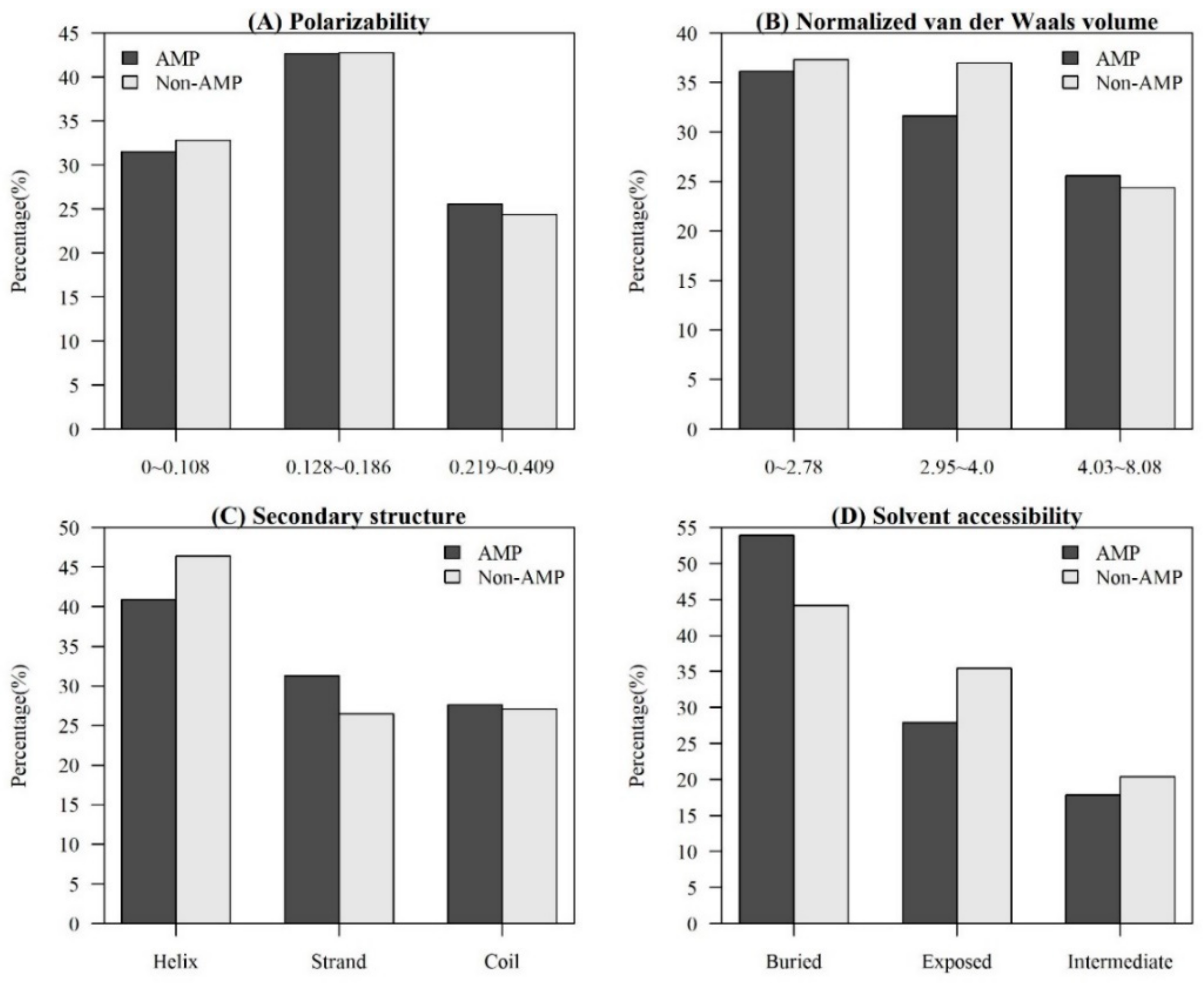
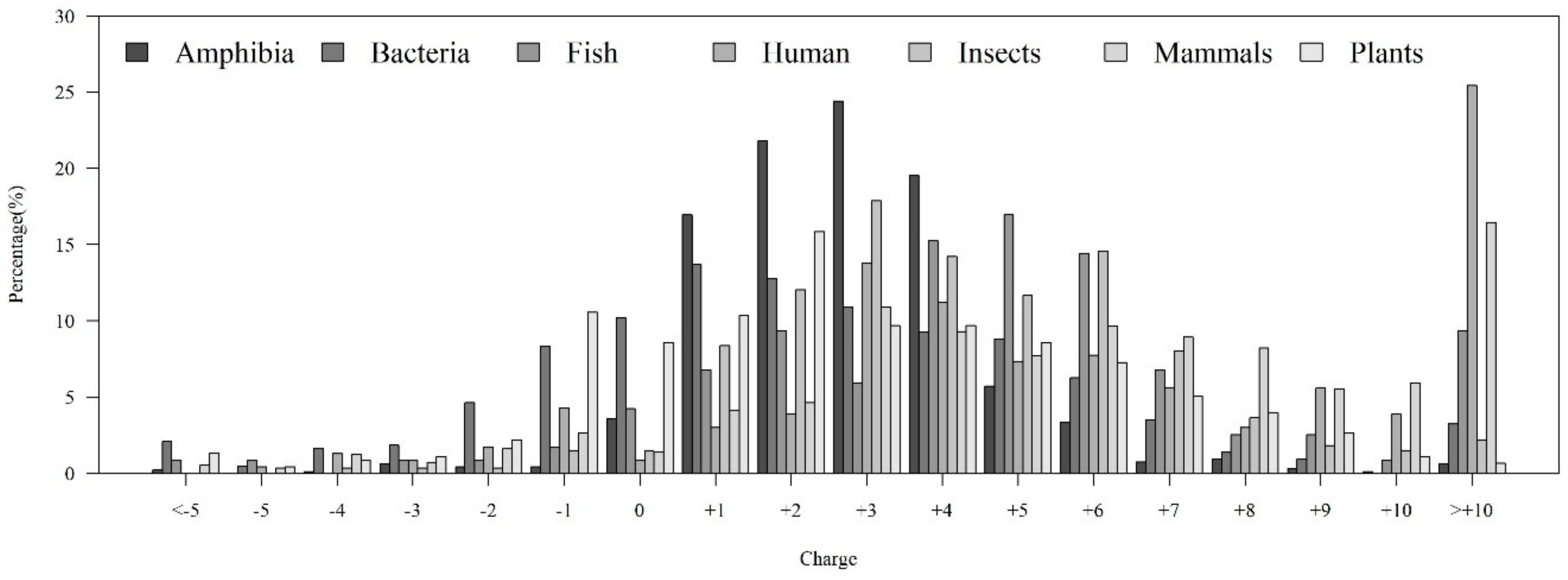
2.1.2. Investigation of Physicochemical Properties
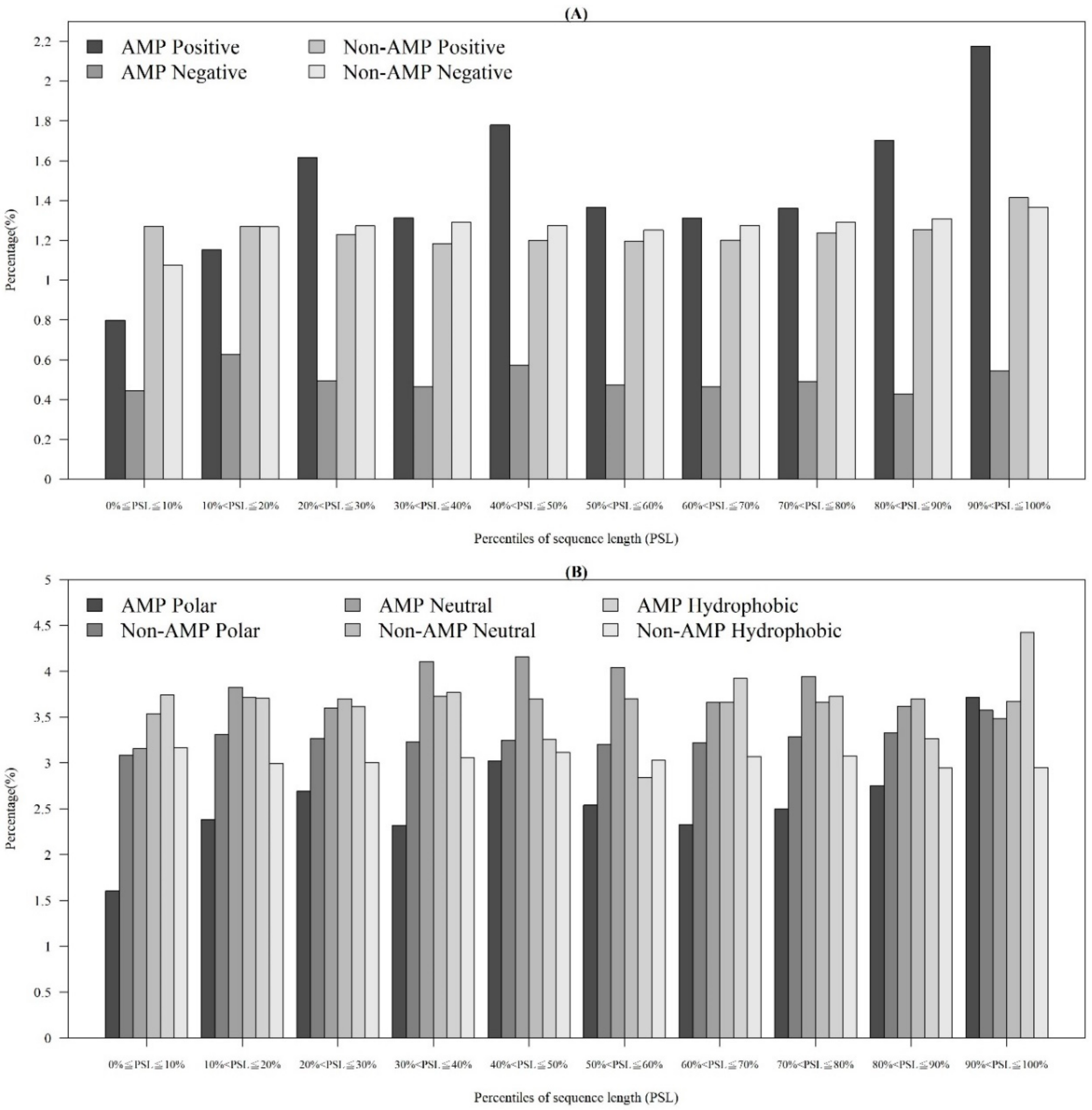
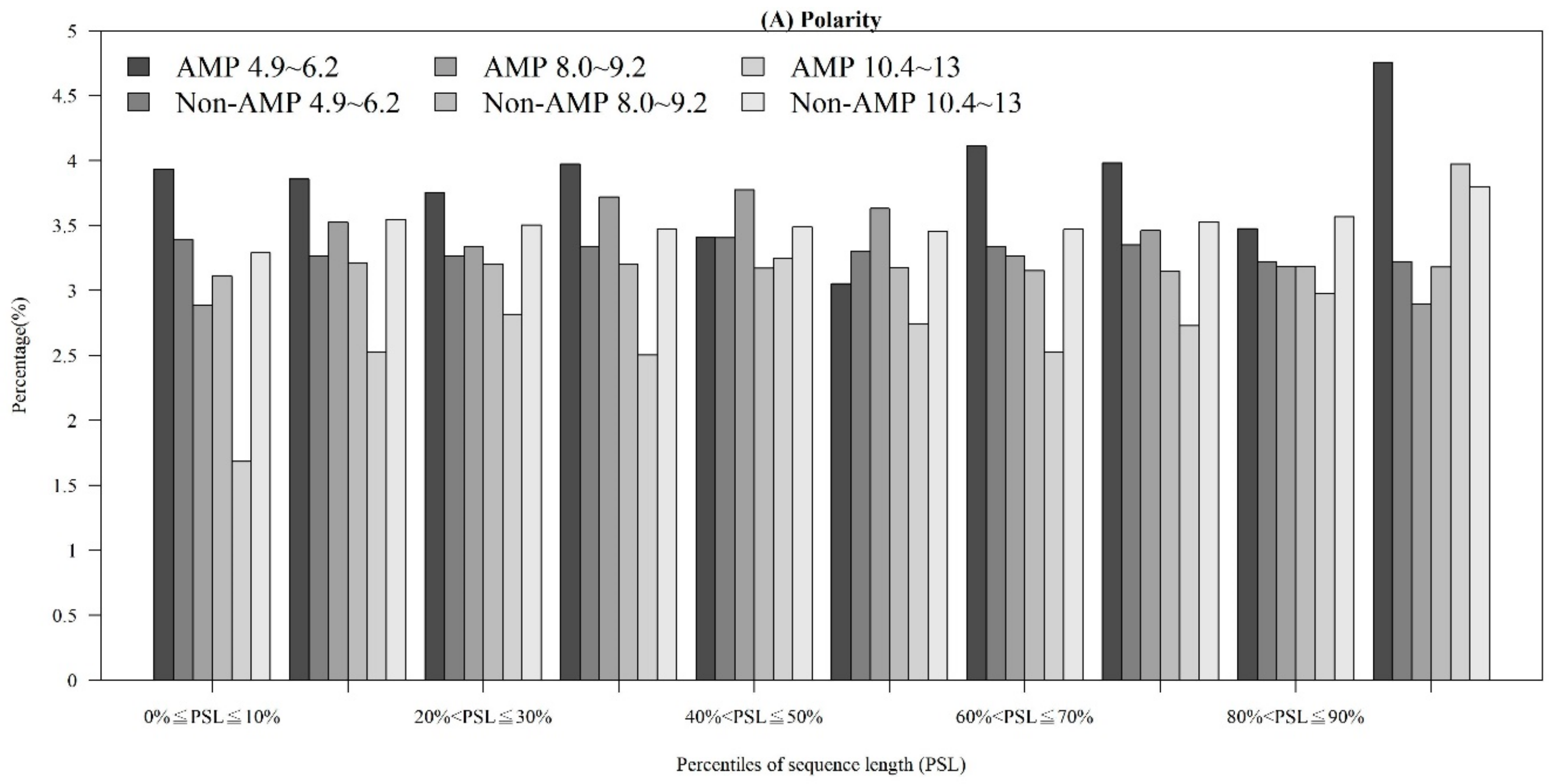
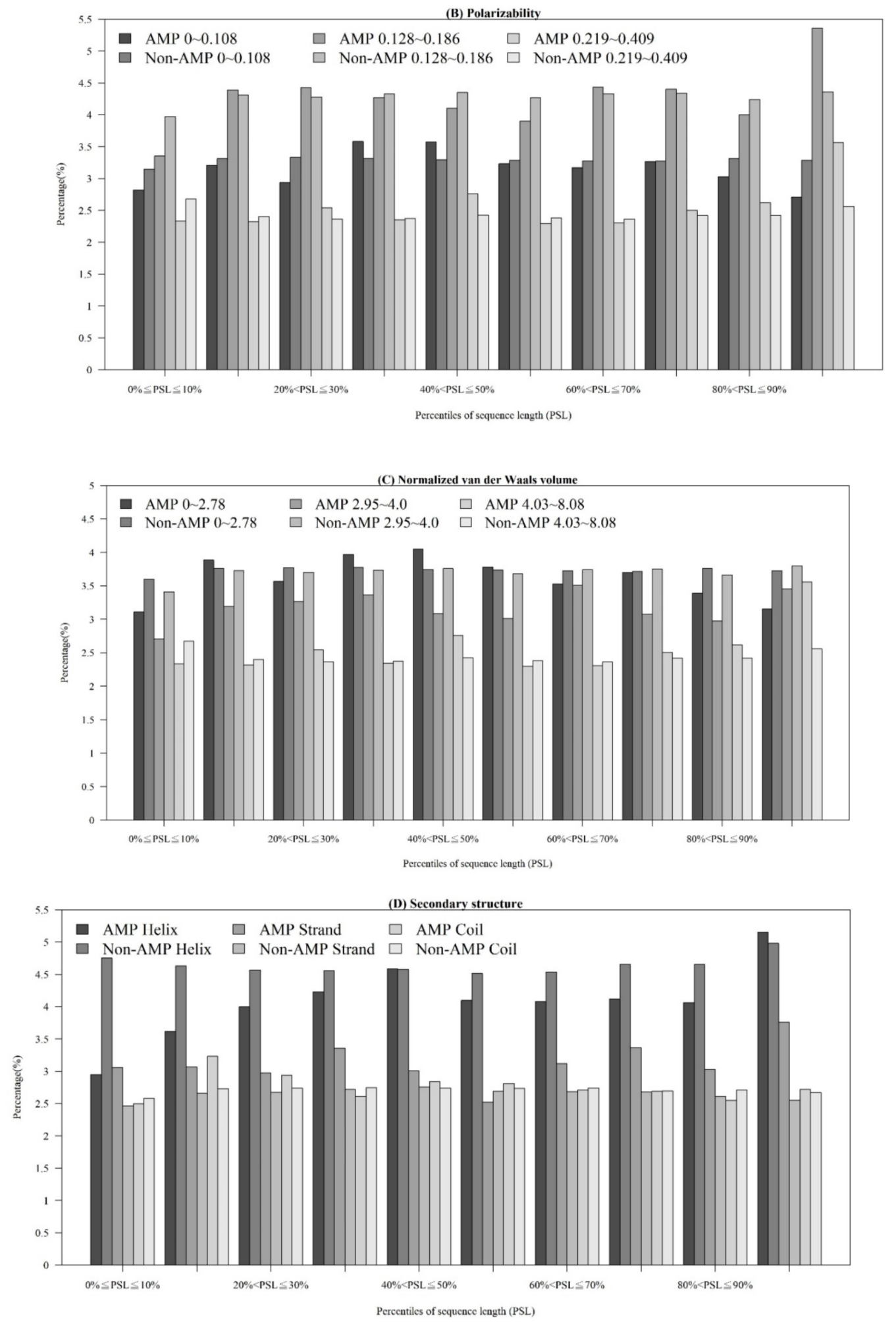
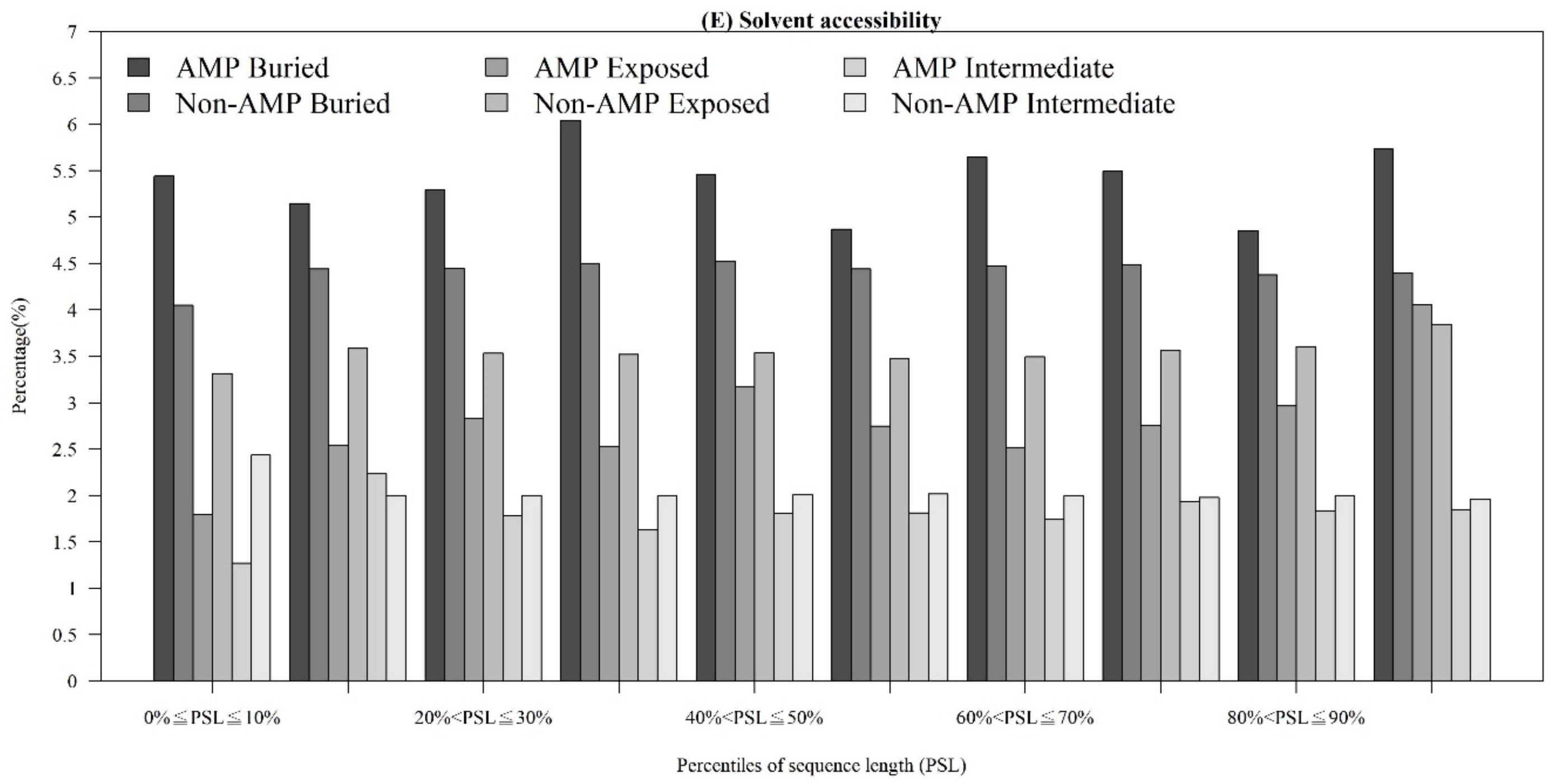
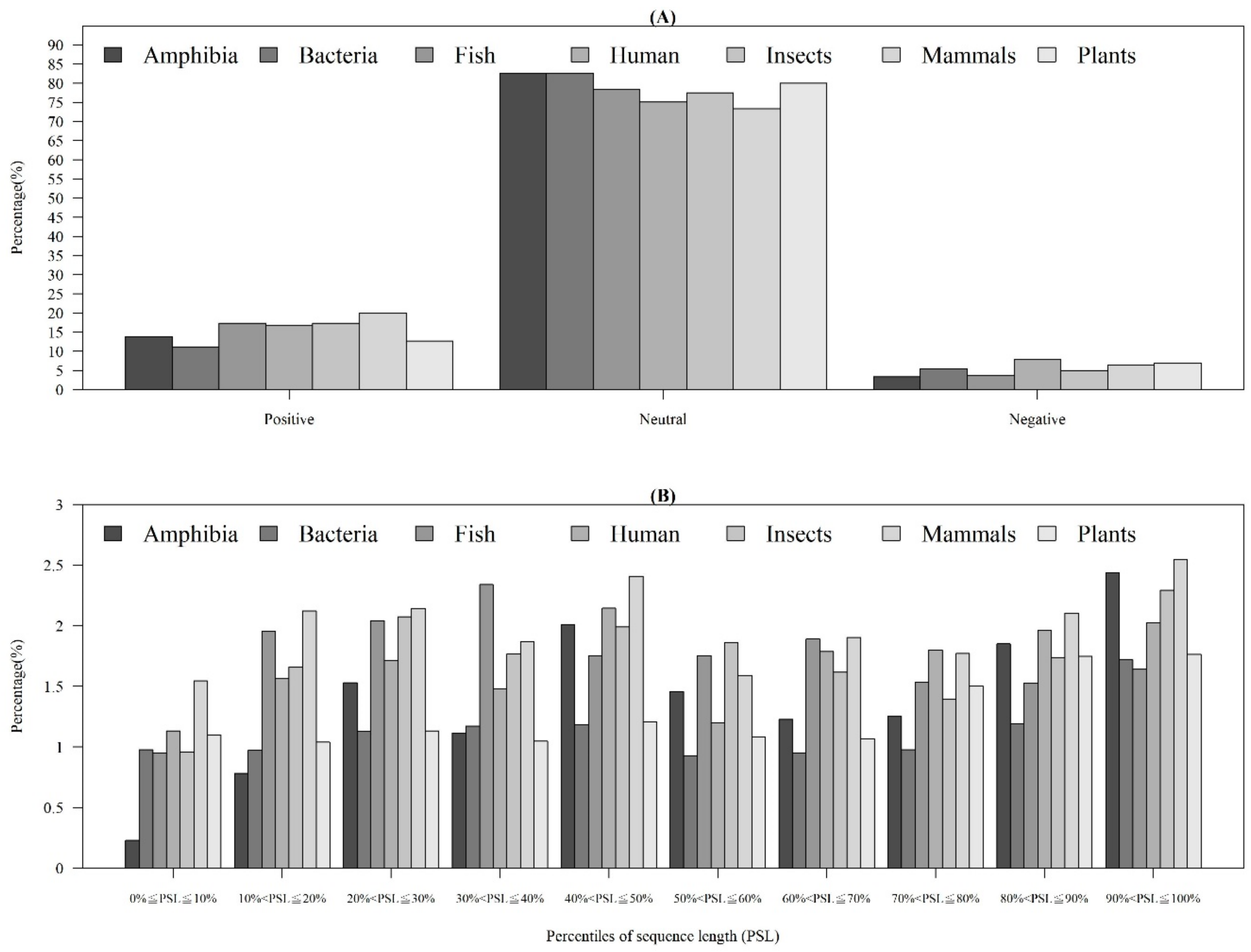
2.1.3. Physicochemical Properties with Respect to Different Sequence Lengths
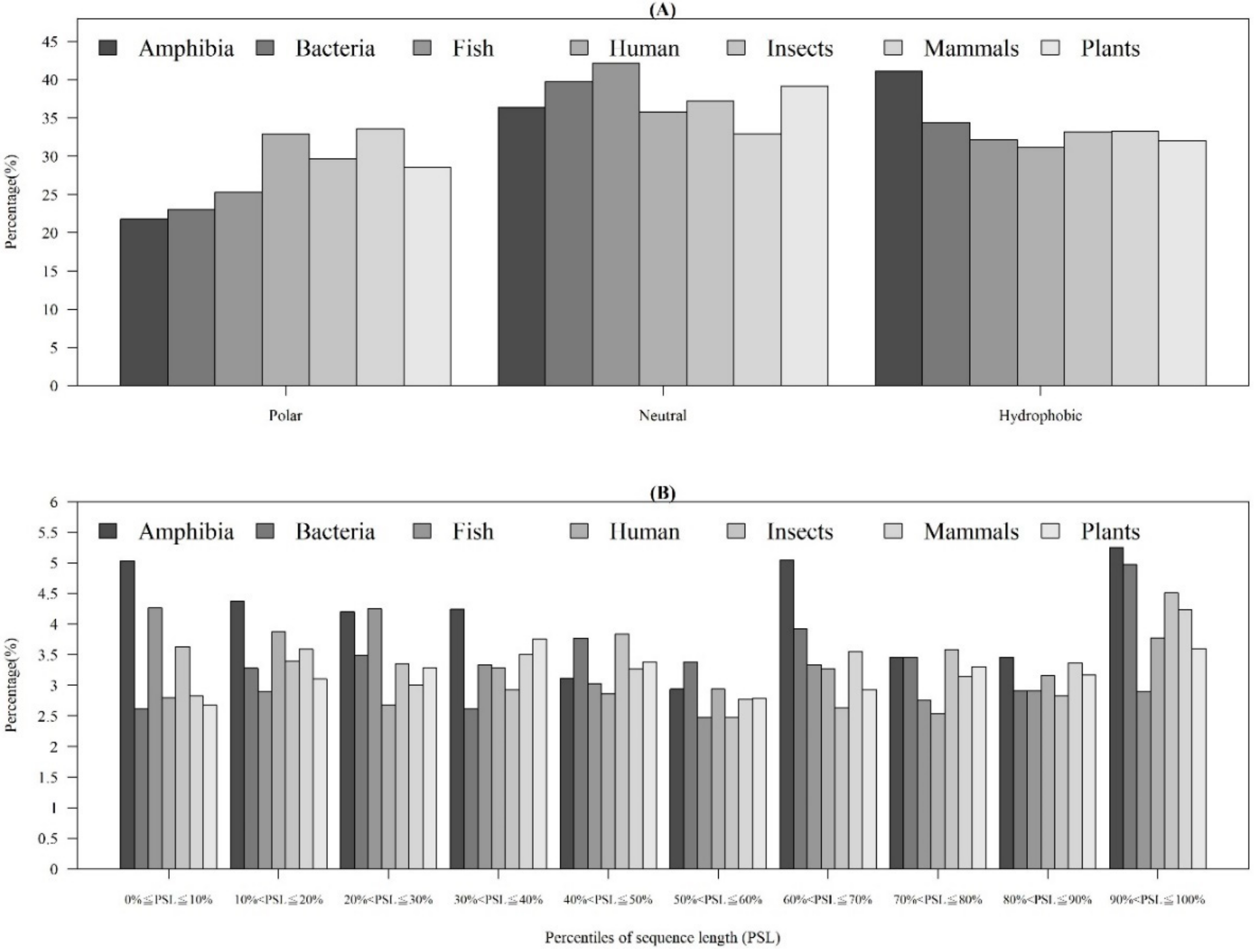
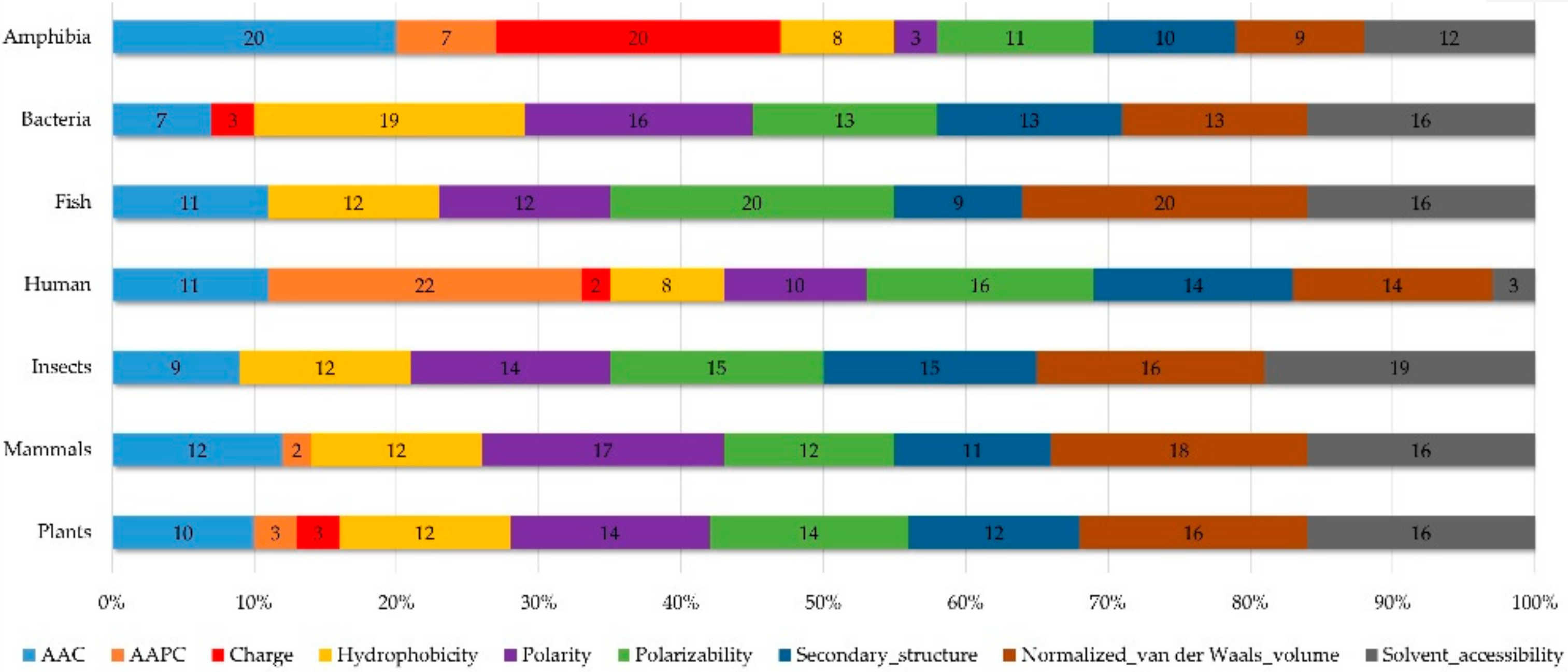
2.1.4. Physicochemical Properties of AMPs with Respect to Different Categories of Organism
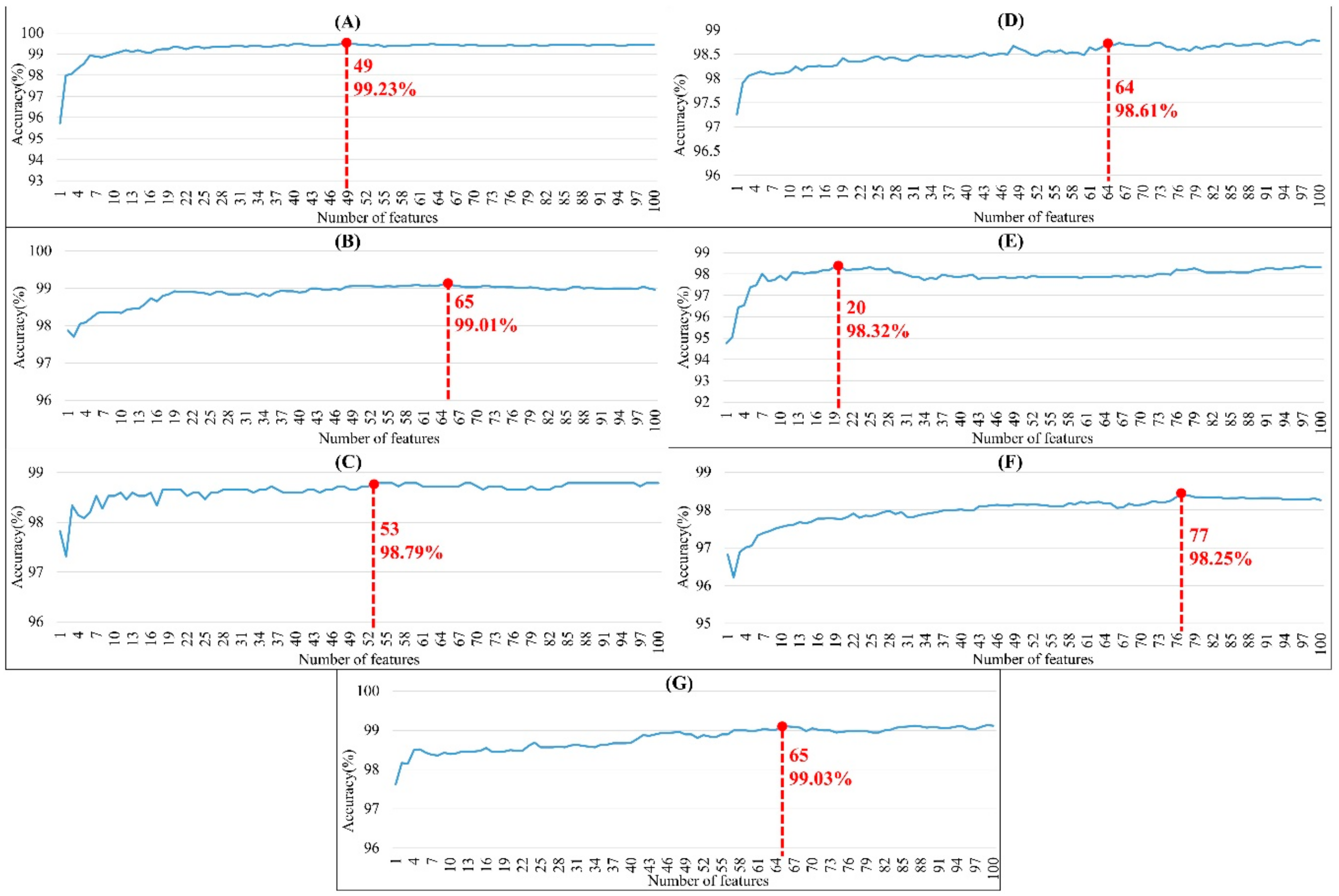
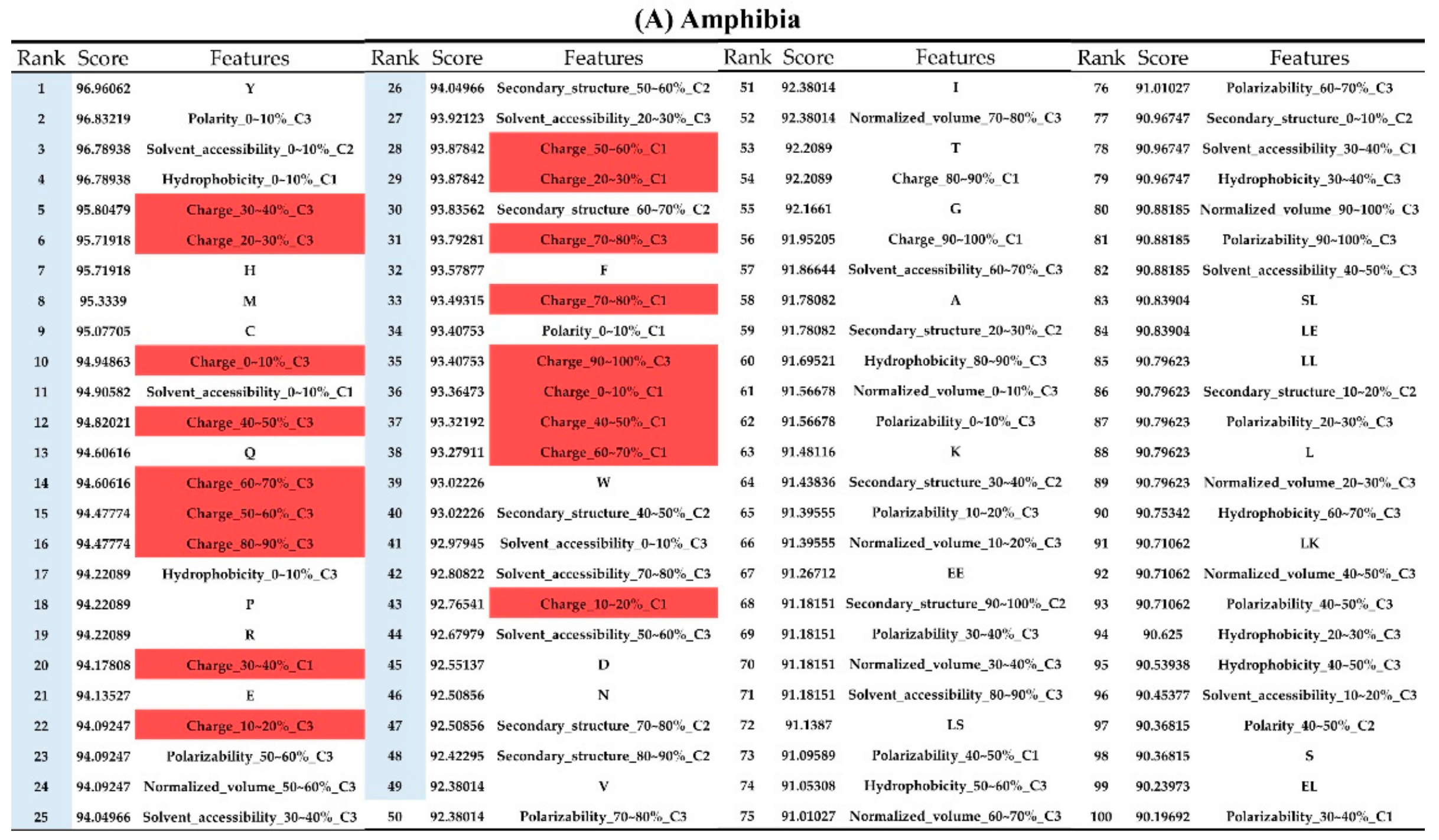
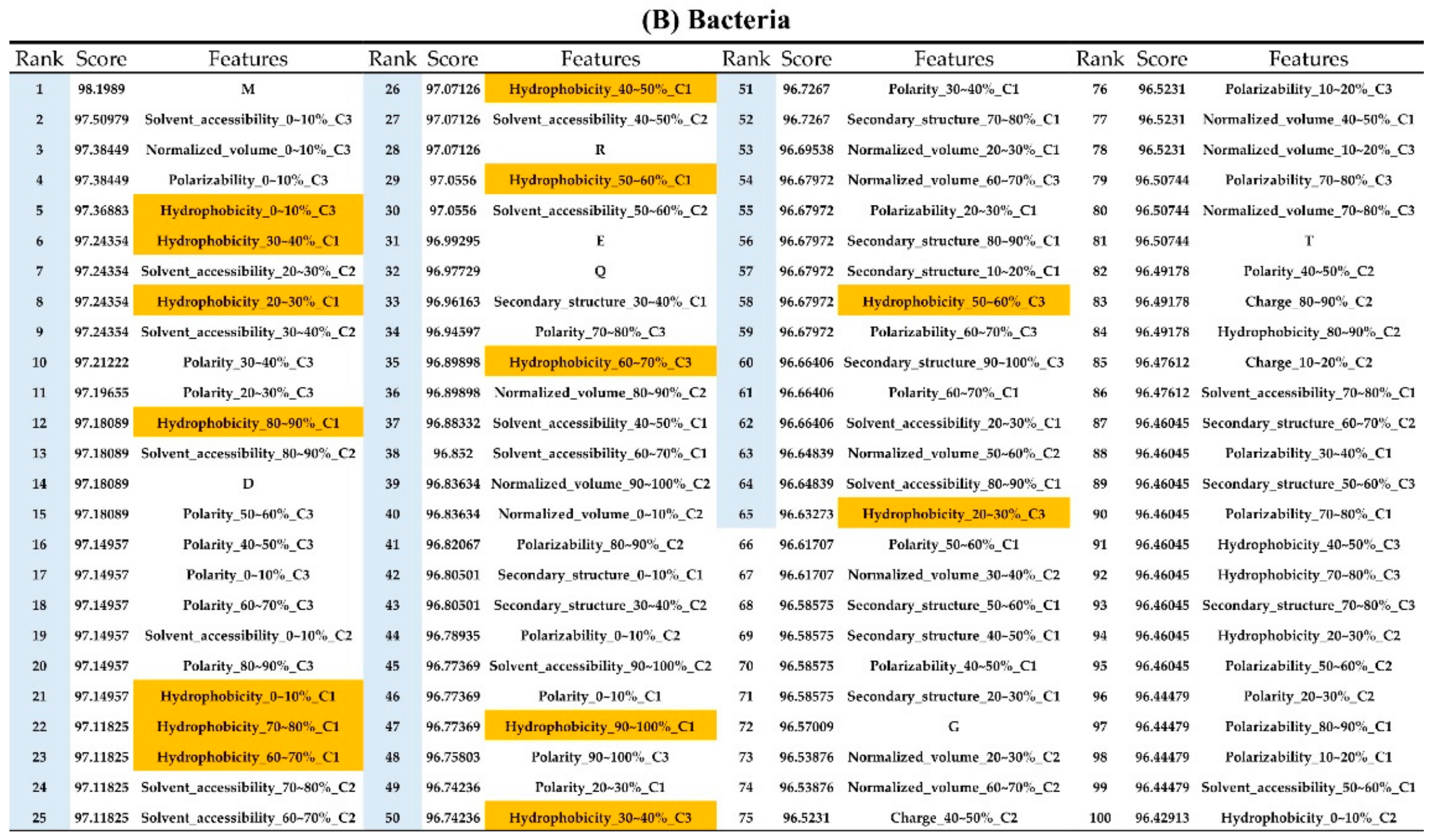
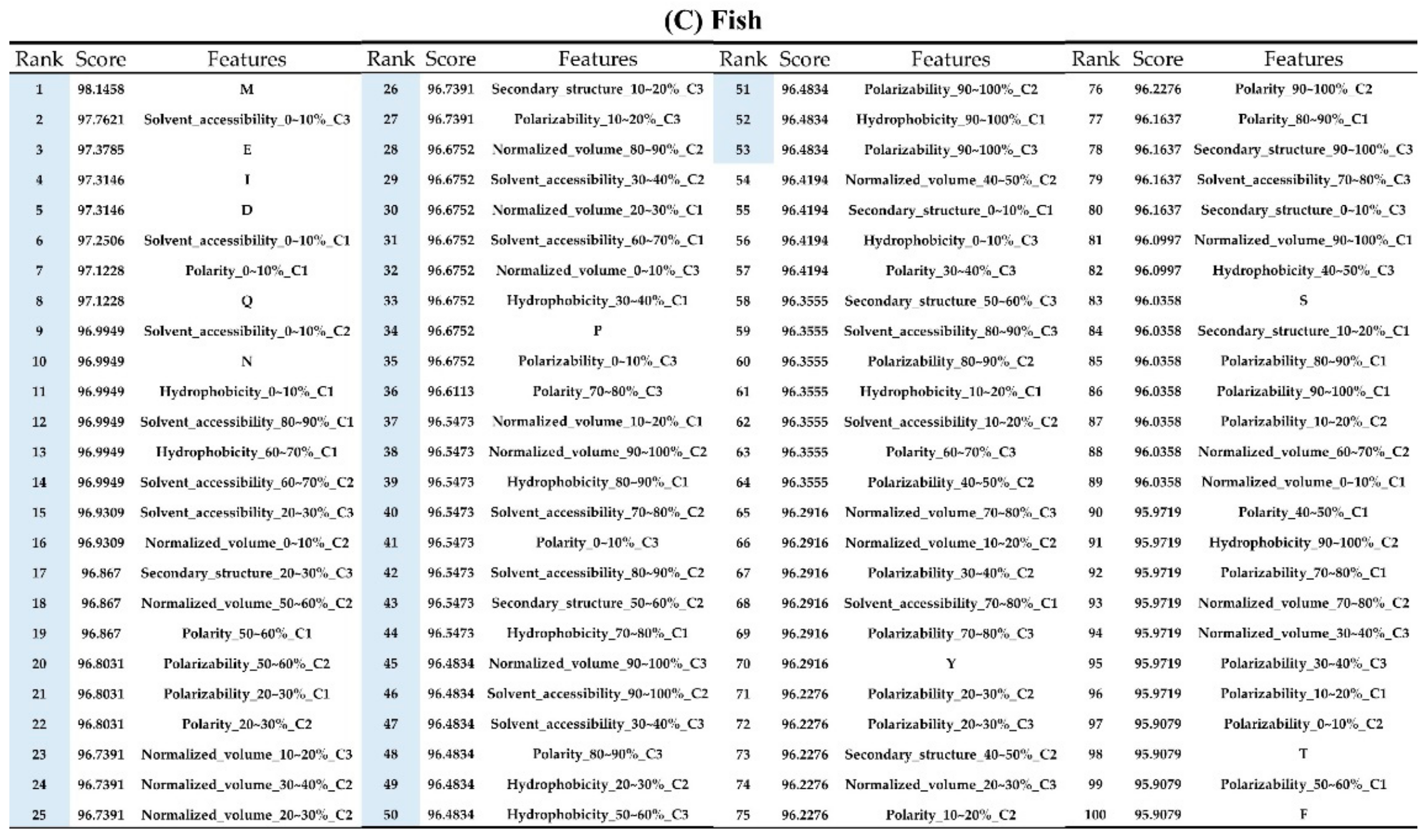
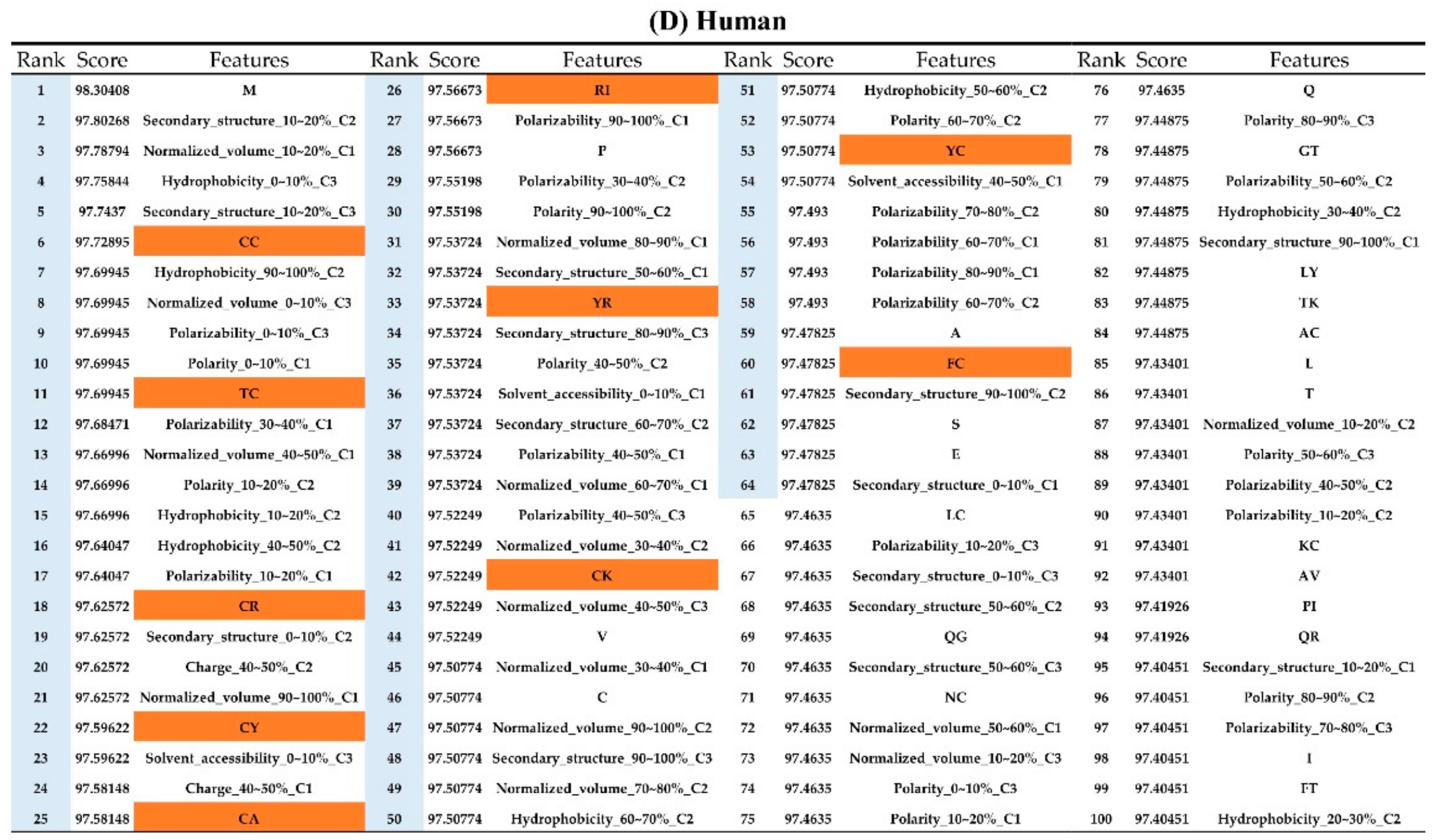
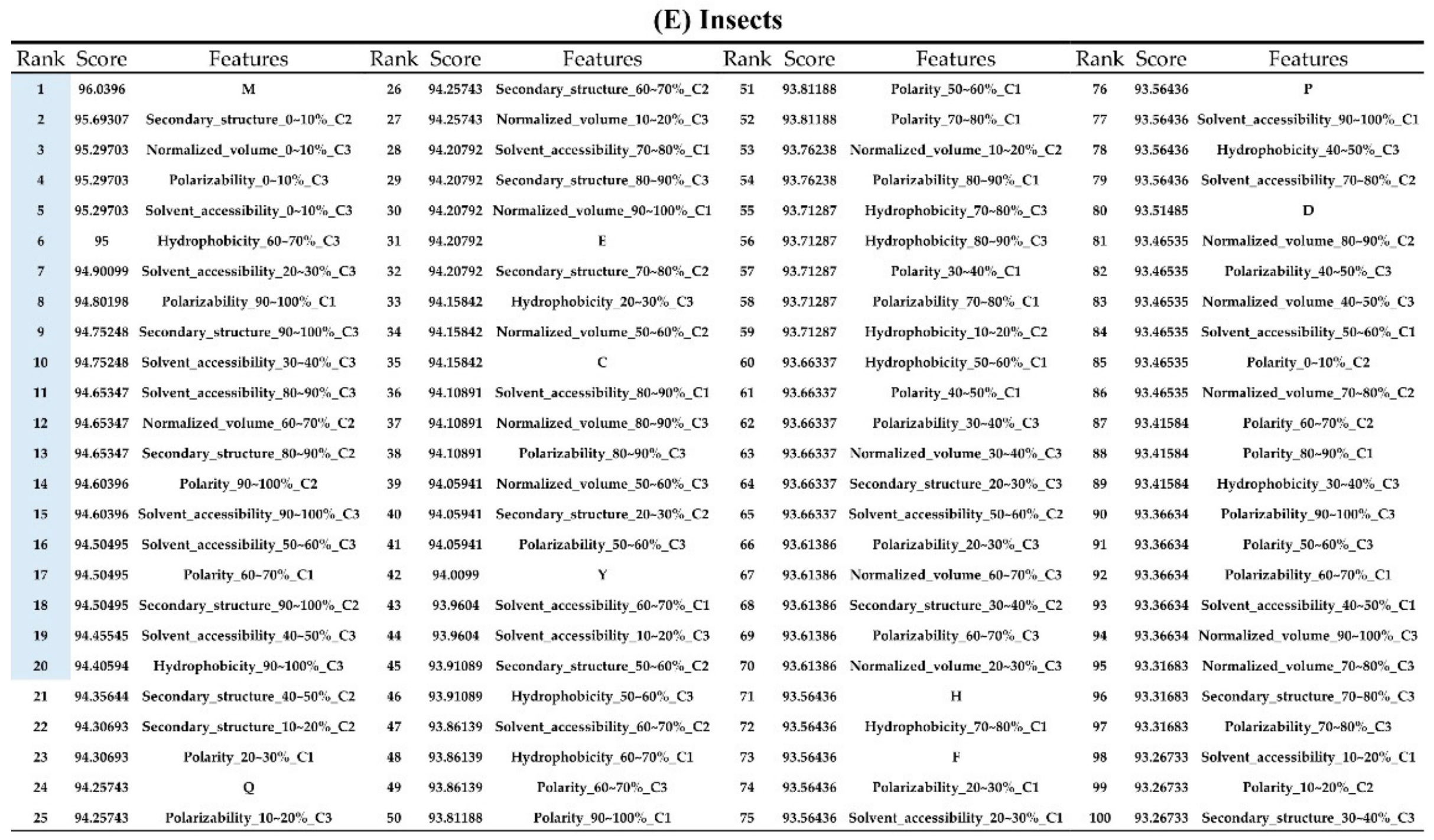
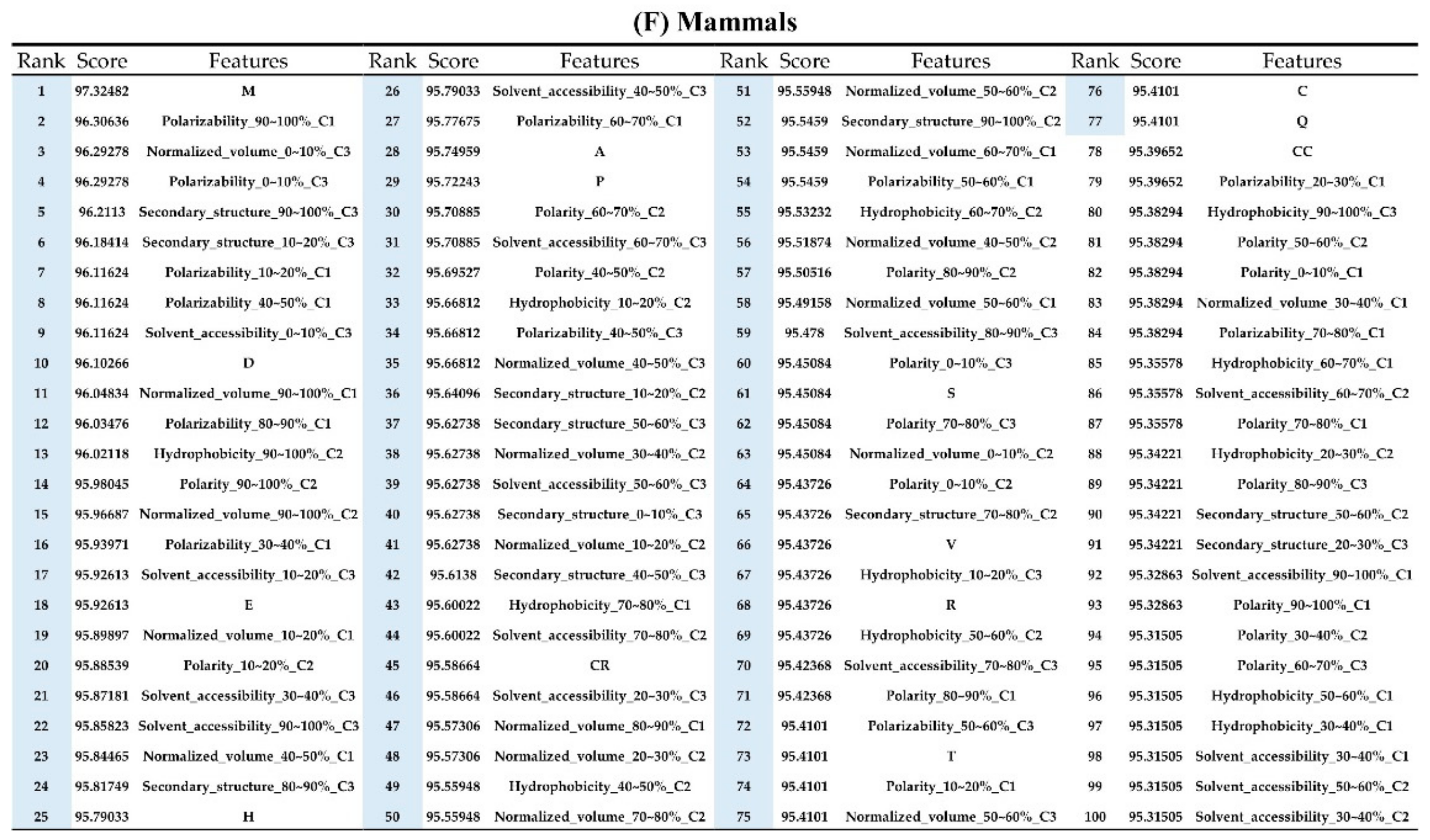
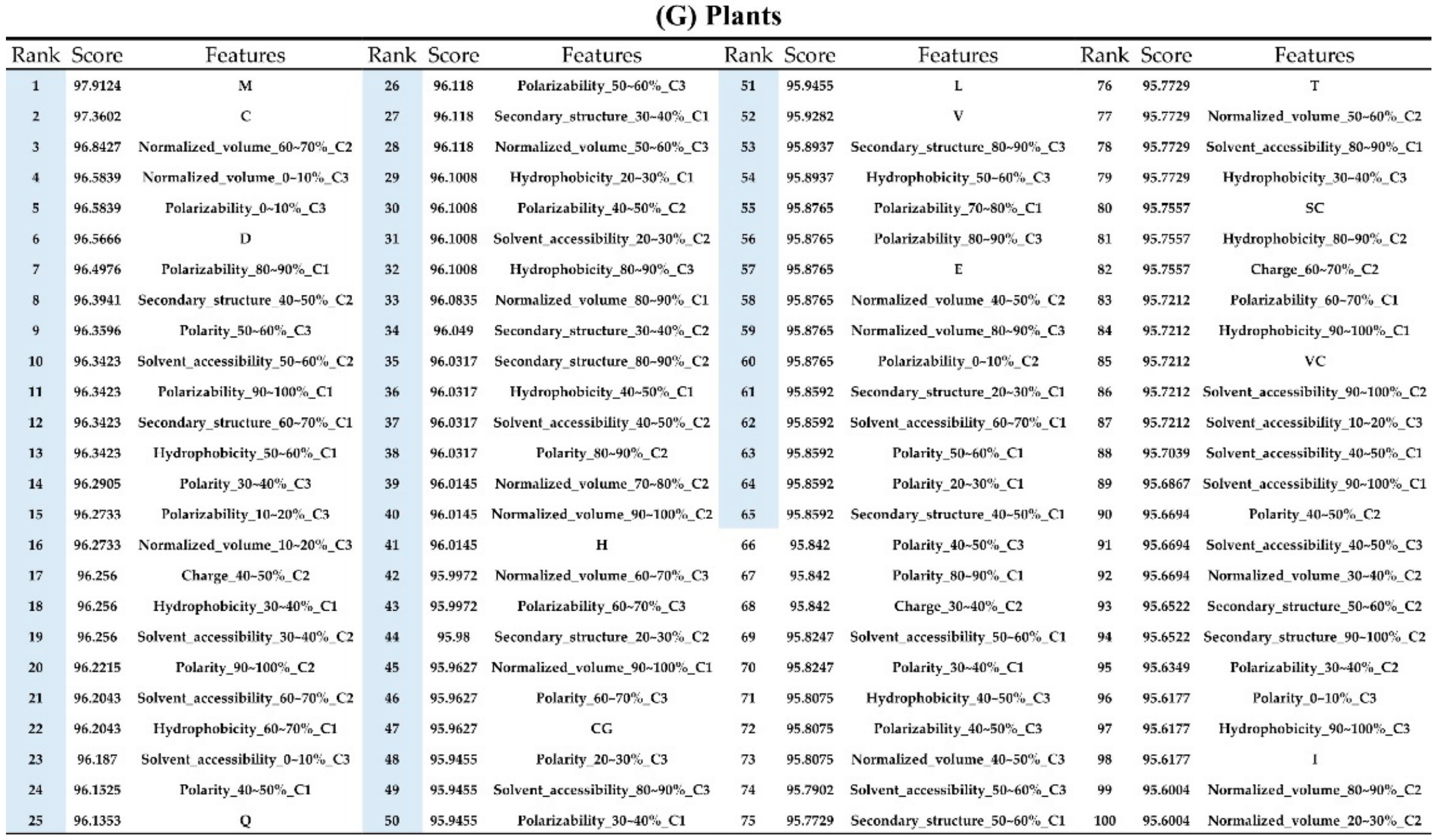
2.2. The Identification of Important Features
2.3. Prediction Performance
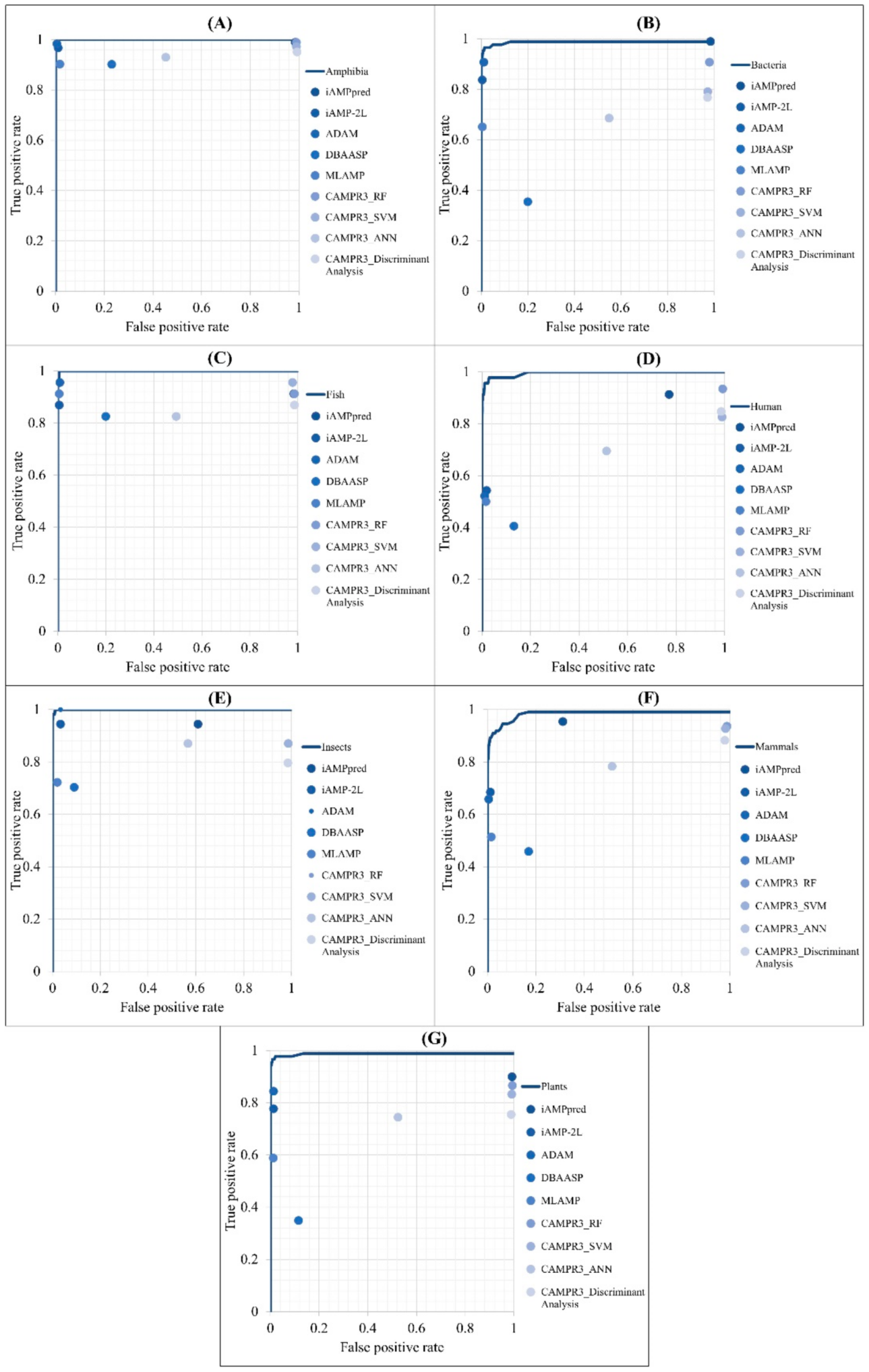
2.4. Comparison with Other AMP Prediction Tools
3. Discussion and Conclusions
4. Materials and Methods
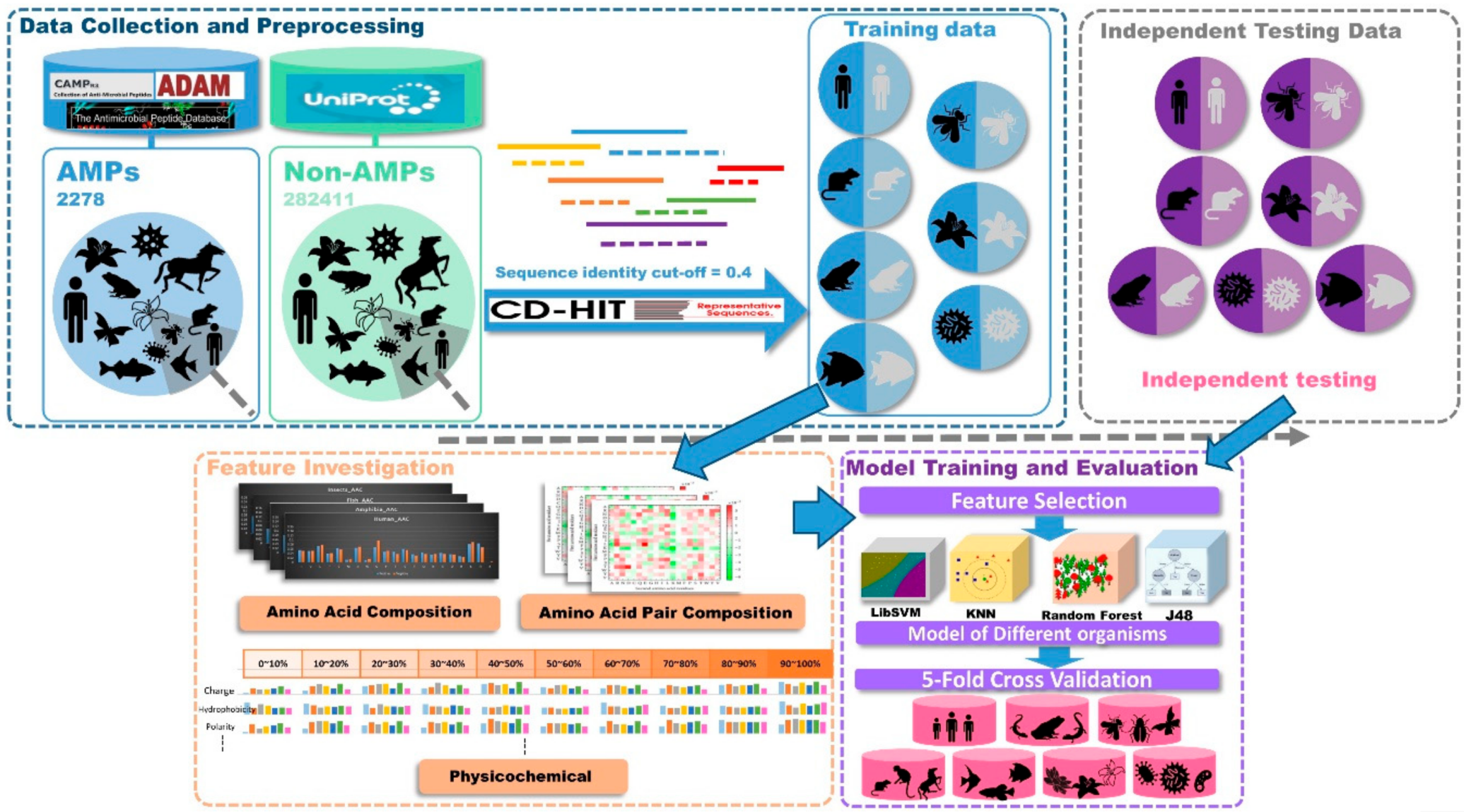
4.1. Data Collection and Preprocessing
4.2. Feature Constructions
4.3. Model Construction and Feature Selection Methods
4.4. Evaluation Matrics
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AMPs | Antimicrobial peptides |
| AACs | Amino acid compositions |
| ML | Machine learning |
| SVM | Support vector machine |
| PseAAC | Pseudo amino acid composition |
| FKNN | Fuzzy K-nearest neighbor |
| ANN | Artificial neural network |
| SMOTE | Synthetic minority oversampling technique |
| RF | Random forest |
| DA | Discriminant analysis |
| AAPC | Amino acid pair composition |
| OneR | One rule attribute evaluation |
| KNN | K-nearest neighbor models |
| Sn | Sensitivity |
| Sp | Specificity |
| Acc | Accuracy |
| MCC | Matthews correlation coefficient |
| TP | True positives |
| TN | True negatives |
| FP | False positives |
| FN | False negatives |
Appendix A

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organisms | Classifier | Sensitivity | Specificity | Accuracy | Matthews Correlation Coefficient |
|---|---|---|---|---|---|
| Amphibia | RF | 99.19% | 99.18% | 99.19% | 0.981 |
| DT | 97.84% | 98.81% | 98.50% | 0.965 | |
| KNN | 96.76% | 99.81% | 98.84% | 0.973 | |
