Transcriptomic Analysis of Ficus carica Peels with a Focus on the Key Genes for Anthocyanin Biosynthesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Anthocyanin Contents in Fig Fruit Peels
2.2. Library Construction, De novo Assembly, and Annotation
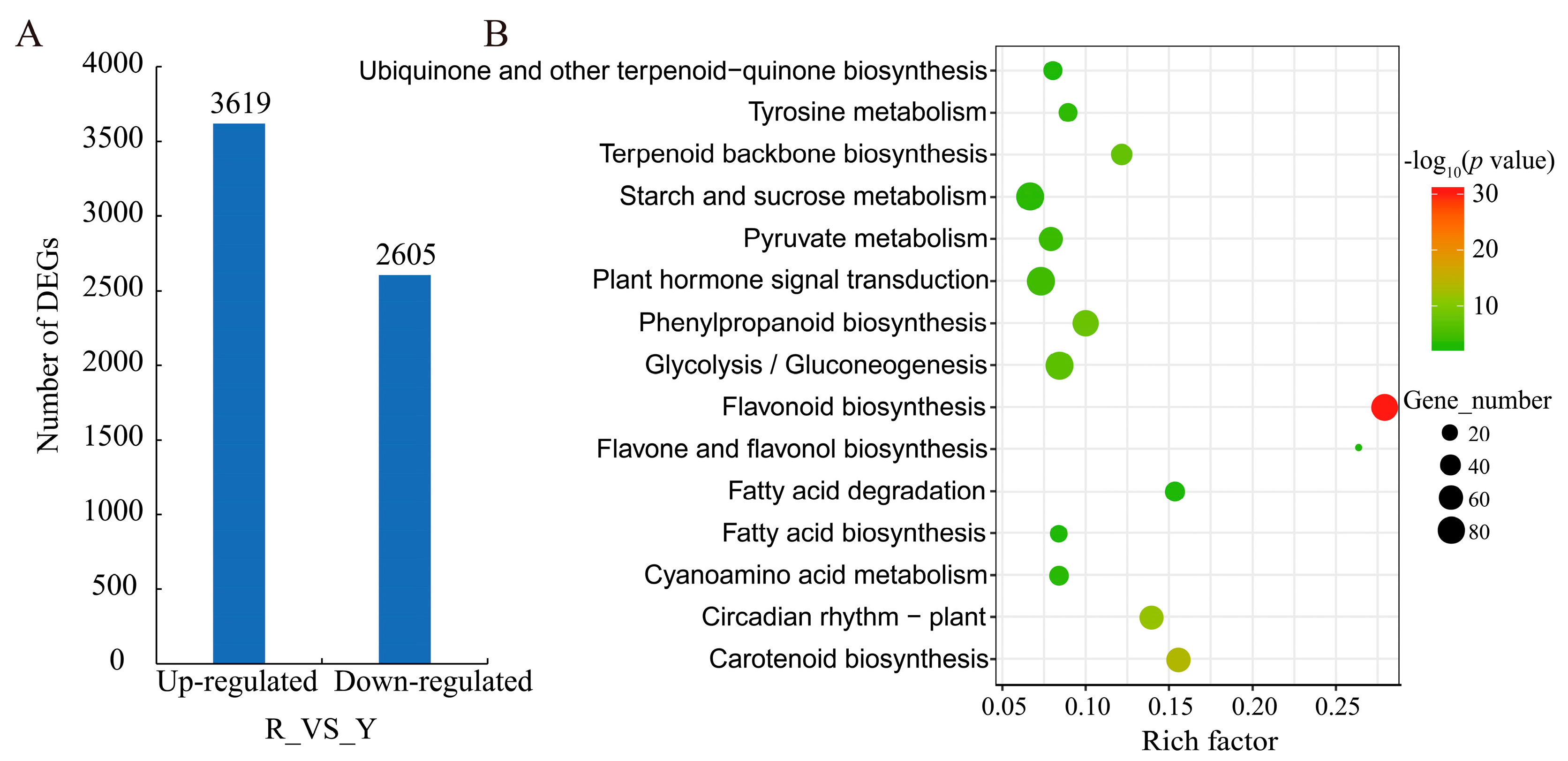
2.3. Analysis of Differentially Expressed Genes
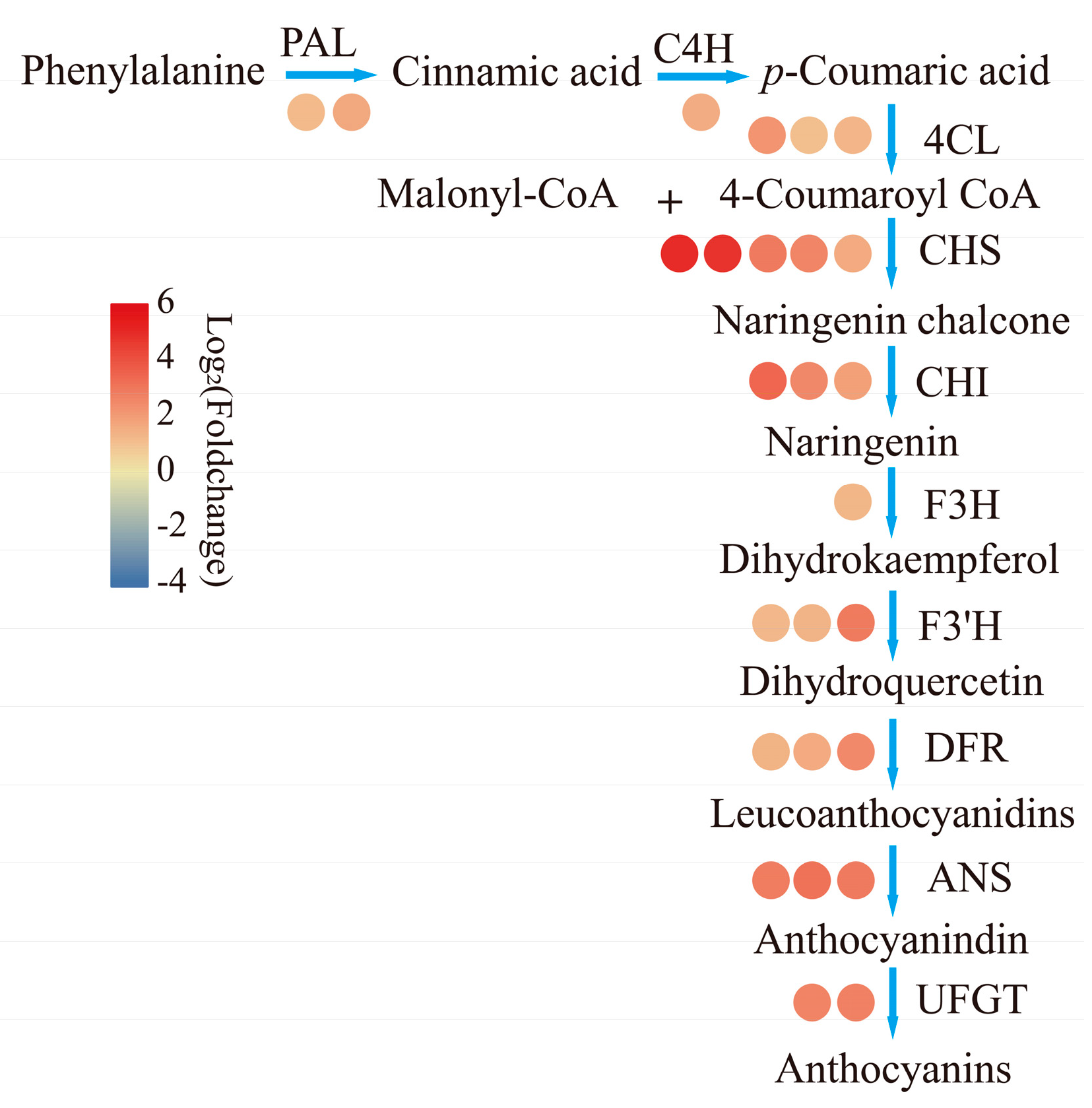
2.4. DEGs Related to Anthocyanin Biosynthesis
2.5. Candidate MYBs of Anthocyanin Biosynthesis
2.6. Molecular Characteristics and Sequence Analysis of FcCHS1, FcCHI1 and FcDFR1
2.7. Sequence Characterization of FcMYB21 and FcMYB123
2.8. The Function of FcMYB21 and FcMYB123 in Apple Peels and Calli
3. Discussion
3.1. De novo Assembled the Peels Transcriptome during Coloration
3.2. Candidate and Cloned Structural Genes of Fig Peels
3.3. Candidate and Validated Functions of MYBs of Fig Peels
4. Materials and Methods
4.1. Plant Materials
4.2. Determination of Anthocyanin Contents
4.3. Library Construction, Sequence Assembly and Gene Annotation
4.4. Differentially Expressed Genes Analysis
4.5. Sequences Alignment, Phylogenetic Relationship and Bioinformatics Analysis
4.6. Gene Clone and Construct Expression Vectors
4.7. Agrobacterium-Mediated Transformation System of Apple Peels and Apple Fruit Calli
4.8. RNA Extraction and Gene Expression Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BEN1 | BRI1-5 ENHANCED 1 |
| bHLH | basic helix-loop-helix |
| DEGs | Differentially expressed genes |
| FPKM | Fragments per kilobase of gene per million mapped reads |
| GO | Gene ontology |
| KEGG | Kyoto encyclopedia of genes and genomes |
| KOG | Clusters of orthologous groups of proteins |
| MS | Murashige and Skoog medium |
| Mw | Molecular weight |
| Nr | The NCBI non-redundant protein sequences |
| Nt | The NCBI non-redundant nucleotide sequences |
| ORF | Open reading frame |
| Pfam | Protein family |
| pI | Isoelectric point |
| RNA-seq | RNA-sequencing |
| SA | Salicylic acid |
References
- Kislev, M.E.; Hartmann, A.; Bar, Y.O. Early domesticated fig in the Jordan Valley. Science 2006, 312, 1372–1374. [Google Scholar] [CrossRef] [PubMed]
- Aradhya, M.K.; Stover, E.; Velasco, D.; Koehmstedt, A. Genetic structure and differentiation in cultivated fig (Ficus carica L.). Genetica 2010, 138, 681–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khadivi, A.; Anjam, R.; Anjam, K. Morphological and pomological characterization of edible fig (Ficus carica L.) to select the superior trees. Sci. Hortic. 2018, 238, 66–74. [Google Scholar] [CrossRef]
- Dueñas, M.; Pérez, A.J.J.; Santos, B.C.; Escribano, B.T. Anthocyanin composition in fig (Ficus carica L.). J. Food Compos. Anal. 2008, 21, 107–115. [Google Scholar]
