Immunoglobulin Abnormalities in Gaucher Disease: an Analysis of 278 Patients Included in the French Gaucher Disease Registry

 , ,
, ,  , , ,
, , ,  , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Study Population
2.2. Prevalence of PG and MG
2.3. Risk Factors of PG and MG
2.4. Associations with Bone Events and Severe Thrombocytopenia
2.5. Evolution of PG and MG
2.6. Malignant Hemopathies
3. Discussion
4. Materials and Methods
4.1. The French GD Registry
4.2. Study Population
4.3. Baseline Characteristics and Collected Data
4.4. Outcomes
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| GD | Gaucher disease |
| PG | polyclonal gammopathy |
| MG | monoclonal gammopathy |
| MGUS | monoclonal gammopathy of unknown significance |
| MM | multiple myeloma |
| NHL | non-Hodgkin lymphoma |
| FGDR | French Gaucher disease registry |
| SD | standard derivation |
| IQR | interquartile range |
| HR | hazard ratio |
| CI | confident interval |
| ERT | enzyme replacement therapy |
| SRT | substrate reduction therapy |
Appendix A


References
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E. Gaucher disease: complexity in a “simple” disorder. Mol. Genet. Metab. 2004, 83, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, B.; Balwani, M.; Bronstein, J.M.; Kolodny, E.; Sathe, S.; Gwosdow, A.R.; Taylor, J.S.; Cole, J.A.; Zimran, A.; Weinreb, N.J. The incidence of Parkinsonism in patients with type 1 Gaucher disease: data from the ICGG Gaucher Registry. Blood Cells Mol. Dis. 2011, 46, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Regenboog, M.; van Dussen, L.; Verheij, J.; Weinreb, N.J.; Santosa, D.; vom Dahl, S.; Häussinger, D.; Müller, M.N.; Canbay, A.; Rigoldi, M.; et