| SVM | 98.92% | 98.93% | 98.93% | 0.975 | |
| Bacteria | RF | 95.94% | 96.18% | 96.16% | 0.735 |
| DT | 86.67% | 97.95% | 97.34% | 0.769 | |
| KNN | 73.62% | 99.44% | 98.04% | 0.7959 | |
| SVM | 95.94% | 95.94% | 95.94% | 0.725 | |
| Fish | RF | 96.84% | 96.87% | 96.87% | 0.789 |
| DT | 73.68% | 98.43% | 96.93% | 0.728 | |
| KNN | 68.42% | 99.52% | 97.63% | 0.774 | |
| SVM | 82.11% | 99.86% | 98.79% | 0.889 | |
| Human | RF | 94.09% | 93.07% | 93.10% | 0.489 |
| DT | 74.19% | 98.15% | 97.49% | 0.615 | |
| KNN | 68.28% | 98.94% | 98.10% | 0.654 | |
| SVM | 88.17% | 87.82% | 87.83% | 0.354 | |
| Insects | RF | 96.36% | 96.33% | 96.34% | 0.838 |
| DT | 91.36% | 97.56% | 96.88% | 0.849 | |
| KNN | 85.91% | 98.28% | 96.93% | 0.842 | |
| SVM | 95.00% | 95.11% | 95.10% | 0.793 | |
| Mammals | RF | 94.42% | 95.24% | 95.19% | 0.708 |
| DT | 83.71% | 92.60% | 92.06% | 0.560 | |
| KNN | 74.55% | 98.92% | 97.43% | 0.767 | |
| SVM | 93.97% | 93.97% | 93.97% | 0.662 | |
| Plants | RF | 97.53% | 97.39% | 97.39% | 0.822 |
| DT | 88.74% | 98.82% | 98.19% | 0.851 | |
| KNN | 80.49% | 99.45% | 98.26% | 0.845 | |
| SVM | 96.70% | 96.70% | 96.70% | 0.786 |
| Organisms | Classifier | Sensitivity | Specificity | Accuracy | Matthews Correlation Coefficient |
|---|---|---|---|---|---|
| Amphibia | Our method | 100.00% | 98.24% | 98.80% | 0.973 |
| iAMPpred | 98.92% | 1.51% | 32.42% | 0.017 | |
| iAMP-2L | 96.76% | 98.99% | 98.28% | 0.960 | |
| ADAM | 98.38% | 99.50% | 99.14% | 0.980 | |
| DBAASP | 90.22% | 76.92% | 89.34% | 0.477 | |
| MLAMP | 90.27% | 98.24% | 95.71% | 0.900 | |
| CAMPR3_RF | 98.92% | 1.01% | 32.08% | −0.004 | |
| CAMPR3_SVM | 97.30% | 1.01% | 31.56% | −0.064 | |
| CAMPR3_ANN | 92.97% | 54.77% | 66.90% | 0.454 | |
| CAMPR3_DA | 95.14% | 0.75% | 30.70% | −0.135 | |
| Bacteria | Our method | 96.51% | 96.36% | 96.36% | 0.746 |
| iAMPpred | 84.88% | 1.99% | 6.46% | −0.183 | |
| iAMP-2L | 83.72% | 99.54% | 98.68% | 0.867 | |
| ADAM | 90.70% | 98.87% | 98.43% | 0.855 | |
| DBAASP | 35.44% | 80.00% | 57.86% | 0.173 | |
| MLAMP | 65.12% | 99.47% | 97.62% | 0.743 | |
| CAMPR3_RF | 90.70% | 1.99% | 6.77% | −0.108 | |
| CAMPR3_SVM | 79.07% | 2.72% | 6.83% | −0.218 | |
| CAMPR3_ANN | 68.60% | 45.00% | 46.27% | 0.062 | |
| CAMPR3_DA | 76.74% | 2.78% | 6.77% | −0.239 | |
| Fish | Our method | 100.00% | 97.00% | 97.18% | 0.810 |