- Ercisli, S.; Tosun, M.; Karlidag, H.; Dzubur, A.; Hadziabulic, S.; Aliman, Y. Color and antioxidant characteristics of some fresh fig (Ficus carica L.) genotypes from northeastern turkey. Plant Foods Hum. Nutr. 2012, 67, 271–276. [Google Scholar] [CrossRef]
- Sun, C.D.; Huang, H.Z.; Xu, C.J.; Li, X.; Chen, K.S. Biological activities of extracts from Chinese bayberry (Myrica rubra Sieb. et Zucc.). Plant Foods Hum. Nutr. 2013, 68, 97–106. [Google Scholar] [CrossRef]
- Tanaka, Y.; Sasaki, N.; Ohmiya, A. Biosynthesis of plant pigments: Anthocyanins, betalains and carotenoids. Plant J. 2008, 54, 733–749. [Google Scholar] [CrossRef]
- Lai, B.; Hu, B.; Qin, Y.H.; Zhao, J.T.; Wang, H.C.; Hu, G.B. Transcriptomic analysis of Litchi chinensis pericarp during maturation with a focus on chlorophyll degradation and flavonoid biosynthesis. BMC Genom. 2015, 16, 225–243. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.Y.; Chen, X.; Chen, W.; Zheng, Y.H.; Yang, Z.F. Comparative transcriptomic analysis of white and red Chinese bayberry (Myrica rubra) fruits reveals flavonoid biosynthesis regulation. Sci. Hortic. 2018, 235, 9–20. [Google Scholar] [CrossRef]
- Allan, A.C.; Hellens, R.P.; Laing, W.A. MYB transcription factors that colour our fruit. Trends Plant Sci. 2008, 13, 99–102. [Google Scholar] [CrossRef]
- Borevitz, J.O.; Xia, Y.; Blount, J.; Lamb, D.C. Activation tagging identifies a conserved MYB regulator of phenylpropanoid biosynthesis. Plant Cell 2000, 12, 2383–2393. [Google Scholar] [CrossRef] [Green Version]
- Ban, Y.; Honda, C.; Hatsuyama, Y.; Igarashi, M.; Bessho, H.; Moriguchi, T. Isolation and functional analysis of a MYB transcription factor gene that is a key regulator for the development of red coloration in apple skin. Plant Cell Physiol. 2007, 48, 958–970. [Google Scholar] [CrossRef]
- Espley, R.V.; Hellens, R.P.; Putterill, J.; Stevenson, D.E.; Kutty-Amma, S.; Allan, A.C. Red colouration in apple fruit is due to the activity of the MYB transcription factor, MdMYB10. Plant J. 2007, 49, 414–427. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Gotoyamamoto, N.; Hirochika, H. Retrotransposon-induced mutations in grape skin color. Science 2004, 304, 982. [Google Scholar] [CrossRef]
- Walker, A.R.; Lee, E.; Bogs, J.; Mcdavid, D.A.J.; Thomas, M.R.; Robinson, S.P. White grapes arose through the mutation of two similar and adjacent regulatory genes. Plant J. 2007, 49, 772–785. [Google Scholar] [CrossRef]
- Barolo, M.I.; Ruiz, M.N.; López, S.N. Ficus carica L. (Moraceae): An ancient source of food and health. Food Chem. 2014, 164, 119–127. [Google Scholar] [CrossRef]
- Sun, R.; Jia, M.; Sun, L. World figs resources development and applied research. World For. Res. 2015, 28, 31–36. [Google Scholar]
- Cao, L.C.; Xu, X.Y.; Chen, S.W.; Ma, H.Q. Cloning and expression analysis of Ficus carica anthocyanidin synthase 1 gene. Sci. Hortic. 2016, 211, 369–375. [Google Scholar] [CrossRef]