al. Hepatocellular carcinoma in Gaucher disease: an international case series. J. Inherit. Metab. Dis. 2018, 14, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Dispenzieri, A.; Gertz, M.A.; Therneau, T.M.; Kyle, R.A. Retrospective cohort study of 148 patients with polyclonal gammopathy. Mayo Clin. Proc. 2001, 76, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Larson, D.R.; Therneau, T.M.; Dispenzieri, A.; Kumar, S.; Cerhan, J.R.; Rajkumar, S.V. Long-Term Follow-up of Monoclonal Gammopathy of Undetermined Significance. N. Engl. J. Med. 2018, 378, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Pratt, P.W.; Estren, S.; Kochwa, S. Immunoglobulin Abnormalities in Gaucher’s Disease Report of 16 Cases. Blood 1968, 31, 633–640. [Google Scholar] [CrossRef]
- Shoenfeld, Y.; Gallant, L.A.; Shaklai, M.; Livni, E.; Djaldetti, M.; Pinkhas, J. Gaucher’s disease: a disease with chronic stimulation of the immune system. Arch. Pathol. Lab. Med. 1982, 106, 388–391. [Google Scholar]
- Marti, G.E.; Ryan, E.T.; Papadopoulos, N.M.; Filling-Katz, M.; Barton, N.; Fleischer, T.A.; Rick, M.; Gralnick, H.R. Polyclonal B-cell lymphocytosis and hypergammaglobulinemia in patients with Gaucher disease. Am. J. Hematol. 1988, 29, 189–194. [Google Scholar] [CrossRef]
- Allen, M.J.; Myer, B.J.; Khokher, A.M.; Rushton, N.; Cox, T.M. Pro-inflammatory cytokines and the pathogenesis of Gaucher’s disease: increased release of interleukin-6 and interleukin-10. QJM Int. J. Med. 1997, 90, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Brautbar, A.; Elstein, D.; Pines, G.; Abrahamov, A.; Zimran, A. Effect of enzyme replacement therapy on gammopathies