| iAMPpred | 91.30% | 1.63% | 6.92% | −0.117 | |
| iAMP-2L | 86.96% | 99.46% | 98.72% | 0.882 | |
| ADAM | 95.65% | 99.18% | 98.97% | 0.912 | |
| DBAASP | 82.61% | 80.00% | 81.58% | 0.620 | |
| MLAMP | 91.30% | 99.46% | 98.97% | 0.908 | |
| CAMPR3_RF | 91.30% | 1.36% | 6.67% | −0.130 | |
| CAMPR3_SVM | 95.65% | 2.18% | 7.69% | −0.034 | |
| CAMPR3_ANN | 82.61% | 50.68% | 52.56% | 0.157 | |
| CAMPR3_DA | 86.96% | 1.36% | 6.41% | −0.194 | |
| Human | Our method | 97.83% | 92.17% | 92.33% | 0.482 |
| iAMPpred | 91.30% | 22.88% | 24.73% | 0.055 | |
| iAMP-2L | 54.35% | 98.18% | 96.99% | 0.482 | |
| ADAM | 52.17% | 98.91% | 97.64% | 0.534 | |
| DBAASP | 40.54% | 86.84% | 64.00% | 0.310 | |
| MLAMP | 50.00% | 98.36% | 97.05% | 0.464 | |
| CAMPR3_RF | 93.48% | 0.85% | 3.36% | −0.092 | |
| CAMPR3_SVM | 82.61% | 1.09% | 3.31% | −0.215 | |
| CAMPR3_ANN | 69.57% | 48.67% | 49.23% | 0.059 | |
| CAMPR3_DA | 84.78% | 1.46% | 3.72% | −0.167 | |
| Insects | Our method | 100.00% | 97.56% | 97.82% | 0.900 |
| iAMPpred | 94.44% | 39.11% | 45.04% | 0.217 | |
| iAMP-2L | 94.44% | 96.67% | 96.43% | 0.835 | |
| ADAM | 100.00% | 96.67% | 97.02% | 0.870 | |
| DBAASP | 70.37% | 90.91% | 73.85% | 0.469 | |
| MLAMP | 72.22% | 98.00% | 95.24% | 0.740 | |
| CAMPR3_RF | 87.04% | 1.33% | 10.52% | −0.227 | |
| CAMPR3_SVM | 87.04% | 1.33% | 10.52% | −0.227 | |
| CAMPR3_ANN | 87.04% | 43.33% | 48.02% | 0.192 | |
| CAMPR3_DA | 79.63% | 1.56% | 9.92% | −0.314 | |
| Mammals | Our method | 92.79% | 94.56% | 94.46% | 0.673 |
| iAMPpred | 95.50% | 68.94% | 70.54% | 0.322 | |
| iAMP-2L | 68.47% | 98.73% | 96.90% | 0.712 | |
| ADAM | 65.77% | 99.48% | 97.45% | 0.753 | |
| DBAASP | 45.88% | 83.02% | 60.14% | 0.295 | |
| MLAMP | 51.35% | 98.44% | 95.60% | 0.568 | |
| CAMPR3_RF | 93.69% | 1.27% | 6.85% | −0.096 | |
| CAMPR3_SVM | 92.79% | 1.91% | 7.39% | −0.085 | |
| CAMPR3_ANN | 78.38% | 48.58% | 50.38% | 0.129 | |
| CAMPR3_DA | 88.29% | 2.14% | 7.34% | −0.140 | |
| Plants | Our method | 97.78% | 97.94% | 97.93% | 0.851 |
| iAMPpred | 90.00% | 0.81% | 6.35% | −0.190 | |
| iAMP-2L | 77.78% | 98.67% | 97.38% | 0.773 | |
| ADAM | 84.44% | 98.67% | 97.79% | 0.815 | |
| DBAASP | 34.94% | 88.46% | 47.71% | 0.219 | |
| MLAMP | 58.89% | 98.82% | 96.34% | 0.654 | |
| CAMPR3_RF | 86.67% | 0.59% | 5.94% | −0.264 | |
| CAMPR3_SVM | 83.33% | 0.88% | 6.01% | −0.282 | |