- An, X.H.; Tian, Y.; Chen, K.Q.; Liu, X.J.; Liu, D.D.; Xie, X.B.; Cheng, C.G.; Cong, P.H.; Hao, Y.J. MdMYB9 and MdMYB11 are involved in the regulation of the JA-induced biosynthesis of anthocyanin and proanthocyanidin in apples. Plant Cell Physiol. 2015, 56, 650–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nesi, N.; Jond, C.; Debeaujon, I.; Caboche, M.; Lepiniec, L. The Arabidopsis TT2 gene encodes an R2R3 MYB domain protein that acts as a key determinant for proanthocyanidin accumulation in developing seed. Plant Cell 2001, 13, 2099–2114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, S.H.; Qi, T.C.; Huang, H.; Ren, Q.C.; Wu, D.W.; Chang, C.Q.; Peng, W.; Liu, Y.L.; Peng, J.R.; Xie, D.X. The jasmonate-zim domain proteins interact with the R2R3-MYB transcription factors MYB21 and MYB24 to affect jasmonate-regulated stamen development in Arabidopsis. Plant Cell 2011, 23, 1000–1013. [Google Scholar] [CrossRef] [Green Version]
- Moyano, E.; Martine, G.J.F.; Martin, C. Apparent Redundancy in MYB Gene function provides gearing for the control of flavonoid biosynthesis in Antirrhinum flowers. Plant Cell 1996, 8, 1519–1532. [Google Scholar]
- Marioni, J.C.; Mason, C.E.; Mane, S.M.; Stephens, M.; Gilad, Y. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008, 18, 1509–1517. [Google Scholar] [CrossRef] [Green Version]
- Fang, Z.Z.; Zhou, D.R.; Ye, X.F.; Jiang, C.C.; Pan, S.L. Identification of candidate anthocyanin-related genes by transcriptomic analysis of ‘Furongli’ plum (Prunus salicina Lindl.) during fruit ripening using RNA-seq. Front. Plant Sci. 2016, 7, 1338–2353. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.R.; Cui, Y.Y.; Vainstein, A.; Chen, S.W.; Ma, H.Q. Regulation of fig (Ficus carica L.) fruit color: Metabolomic and transcriptomic analyses of the flavonoid biosynthetic pathway. Front. Plant Sci. 2017, 8, 1990–2005. [Google Scholar] [CrossRef] [Green Version]
- Ikegami, H.; Habu, T.; Mori, K.; Nogata, H.; Hirata, C.; Hirashima, K.; Tashiro, K.; Kuhara, S. De novo sequencing and comparative analysis of expressed sequence tags from gynodioecious fig ( Ficus carica L.) fruits: Caprifig and common Figure. Tree Genet. Genomes 2013, 9, 1075–1088. [Google Scholar]
- Mori, K.; Shirasawa, K.; Nogata, H.; Hirata, C.; Tashiro, K.; Habu, T.; Kim, S.; Himeno, S.; Kuhara, S.; Ikegami, H. Identification of RAN1 orthologue associated with sex determination through whole genome sequencing analysis in fig (Ficus carica L.). Sci. Rep. 2017, 7, 41124–41136. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.X.; An, Y.Y.; Zheng, J.; Sun, M.; Wang, L.J. Proteomics and SSH analyses of ALA-promoted fruit coloration and evidence for the involvement of a MADS-box gene, MdMADS1. Front. Plant Sci. 2016, 7, 1615–1634. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.W.; Kim, H.J.; Chen, H.B.; Lu, X.Y.; Zhou, B.Y. Identification of MV-generated ROS responsive EST clones in floral buds of Litchi chinensis Sonn. Plant Cell Rep. 2013, 32, 1361–1372. [Google Scholar] [CrossRef]