in Gaucher disease. Blood Cells Mol. Dis. 2004, 32, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Wine, E.; Yaniv, I.; Cohen, I.J. Hyperimmunoglobulinemia in pediatric-onset type 1 Gaucher disease and effects of enzyme replacement therapy. J. Pediatr. Hematol. Oncol. 2007, 29, 451–457. [Google Scholar] [CrossRef] [PubMed]
- De Fost, M.; Out, T.A.; de Wilde, F.A.; Tjin, E.P.M.; Pals, S.T.; van Oers, M.H.J.; Boot, R.G.; Aerts, J.F.M.G.; Maas, M.; vom Dahl, S.; et al. Immunoglobulin and free light chain abnormalities in Gaucher disease type I: data from an adult cohort of 63 patients and review of the literature. Ann. Hematol. 2008, 87, 439–449. [Google Scholar] [CrossRef] [Green Version]
- Grosbois, B.; Rose, C.; Noël, E.; de Serratrice, C.R.; Dobbelaere, D.; Gressin, V.; Chérin, P.; Hartmann, A.; Javier, R.M.; Clerson, P.; et al. Gaucher disease and monoclonal gammopathy: a report of 17 cases and impact of therapy. Blood Cells Mol. Dis. 2009, 43, 138–139. [Google Scholar] [CrossRef]
- Taddei, T.H.; Kacena, K.A.; Yang, M.; Yang, R.; Malhotra, A.; Boxer, M.; Aleck, K.A.; Rennert, G.; Pastores, G.M.; Mistry, P.K. The underrecognized progressive nature of N370S Gaucher disease and assessment of cancer risk in 403 patients. Am. J. Hematol. 2009, 84, 208–214. [Google Scholar] [CrossRef] [Green Version]
- Jurecka, A.; Gregorek, H.; Kleinotiene, G.; Czartoryska, B.; Tylki-Szymanska, A. Gaucher disease and dysgammaglobulinemia: a report of 61 patients, including 18 with GD type III. Blood Cells Mol. Dis. 2011, 46, 85–87. [Google Scholar] [CrossRef]