| CAMPR3_ANN | 74.44% | 47.57% | 49.24% | 0.107 | |
| CAMPR3_DA | 75.56% | 1.10% | 5.73% | −0.357 |
| Physicochemical Properties | Group | ||
|---|---|---|---|
| Class 1 | Class 2 | Class 3 | |
| Charge | Positive K, R | Neutral A, N, C, Q, G, H, I, L, M, F, P, S, T, W, Y, V | Negative D, E |
| Hydrophobicity | Polar R, K, F, D, Q, N | Neutral G, A, S, T, P, H, Y | Hydrophobic C, L, V, I, M, F, W |
| Polarity | Polarity value 4.9~6.2 L, I, F, W, C, M, V, Y | Polarity value 8.0~9.2 P, A, T, G, S | Polarity value 10.4~13 H, Q, R, K, N, E, D |
| Polarizability | Polarizability value 0~0.108 G, A, S, D, T | Polarizability value 0.128~0.186 C, P, N, V, E, Q, I, L | Polarizability value 0.219~0.409 K, M, H, F, R, Y, W |
| Secondary Structure | Helix E, A, L, M, Q, K, R, H | Strand V, I, Y, C, W, F, T | Coil G, N, P, S, D |
| Normalized van der Waals volume | Volume range 0~2.78 G, A, S, T, P, D | Volume range 2.95~4.0 N, V, E, Q, I, L | Volume range 4.03~8.08 M, H, K, F, R, Y, W |
| Solvent accessibility | Buried A, L, F, C, G, I, V, W | Exposed R, K, Q, E, N, D | Intermediate M, P, S, T, H, Y |
References
- Huang, K.Y.; Chang, T.H.; Jhong, J.H.; Chi, Y.H.; Li, W.C.; Chan, C.L.; Robert Lai, K.; Lee, T.Y. Identification of natural antimicrobial peptides from bacteria through metagenomic and metatranscriptomic analysis of high-throughput transcriptome data of Taiwanese oolong teas. BMC Syst. Biol. 2017, 11, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waghu, F.H.; Barai, R.S.; Gurung, P.; Idicula-Thomas, S. CAMPR3: A database on sequences, structures and signatures of antimicrobial peptides. Nucleic Acids Res. 2016, 44, D1094–D1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabere, M.N.; Noble, W.S. Empirical comparison of web-based antimicrobial peptide prediction tools. Bioinformatics 2017, 33, 1921–1929. [Google Scholar] [CrossRef] [PubMed]
- Lata, S.; Sharma, B.K.; Raghava, G.P. Analysis and prediction of antibacterial peptides. BMC Bioinform. 2007, 8, 263. [Google Scholar] [CrossRef] [Green Version]
- Lata, S.; Mishra, N.K.; Raghava, G.P. AntiBP2: Improved version of antibacterial peptide prediction. BMC Bioinform. 2010, 11 (Suppl. 1), S19. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.; Karnik, S.; Barai, R.S.; Jayaraman, V.K.; Idicula-Thomas, S. CAMP: A useful resource for research on antimicrobial peptides. Nucleic Acids Res. 2010, 38, D774–D780. [Google Scholar] [CrossRef] [Green Version]