- Liu, R.; Xu, S.; Li, J.; Hu, Y.; Lin, Z. Expression profile of a PAL gene from Astragalus membranaceus var. Mongholicus and its crucial role in flux into flavonoid biosynthesis. Plant Cell Rep. 2006, 25, 705–710. [Google Scholar]
- Wanner, L.A.; Li, G.; Ware, D.; Somssich, I.E.; Davis, K.R. The phenylalanine ammonia-lyase gene family in Arabidopsis thaliana. Plant Mol. Biol. 1995, 27, 327–338. [Google Scholar] [CrossRef]
- Shi, R.; Shuford, C.M.; Wang, J.P.; Sun, Y.H.; Yang, Z.C.; Chen, H.C.; Tunlaya, A.S.; Li, Q.Z.; Liu, J.; Muddiman, D.C.; et al. Regulation of phenylalanine ammonia-lyase (PAL) gene family in wood forming tissue of Populus trichocarpa. Planta 2013, 238, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Lepelley, M.; Mahesh, V.; Mccarthy, J.; Rigoreau, M.; Crouzillat, D.; Chabrillange, N.; Kochko, A.D.; Campa, C. Characterization, high-resolution mapping and differential expression of three homologous genes in Coffea canephora Pierre (Rubiaceae). Planta 2012, 236, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Kim, J.I.; Pysh, L.; Chapple, C. Four isoforms of Arabidopsis thaliana 4-coumarate: CoA ligase (4CL) have overlapping yet distinct roles in phenylpropanoid metabolism. Plant Physiol. 2015, 169, 2409–2421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindermayr, C.; Britta, M.; Fliegmann, J.; Uhlmann, A.; Lottspeich, F.; Meimberg, H.; Ebel, J. Divergent members of a soybean (Glycine max L.) 4-coumarate: Coenzyme a ligase gene family: Primary structures, catalytic properties, and differential expression. Eur. J. Biochem. 2002, 269, 1304–1315. [Google Scholar] [CrossRef] [PubMed]
- Gui, J.S.; Shen, J.H.; Li, L.G. Functional characterization of evolutionarily divergent 4-coumarate: Coenzyme a ligases in rice. Plant Physiol. 2011, 157, 574–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.H.; Yu, J.; Cai, Y.X.; Zhu, P.P.; Liu, C.Y.; Zhao, A.C.; Lü, R.H.; Li, M.J.; Xu, F.X.; Yu, M.D. Characterization and functional analysis of 4-coumarate: CoA ligase genes in mulberry. PLoS ONE 2016, 11, e0155814. [Google Scholar]
- Bell, L.D.A.; Cusumano, J.C.; Meyer, K.; Chapple, C. Cinnamate-4-hydroxylase expression in Arabidopsis. regulation in response to development and the environment. Plant Physiol. 1997, 113, 729–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koopmann, E.; Logemann, E.; Hahlbrock, K. Regulation and functional expression of cinnamate 4-hydroxylase from parsley. Plant Physiol. 1999, 119, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Pang, Y.Z.; Shen, G.A.; Wu, W.S.; Liu, X.F.; Lin, J.; Tan, F.; Sun, X.F.; Tang, K.X. Characterization and expression of chalcone synthase gene from Ginkgo biloba. Plant Sci. 2005, 168, 1525–1531. [Google Scholar] [CrossRef]