- Arends, M.; van Dussen, L.; Biegstraaten, M.; Hollak, C.E.M. Malignancies and monoclonal gammopathy in Gaucher disease; a systematic review of the literature. Br. J. Haematol. 2013, 161, 832–842. [Google Scholar] [CrossRef]
- De Fost, M.; vom Dahl, S.; Weverling, G.J.; Brill, N.; Brett, S.; Häussinger, D.; Hollak, C.E.M. Increased incidence of cancer in adult Gaucher disease in Western Europe. Blood Cells Mol. Dis. 2006, 36, 53–58. [Google Scholar] [CrossRef]
- Landgren, O.; Turesson, I.; Gridley, G.; Caporaso, N.E. Risk of Malignant Disease Among 1525 Adult Male US Veterans With Gaucher Disease. Arch. Intern. Med. 2007, 167, 1189–1194. [Google Scholar] [CrossRef] [Green Version]
- Nair, S.; Boddupalli, C.S.; Verma, R.; Liu, J.; Yang, R.; Pastores, G.M.; Mistry, P.K.; Dhodapkar, M.V. Type II NKT-TFH cells against Gaucher lipids regulate B-cell immunity and inflammation. Blood 2015, 125, 1256–1271. [Google Scholar] [CrossRef] [Green Version]
- Nair, S.; Branagan, A.R.; Liu, J.; Boddupalli, C.S.; Mistry, P.K.; Dhodapkar, M.V. Clonal Immunoglobulin against Lysolipids in the Origin of Myeloma. N. Engl. J. Med. 2016, 374, 555–561. [Google Scholar] [CrossRef]
- Preuss, K.-D.; Hollak, C.E.M.; Fadle, N.; van Oers, M.; Regitz, E.; Pfreundschuh, M. Saposin C is a frequent target of paraproteins in Gaucher disease-associated MGUS/multiple myeloma. Br. J. Haematol. 2019, 184, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Therneau, T.M.; Rajkumar, S.V.; Larson, D.R.; Plevak, M.F.; Offord, J.R.; Dispenzieri, A.; Katzmann, J.A.; Melton, L.J. Prevalence of monoclonal gammopathy of undetermined significance. N. Engl. J. Med. 2006, 354, 1362–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landgren, O.; Graubard, B.I.; Kumar, S.; Kyle, R.A.; Katzmann, J.A.; Murata, K.; Costello, R.; Dispenzieri, A.; Caporaso, N.; Mailankody, S.; et