- Joseph, S.; Karnik, S.; Nilawe, P.; Jayaraman, V.K.; Idicula-Thomas, S. ClassAMP: A prediction tool for classification of antimicrobial peptides. IEEE/ACM Trans. Comput. Biol. Bioinform. 2012, 9, 1535–1538. [Google Scholar] [CrossRef]
- Thakur, N.; Qureshi, A.; Kumar, M. AVPpred: Collection and prediction of highly effective antiviral peptides. Nucleic Acids Res. 2012, 40, W199–W204. [Google Scholar] [CrossRef] [Green Version]
- Fjell, C.D.; Hancock, R.E.; Cherkasov, A. AMPer: A database and an automated discovery tool for antimicrobial peptides. Bioinformatics 2007, 23, 1148–1155. [Google Scholar] [CrossRef]
- Xiao, X.; Wang, P.; Lin, W.Z.; Jia, J.H.; Chou, K.C. iAMP-2L: A two-level multi-label classifier for identifying antimicrobial peptides and their functional types. Anal. Biochem. 2013, 436, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Meher, P.K.; Sahu, T.K.; Saini, V.; Rao, A.R. Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou’s general PseAAC. Sci. Rep. 2017, 7, 42362. [Google Scholar] [CrossRef] [PubMed]
- Bhadra, P.; Yan, J.; Li, J.; Fong, S.; Siu, S.W.I. AmPEP: Sequence-based prediction of antimicrobial peptides using distribution patterns of amino acid properties and random forest. Sci. Rep. 2018, 8, 1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veltri, D.; Kamath, U.; Shehu, A. Improving Recognition of Antimicrobial Peptides and Target Selectivity through Machine Learning and Genetic Programming. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017, 14, 300–313. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef] [Green Version]
- Hammami, R.; Ben Hamida, J.; Vergoten, G.; Fliss, I. PhytAMP: A database dedicated to antimicrobial plant peptides. Nucleic Acids Res 2009, 37, D963–D968. [Google Scholar] [CrossRef] [Green Version]
- Mishra, B.; Wang, G. The Importance of Amino Acid Composition in Natural AMPs: An Evolutional, Structural, and Functional Perspective. Front. Immunol. 2012, 3, 221. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.R.; Kuo, T.R.; Wu, L.C.; Lee, T.Y.; Horng, J.T. Characterization and identification of antimicrobial peptides with different functional activities. Brief Bioinform. 2019. [Google Scholar] [CrossRef]