- Cheng, A.X.; Zhang, X.B.; Han, X.J.; Zhang, Y.Y.; Gao, S.; Liu, C.J.; Luo, H.X. Identification of chalcone isomerase in the basal land plants reveals an ancient evolution of enzymatic cyclization activity for synthesis of flavonoids. New Phytol. 2017, 217, 909–924. [Google Scholar] [CrossRef] [Green Version]
- Shimada, N.; Aoki, T.; Sato, S.; Nakamura, Y.; Tabata, S.; Ayabe, T.S.I. A cluster of genes encodes the two types of chalcone isomerase involved in the biosynthesis of general flavonoids and Legume-specific 5-deoxy (iso) flavonoids in Lotus japonicus. Plant Physiol. 2003, 131, 941–951. [Google Scholar] [CrossRef] [Green Version]
- Zuker, A.; Tzfira, T.; Ben, M.H.; Ovadis, M.; Shklarman, E.; Itzhaki, H.; Forkmann, G.; Martens, S.; Nete, S.I.; Weiss, D.; et al. Modification of flower color and fragrance by antisense suppression of the flavanone 3-hydroxylase gene. Mol. Breed. 2002, 9, 33–41. [Google Scholar] [CrossRef]
- Schoenbohm, C.; Martens, S.; Eder, C.; Forkmann, G.; Weisshaar, B. Identification of the Arabidopsis thaliana flavonoid 3′-hydroxylase gene and functional expression of the encoded P450 enzyme. Biol. Chem. 2000, 381, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Feng, F.J.; Li, M.J.; Ma, F.W.; Cheng, L.L. Phenylpropanoid metabolites and expression of key genes involved in anthocyanin biosynthesis in the shaded peel of apple fruit in response to sun exposure. Plant Physiol. Biochem. 2013, 69, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.N.; Yao, G.F.; Zheng, D.M.; Zhang, S.L.; Wang, C.; Zhang, M.Y.; Wu, J. Expression differences of anthocyanin biosynthesis genes reveal regulation patterns for red pear coloration. Plant Cell Rep. 2014, 34, 189–198. [Google Scholar] [CrossRef]
- Xu, W.P.; Peng, H.; Yang, T.B.; Whitaker, B.; Huang, L.H.; Sun, J.H.; Chen, P. Effect of calcium on strawberry fruit flavonoid pathway gene expression and anthocyanin accumulation. Plant Physiol. Biochem. 2014, 82, 289–298. [Google Scholar] [CrossRef]
- Wu, X.X.; Gong, Q.H.; Ni, X.P.; Zhou, Y.; Gao, Z.H. UFGT: The key enzyme associated with the petals variegation in Japanese apricot. Front. Plant Sci. 2017, 8, 108–122. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.M.; Zhu, P.P.; Li, J.; Zhao, A.C.; Liu, C.Y.; Diane, U.; Li, Z.G.; Lu, C.; Yu, M.D. Identification of MaUFGTs from mulberry and function analysis of the major gene. Acta Hortic. Sin. 2019, 42, 1919–1930. [Google Scholar]