al. Prevalence of myeloma precursor state monoclonal gammopathy of undetermined significance in 12372 individuals 10-49 years old: a population-based study from the National Health and Nutrition Examination Survey. Blood Cancer J. 2017, 7, e618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, T.M.; Rosenbloom, B.E.; Barker, R.A. Gaucher disease and comorbidities: B-cell malignancy and parkinsonism. Am. J. Hematol. 2015, 90, S25–S28. [Google Scholar] [CrossRef] [PubMed]
- Murugesan, V.; Chuang, W.-L.; Liu, J.; Lischuk, A.; Kacena, K.; Lin, H.; Pastores, G.M.; Yang, R.; Keutzer, J.; Zhang, K.; et al. Glucosylsphingosine is a key biomarker of Gaucher disease. Am. J. Hematol. 2016, 91, 1082–1089. [Google Scholar] [CrossRef] [Green Version]
- Arkadir, D.; Dinur, T.; Revel-Vilk, S.; Cohen, M.B.; Cozma, C.; Hovakimyan, M.; Eichler, S.; Rolfs, A.; Zimran, A. Glucosylsphingosine is a reliable response biomarker in Gaucher disease. Am. J. Hematol. 2018, 93, E140–E142. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Redondo, C.; Ortuño, F.J.; Lozano, M.L.; Jerez, A.; del Mar Osma, M.; Giraldo, P.; Vicente, V. IgM monoclonal component associated with type I Gaucher disease resolved after enzyme replacement therapy: a case report. J. Inherit. Metab. Dis. 2009, 32 (Suppl. 1), S265–S267. [Google Scholar] [CrossRef]
- Stirnemann, J.; Vigan, M.; Hamroun, D.; Heraoui, D.; Rossi-Semerano, L.; Berger, M.G.; Rose, C.; Camou, F.; de Roux-Serratrice, C.; Grosbois, B.; et al. The French Gaucher’s disease registry: clinical characteristics, complications and treatment of 562 patients. Orphanet J. Rare Dis. 2012, 7, 77. [Google Scholar] [CrossRef] [Green Version]