- Lee, H.T.; Lee, C.C.; Yang, J.R.; Lai, J.Z.; Chang, K.Y. A large-scale structural classification of antimicrobial peptides. Biomed. Res. Int. 2015, 2015, 475062. [Google Scholar] [CrossRef]
- Vishnepolsky, B.; Pirtskhalava, M. Prediction of Linear Cationic Antimicrobial Peptides Based on Characteristics Responsible for Their Interaction with the Membranes. J. Chem. Inf. Model. 2014, 54, 1512–1523. [Google Scholar] [CrossRef]
- Fan, L.; Sun, J.; Zhou, M.; Zhou, J.; Lao, X.; Zheng, H.; Xu, H. DRAMP: A comprehensive data repository of antimicrobial peptides. Sci. Rep. 2016, 6, 24482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, K.Y.; Lin, T.P.; Shih, L.Y.; Wang, C.K. Analysis and prediction of the critical regions of antimicrobial peptides based on conditional random fields. PLoS ONE 2015, 10, e0119490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavares, L.S.; Rettore, J.V.; Freitas, R.M.; Porto, W.F.; Duque, A.P.; Singulani Jde, L.; Silva, O.N.; Detoni Mde, L.; Vasconcelos, E.G.; Dias, S.C.; et al. Antimicrobial activity of recombinant Pg-AMP1, a glycine-rich peptide from guava seeds. Peptides 2012, 37, 294–300. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, K. Control of cell selectivity of antimicrobial peptides. Biochim. Biophys. Acta 2009, 1788, 1687–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tadeg, H.; Mohammed, E.; Asres, K.; Gebre-Mariam, T. Antimicrobial activities of some selected traditional Ethiopian medicinal plants used in the treatment of skin disorders. J. Ethnopharmacol. 2005, 100, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Hilpert, K.; Elliott, M.; Jenssen, H.; Kindrachuk, J.; Fjell, C.D.; Korner, J.; Winkler, D.F.; Weaver, L.L.; Henklein, P.; Ulrich, A.S.; et al. Screening and characterization of surface-tethered cationic peptides for antimicrobial activity. Chem. Biol. 2009, 16, 58–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnsen, L.; Fimland, G.; Nissen-Meyer, J. The C-terminal domain of pediocin-like antimicrobial peptides (class IIa bacteriocins) is involved in specific recognition of the C-terminal part of cognate immunity proteins and in determining the antimicrobial spectrum. J. Biol. Chem. 2005, 280, 9243–9250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dathe, M.; Nikolenko, H.; Meyer, J.; Beyermann, M.; Bienert, M. Optimization of the antimicrobial activity of magainin peptides by modification of charge. FEBS Lett. 2001, 501, 146–150. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Guarnieri, M.T.; Vasil, A.I.; Vasil, M.L.; Mant, C.T.; Hodges, R.S. Role of peptide hydrophobicity in the mechanism of action of alpha-helical antimicrobial peptides. Antimicrob. Agents Chemother. 2007, 51, 1398–1406. [Google Scholar] [CrossRef] [Green Version]
- Pirtskhalava, M.; Gabrielian, A.; Cruz, P.; Griggs, H.L.; Squires, R.B.; Hurt, D.E.; Grigolava, M.; Chubinidze, M.; Gogoladze, G.; Vishnepolsky, B.; et al. DBAASP v.2: An enhanced database of structure and antimicrobial/cytotoxic activity of natural and synthetic peptides. Nucleic Acids Res. 2016, 44, D1104–D1112. [Google Scholar] [CrossRef]
- Lin, W.Z.; Xu, D. Imbalanced multi-label learning for identifying antimicrobial peptides and their functional types. Bioinformatics 2016, 32, 3745–3752. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.D.T.; de la Fuente-Nunez, C. Toward computer-made artificial antibiotics. Curr. Opin. Microbiol. 2019, 51, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Porto, W.F.; Irazazabal, L.; Alves, E.S.; Ribeiro, S.M.; Matos, C.O.; Pires, Á.S.; Fensterseifer, I.C.; Miranda, V.J.; Haney, E.F.; Humblot, V. In silico optimization of a guava antimicrobial peptide enables combinatorial exploration for peptide design. Nature Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Hu, L.; Liu, G.; Jiang, N.; Chen, X.; Xu, J.; Zheng, W.; Li, L.; Tan, M.; Chen, Z.; et al. Prediction of antimicrobial peptides based on sequence alignment and feature selection methods. PLoS ONE 2011, 6, e18476. [Google Scholar] [CrossRef]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef]
- Hall, M.; Frank, E.; Holmes, G.; Pfahringer, B.; Reutemann, P.; Witten, I.H. The WEKA data mining software: An update. ACM SIGKDD explorations newsletter. ACM SIGKDD Explor. Newsl. 2009, 11, 10–18. [Google Scholar] [CrossRef]
- Vapnik, V.N. An overview of statistical learning theory. IEEE Trans. Neural. Netw. 1999, 10, 988–999. [Google Scholar] [CrossRef] [Green Version]
- Salzberg, S. Locating protein coding regions in human DNA using a decision tree algorithm. J. Comput. Biol. 1995, 2, 473–485. [Google Scholar] [CrossRef] [Green Version]







| Organisms | Number of Peptides with Length L | ||||||
|---|---|---|---|---|---|---|---|