- Wang, Z.R.; Song, M.Y.; Li, Y.Z.; Chen, S.W.; Ma, H.Q. Differential color development and response to light deprivation of fig (Ficus carica L.) syconia peel and female flower tissues: Transcriptome elucidation. BMC Plant Biol. 2019, 19, 217–236. [Google Scholar] [CrossRef]
- Ohno, S.; Hosokawa, M.; Kojima, M.; Kitamura, Y.; Hoshino, A.; Tatsuzawa, F.; Doi, M.; Yazawa, S. Simultaneous post-transcriptional gene silencing of two different chalcone synthase genes resulting in pure white flowers in the octoploid dahlia. Planta 2011, 234, 945–958. [Google Scholar] [CrossRef] [Green Version]
- Nishihara, M.; Nakatsuka, T.; Hosokawa, K.; Yokoi, T.; Abe, Y.; Mishiba, K.; Yamamura, S. Dominant inheritance of white-flowered and herbicide-resistant traits in trangenic gentian plants. Plant Biotechnol. 2006, 23, 25–31. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Jones, R.; Yoo, K.S.; Pike, L.M. Gold color in onions (Allium cepa): A natural mutation of the chalcone isomerase gene resulting in a premature stop codon. Mol. Genet. Genom. 2004, 272, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Aida, R.; Yoshida, K.; Kondo, T.; Kishimoto, S.; Shibata, M. Copigmentation gives bluer flowers on transgenic torenia plants with the antisense dihydroflavonol-4-reductase gene. Plant Sci. 2000, 160, 49–56. [Google Scholar] [CrossRef]
- Johnson, E.T.; Ryu, S.; Shin, B.; Cheong, H.; Choi, G. Alteration of a single amino acid changes the substrate specificity of dihydroflavonol 4-reductase. Plant J. 2001, 25, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.W.; Qin, S.; Chen, H.; Li, F.; Zeng, Q.W.; He, N.J. Cloning and expression analyses of the anthocyanin biosynthetic genes in mulberry plants. Mol. Genet. Genom. 2014, 289, 783–793. [Google Scholar] [CrossRef]
- Cheng, H.; Li, L.L.; Cheng, S.Y.; Cao, F.; Xu, F.L.; Yuan, H.H.; Wu, C.H. Molecular cloning and characterization of three genes encoding dihydroflavonol-4-reductase from Ginkgo biloba in anthocyanin biosynthetic pathway. PLoS ONE 2013, 8, e72017. [Google Scholar]
- Johnson, E.T.; Yi, H.; Shin, B.; Oh, B.J.; Cheong, H.; Choi, G. Cymbidium hybrida dihydroflavonol 4-reductase does not efficiently reduce dihydrokaempferol to produce orange pelargonidin-type anthocyanins. Plant J. 1999, 19, 81–85. [Google Scholar] [CrossRef]
- Jez, J.M.; Noel, J.P. Mechanism of chalcone synthase: pKa of the catalytic cysteine and the role of the conserved histidine in a plant polyketide synthase. J. Biol. Chem. 2000, 275, 39640–39646. [Google Scholar] [CrossRef] [Green Version]
- Jaakola, L. New insights into the regulation of anthocyanin biosynthesis in fruits. Trends Plant Sci. 2013, 18, 477–483. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Chen, Z.; Kang, J.; Kang, D.; Gu, H.; Qin, G. AtMYB14 regulates cold tolerance in Arabidopsis. Plant Mol. Biol. Rep. 2013, 31, 87–97. [Google Scholar] [CrossRef] [Green Version]