- Szymanowicz, A.; Cartier, B.; Couaillac, J.-P.; Gibaud, C.; Poulin, G.; Rivière, H.; Le Carrer, D.; Groupe de Travail du Collège National de Biochimie des Hôpitaux. A proposal of ready-made interpretative comments applicable to serum protein electrophoresis. Ann. Biol. Clin. (Paris) 2006, 64, 367–380. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population 1 (n = 278) (All Cohort) | Population 2 (n = 235) | Population 3 (n = 187) (At Least One Determination of Presence or Absence of MG) | |||
|---|---|---|---|---|---|
| (At Least One Gammaglobulin Determination) | |||||
| PG– | PG+ | MG– | MG+ | ||
| Characteristics at GD diagnosis | n = 278 | n = 123 | n = 112 | n = 128 | n = 59 |
| Sex = male, n (%) | 132 (47.5) | 62 (50.4) | 47 (42.0) | 59 (46.1) | 31 (52.5) |
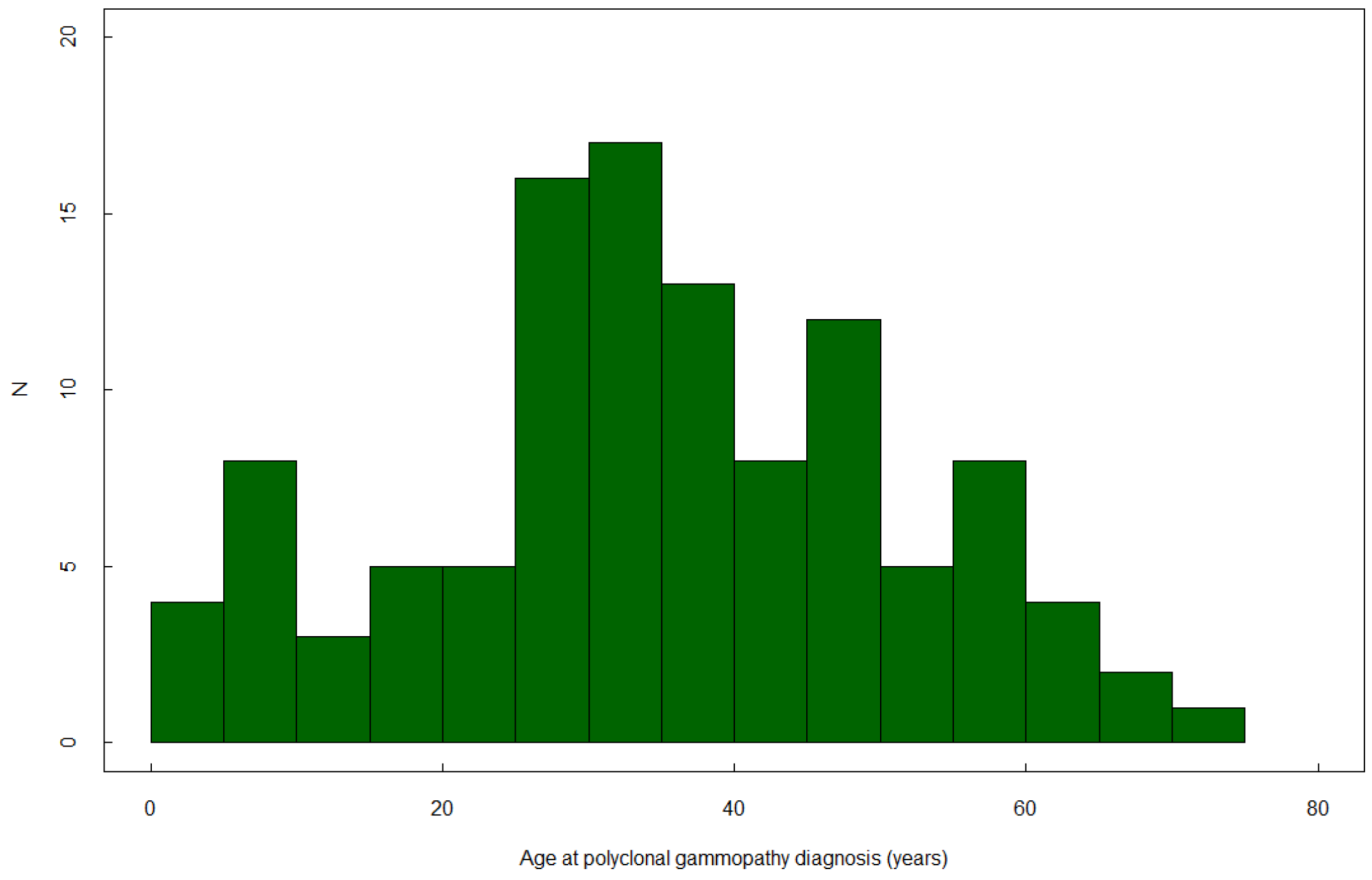
| Age at GD diagnosis, years, n (%) | 24.4 (18.3) | 24.1 (19.8) | 22.0 (15.2) | 22.6 (15.2) | 37.2 (18.5) |
| >30 years old | 91 (32.7) | 43 (35.0) | 28 (25.0) | 37 (28.9) | 36 (61.0) |
| Phenotype, n (%) | |||||
| type 1 GD | 262 (94.2) | 113 (91.9) | 106 (94.6) | 123 (96.1) | 59 (100) |
| type 2 GD | 3 (1.1) | 3 (2.4) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| type 3 GD | 13 (4.7) | 7 (5.7) | 6 (5.4) | 5 (3.9) | 0 (0.0) |
| Genotype, n (%) | |||||
| p.Asn409Ser/p.Asn409Ser | 29 (10.4) | 14 (11.4) | 15 (13.4) | 18 (14.1) | 6 (10.2) |
| p.Asn409Ser/p.Leu483Pro | 45 (16.2) | 15 (12.2) | 20 (17.9) | 26 (20.3) | 9 (15.3) |
| p.Leu483Pro/p.Leu483Pro | 5 (1.8) | 2 (1.6) | 3 (2.7) | 2 (1.6) | 0 (0.0) |
| p.Asn409Ser/other | 68 (24.5) | 41 (33.3) | 21 (18.8) | 31 (24.2) | 16 (27.1) |