| L ≤ 20 | 20 < L ≤ 40 | 40 < L ≤ 60 | 60 < L ≤ 80 | 80 < L ≤ 100 | 100 < L | Total | |
| Amphibia | 269 | 437 | 28 | 3 | 0 | 4 | 741 |
| Bacteria | 117 | 111 | 61 | 16 | 13 | 27 | 345 |
| Fish | 18 | 54 | 10 | 5 | 3 | 5 | 95 |
| Human | 11 | 53 | 13 | 26 | 7 | 76 | 186 |
| Insects | 67 | 94 | 32 | 12 | 7 | 8 | 220 |
| Mammals | 78 | 180 | 51 | 43 | 11 | 85 | 448 |
| Plants | 63 | 153 | 95 | 7 | 14 | 32 | 364 |
| Organisms | Sensitivity | Specificity | Accuracy | Matthews Correlation Coefficient |
|---|---|---|---|---|
| Amphibia | 100.00% | 98.24% | 98.80% | 0.973 |
| Bacteria | 96.51% | 96.36% | 96.36% | 0.746 |
| Fish | 100.00% | 97.00% | 97.18% | 0.810 |
| Human | 97.83% | 92.17% | 92.33% | 0.482 |
| Insects | 100.00% | 97.56% | 97.82% | 0.900 |
| Mammals | 92.79% | 94.56% | 94.46% | 0.673 |
| Plants | 97.78% | 97.94% | 97.93% | 0.851 |
| Organisms | Training Dataset | Testing Dataset | ||
|---|---|---|---|---|
| Positive | Negative | Positive | Negative | |
| Amphibia | 741 | 1595 | 185 | 398 |
| Bacteria | 345 | 6040 | 86 | 1509 |
| Fish | 95 | 1469 | 23 | 367 |
| Human | 186 | 6595 | 46 | 1648 |
| Insects | 220 | 1800 | 54 | 450 |
| Mammals | 448 | 6919 | 111 | 1729 |
| Plants | 364 | 5432 | 90 | 1358 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chung, C.-R.; Jhong, J.-H.; Wang, Z.; Chen, S.; Wan, Y.; Horng, J.-T.; Lee, T.-Y. Characterization and Identification of Natural Antimicrobial Peptides on Different Organisms. Int. J. Mol. Sci. 2020, 21, 986. https://doi.org/10.3390/ijms21030986
Chung C-R, Jhong J-H, Wang Z, Chen S, Wan Y, Horng J-T, Lee T-Y. Characterization and Identification of Natural Antimicrobial Peptides on Different Organisms. International Journal of Molecular Sciences. 2020; 21(3):986. https://doi.org/10.3390/ijms21030986
Chicago/Turabian StyleChung, Chia-Ru, Jhih-Hua Jhong, Zhuo Wang, Siyu Chen, Yu Wan, Jorng-Tzong Horng, and Tzong-Yi Lee. 2020. "Characterization and Identification of Natural Antimicrobial Peptides on Different Organisms" International Journal of Molecular Sciences 21, no. 3: 986. https://doi.org/10.3390/ijms21030986
APA StyleChung, C. -R., Jhong, J. -H., Wang, Z., Chen, S., Wan, Y., Horng, J. -T., & Lee, T. -Y. (2020). Characterization and Identification of Natural Antimicrobial Peptides on Different Organisms. International Journal of Molecular Sciences, 21(3), 986. https://doi.org/10.3390/ijms21030986