- Mandaokar, A.; Browse, M.J. MYB108 acts together with MYB24 to regulate jasmonate-mediated stamen maturation in Arabidopsis. Plant Physiol. 2009, 149, 851–862. [Google Scholar] [CrossRef] [Green Version]
- Jung, C.; Seo, J.S.; Han, S.W.; Koo, Y.J.; Kim, C.H.; Song, S.I.; Nahm, B.H.; Choi, Y.D.; Cheong, J. Overexpression of AtMYB44 enhances stomatal closure to confer abiotic stress tolerance in transgenic Arabidopsis. Plant Physiol. 2007, 146, 623–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christian, D.; Ralf, S.; Erich, G.; Bernd, W.; Cathie, M.; LoïC, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar]
- Gonzalez, A.; Zhao, M.; Leavitt, J.M.; Lloyd, A.M. Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLH/MYB transcriptional complex in Arabidopsis seedlings. Plant J. 2008, 53, 814–827. [Google Scholar] [CrossRef] [PubMed]
- Chagné, D.; Lin, W.K.; Espley, R.V.; Volz, R.K.; How, N.M.; Rouse, S.; Brendolise, C.; Carlisle, C.M.; Kumar, S.; De, S.N.; et al. An ancient duplication of apple MYB transcription factors is responsible for novel red fruit-flesh phenotypes. Plant Physiol. 2013, 161, 225–239. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.Y. Genome-Wide Analysis and Expression Involved in Flavonoids Synthesis of the Mulberry MYB Transcription Factor. Master’s Thesis, Southwest University, Chongqing, China, 2015. [Google Scholar]
- Liu, G.; Ren, G.; Guirgis, A.; Thornburg, R.W. The MYB305 transcription factor regulates expression of nectarin genes in the ornamental tobacco floral nectary. Plant Cell 2009, 21, 2672–2687. [Google Scholar] [CrossRef] [Green Version]
- Uematsu, C.; Katayama, H.; Makino, I.; Inagaki, A.; Arakawa, O.; Martin, C. Peace, a MYB-like transcription factor, regulates petal pigmentation in flowering peach Genpei bearing variegated and fully pigmented flowers. J. Exp. Bot. 2014, 65, 1081–1094. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.H.; Chen, M.; He, N.B.; Chen, X.L.; Wang, N.; Chen, X.S. MdGSTF6, activated by MdMYB1, plays an essential role in anthocyanin accumulation in apple. Hortic. Res. 2019, 6, 40–54. [Google Scholar] [CrossRef] [Green Version]
- Lai, B.; Li, X.J.; Hu, B.; Qin, Y.H.; Huang, X.M.; Wang, H.C.; Hu, G.B. LcMYB1 is a key determinant of differential anthocyanin accumulation among genotypes, tissues, developmental phases and ABA and light stimuli in Litchi chinensis. PLoS ONE 2014, 21, e86293. [Google Scholar] [CrossRef]
- Huang, Y.J.; Song, S.; Andrew, C.A.; Liu, X.F.; Yin, X.R.; Xu, C.J.; Chen, K.S. Differential activation of anthocyanin biosynthesis in Arabidopsis and tobacco over-expressing an R2R3 MYB from Chinese bayberry. Plant Cell Tissue Organ Cult. 2013, 113, 491–499. [Google Scholar] [CrossRef]