| p.Leu483Pro/other | 9 (3.2) | 4 (3.3) | 3 (2.7) | 6 (4.7) | 1 (1.7) |
| NA | 122 (43.9) | 47 (38.2) | 50 (44.6) | 45 (35.2) | 27 (45.8) |
| Splenomegaly at GD diagnosis, n (%) | |||||
| No | 58 (20.9) | 11 (8.9) | 9 (8.0) | 11 (8.6) | 3 (5.1) |
| Yes | 129 (46.4) | 89 (72.4) | 79 (70.5) | 91 (71.1) | 35 (59.3) |
| NA | 64 (23.0) | 23 (18.7) | 24 (21.4) | 26 (20.3) | 21 (35.6) |
| Hepatomegaly at GD diagnosis, n (%) | |||||
| No | 58 (20.9) | 30 (24.4) | 18 (16.1) | 34 (26.6) | 7 (11.9) |
| Yes | 129 (46.4) | 58 (47.2) | 57 (50.9) | 57 (44.5) | 25 (42.4) |
| NA | 91 (32.7) | 35 (28.5) | 37 (33.0) | 37 (28.9) | 27 (45.8) |
| Anemia at GD diagnosis, n (%) | |||||
| No | 131 (47.1) | 73 (59.3) | 39 (34.8) | 68 (53.1) | 28 (47.5) |
| Yes | 37 (13.3) | 12 (9.8) | 20 (17.9) | 15 (11.7) | 6 (10.2) |
| NA | 110 (39.6) | 38 (30.9) | 53 (47.3) | 45 (35.2) | 25 (42.4) |
| Thrombocytopenia at diagnosis, n (%) | |||||
| No | 84 (30.2) | 43 (35.0) | 26 (23.2) | 44 (34.4) | 14 (23.7) |
| Yes | 105 (37.8) | 51 (41.5) | 39 (34.8) | 47 (36.7) | 24 (40.7) |
| NA | 89 (32.0) | 29 (23.6) | 47 (42.0) | 37 (28.9) | 21 (35.6) |
| Characteristics during follow-up, | |||||
| Splenectomy, n (%) | 69 (24.8) | 15 (12.2) | 42 (37.5) | 24 (18.8) | 22 (37.3) |
| ERT/SRT, n (%) | 228 (82.0) | 101 (82.1) | 97 (86.6) | 102 (79.7) | 49 (83.1) |
| Risk of PG | Risk of MG | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Univariate Analysis | Multivariable Analysis | Univariate Analysis | Multivariable Analysis | |||||||||||||
| n | PG | HR | 95%CI | p | HR | 95%CI | p | n | MG | HR | 95%CI | p | HR | 95%CI | p | |
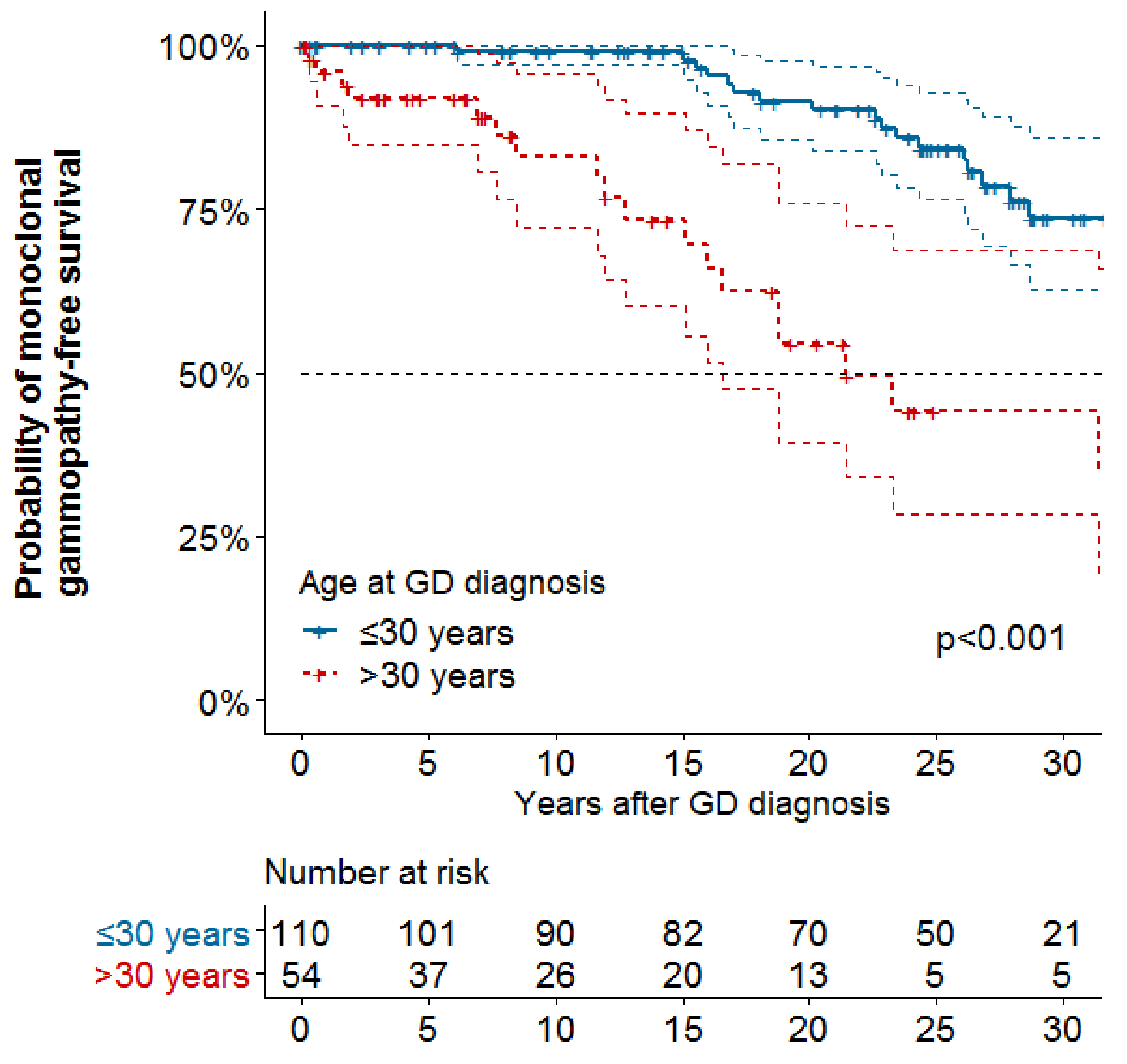