- Qi, T.C.; Huang, H.; Song, S.S.; Xie, D.X. Regulation of jasmonate-mediated stamen development and seed production by a bHLH-MYB complex in Arabidopsis. Plant Cell 2015, 27, 1620–1633. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; An, Y.Y.; Wang, L.J. 24-epibrassinolide enhances 5-ALA -induced anthocyanin and flavonol accumulation in calli of ‘fuji’ apple flesh. Plant Cell Tissue Organ Cult. 2018, 134, 319–330. [Google Scholar] [CrossRef]
- Ma, Z.B.; Cheng, Y.E. Methods for the chemical determination of anthocyanin on the surface of apple fruit. China Fruits 1984, 4, 49–51. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, N.M.; Oshlack, A. Corset: Enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biol. 2014, 15, 410–424. [Google Scholar]
- Götz, S.; García-Gómez, J.M.; Javier, T.; Williams, T.D.; Nagaraj, S.H.; Nagaraj, M.J.; Nueda, M.J.; Robles, M.; Talón, M.; Dopazo, J.; et al. High-throughput functional annotation and data mining with the Blast2go suite. Nucleic Acids Res. 2008, 36, 3420–3435. [Google Scholar] [CrossRef]
- Bo, L.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323–335. [Google Scholar]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van, B.M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DEseq2. Genome Biol. 2014, 15, 550–571. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. Mega5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [Green Version]
- An, J.P.; Yao, J.F.; Xu, R.R.; You, C.X.; Wang, X.F.; Hao, Y.J. Apple bZIP transcription factor MdbZIP44 regulates abscisic acid-promoted anthocyanin accumulation. Plant Cell Environ. 2018, 41, 2678–2692. [Google Scholar] [CrossRef]
- Jaakola, L.; Pirttilä, A.M.; Halonen, M.; Hohtola, A. Isolation of high quality RNA from bilberry (Vaccinium myrtillus L.) fruit. Mol. Biotechnol. 2001, 19, 201–203. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; An, Y.; Wang, L. Transcriptomic Analysis of Ficus carica Peels with a Focus on the Key Genes for Anthocyanin Biosynthesis. Int. J. Mol. Sci. 2020, 21, 1245. https://doi.org/10.3390/ijms21041245
Li J, An Y, Wang L. Transcriptomic Analysis of Ficus carica Peels with a Focus on the Key Genes for Anthocyanin Biosynthesis. International Journal of Molecular Sciences. 2020; 21(4):1245. https://doi.org/10.3390/ijms21041245
Chicago/Turabian StyleLi, Jing, Yuyan An, and Liangju Wang. 2020. "Transcriptomic Analysis of Ficus carica Peels with a Focus on the Key Genes for Anthocyanin Biosynthesis" International Journal of Molecular Sciences 21, no. 4: 1245. https://doi.org/10.3390/ijms21041245
APA StyleLi, J., An, Y., & Wang, L. (2020). Transcriptomic Analysis of Ficus carica Peels with a Focus on the Key Genes for Anthocyanin Biosynthesis. International Journal of Molecular Sciences, 21(4), 1245. https://doi.org/10.3390/ijms21041245