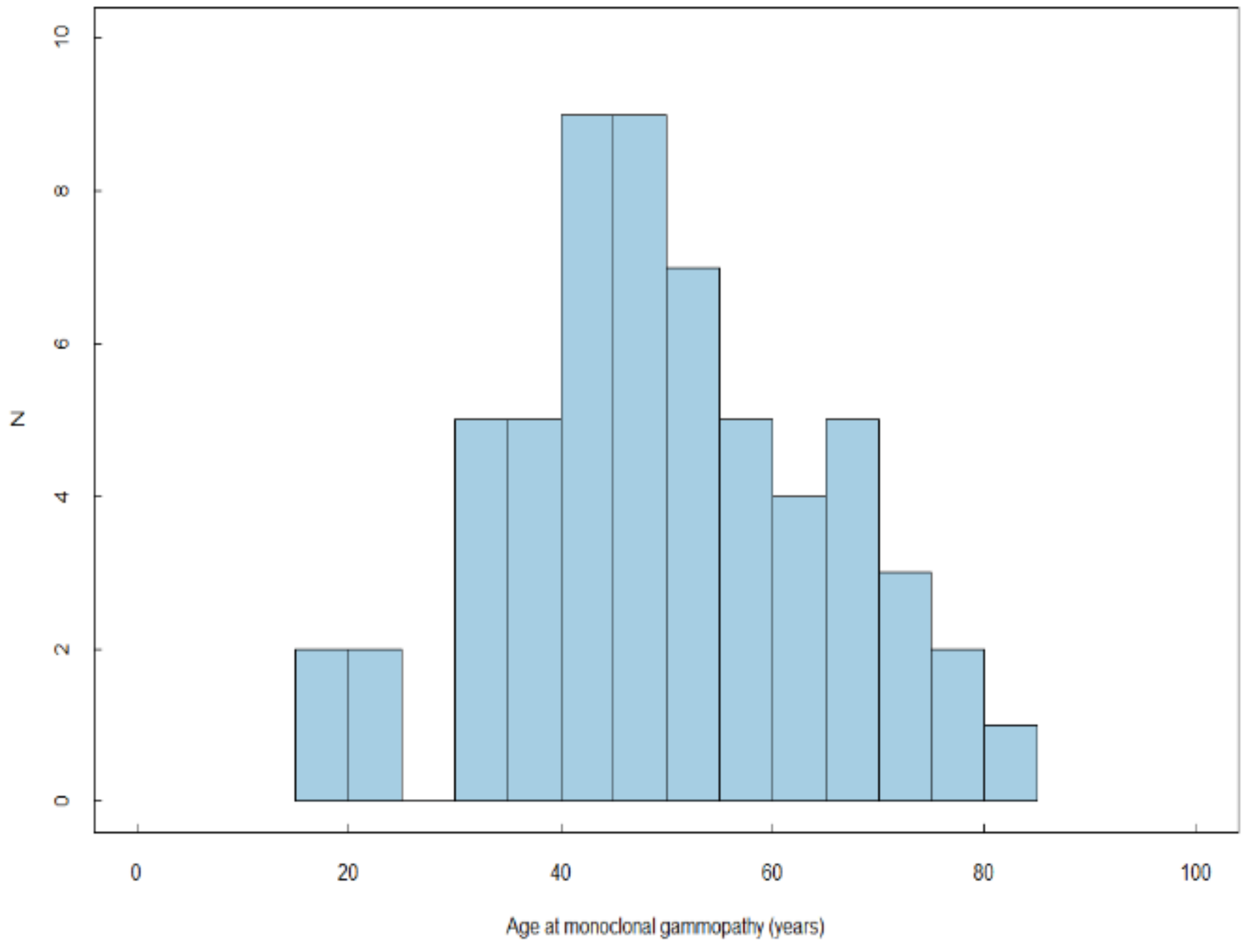
| Age at GD diagnosis, years | 209 | 97 | 1.01 | 1.00–1.03 | 0.034 | 1.01 | 0.99–1.03 | 0.356 | 164 | 39 | 1.08 | 1.05–1.10 | <0.001 | 1.08 | 1.05–1.10 | <0.001 |
| Age at GD diagnosis | 209 | 97 | 164 | 39 | ||||||||||||
| ≤30 years | (ref) | (ref) | ||||||||||||||
| >30 years | 1.48 | 0.92–2.39 | 0.107 | 4.71 | 2.40–9.27 | <0.001 | ||||||||||
| Male | 209 | 97 | 0.92 | 0.61–1.39 | 0.69 | 1.3 | 0.69–2.46 | 0.414 | ||||||||
| Genotype | 126 | 52 | 104 | 21 | ||||||||||||
| p.Asn409Ser/p.Asn409Ser | (ref) | (ref) | ||||||||||||||
| p.Leu483Pro/p.Leu483Pro | 1.15 | 0.25–5.17 | 0.86 | - | - | - | ||||||||||
| p.Asn409Ser/p.Leu483Pro | 0.90 | 0.42–1.95 | 0.797 | 0.59 | 0.14–2.53 | 0.48 | ||||||||||
| Other | 0.71 | 0.20–2.56 | 0.605 | 1.11 | 0.39–3.17 | 0.84 | ||||||||||
| Splenomegaly at diagnosis | 164 | 73 | 0.66 | 0.30–1.45 | 0.302 | 123 | 35 | 1.06 | 0.14–8.02 | 0.954 | ||||||
| Hepatomegaly at diagnosis | 138 | 60 | 1.04 | 0.56–1.92 | 0.91 | 106 | 18 | 2.58 | 0.59–11.27 | 0.207 | ||||||
| Anemia at diagnosis | 120 | 45 | 1.76 | 0.95–3.26 | 0.071 | 1.82 | 0.95–3.51 | 0.072 | 101 | 20 | 0.69 | 0.23–2.07 | 0.506 | |||
| Thrombocytopenia at diagnosis | 135 | 51 | 1.03 | 0.58–1.83 | 0.92 | 110 | 21 | 1.43 | 0.55–3.69 | 0.463 | ||||||
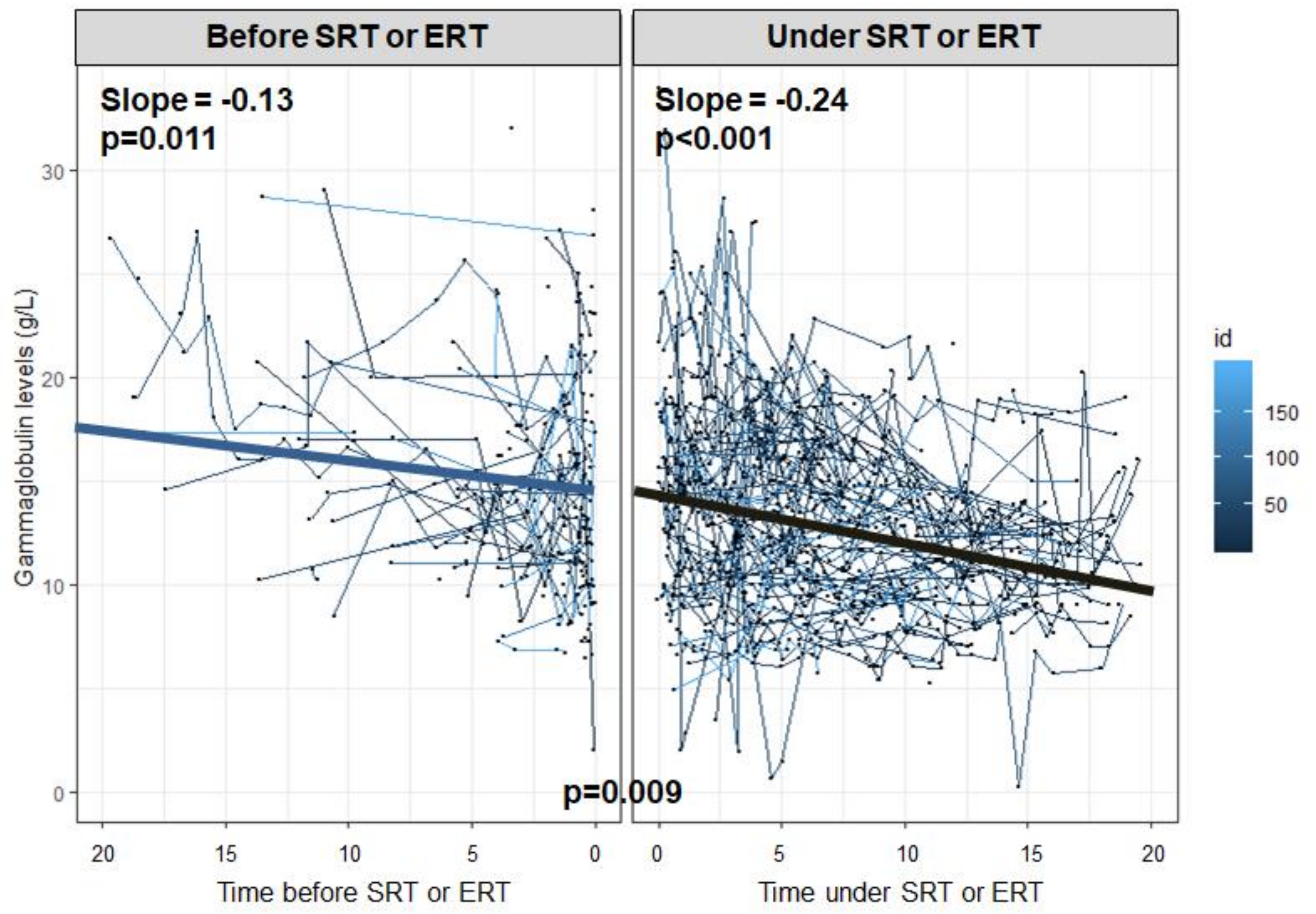
| Treatment during follow-up *§ | 209 | 97 | 0.89 | 0.56–1.43 | 0.635 | 0.80 | 0.40–1.62 | 0.536 | 164 | 39 | 1.35 | 0.67–2.73 | 0.405 | 1.25 | 0.61–2.60 | 0.543 |
| Splenectomy during follow-up § | 209 | 97 | 1.17 | 0.76–1.82 | 0.471 | 1.22 | 0.57–2.61 | 0.602 | 164 | 39 | 1.04 | 0.52–2.05 | 0.917 | 0.98 | 0.49–1.98 | 0.963 |
| MG § | 135 | 63 | 0.81 | 0.34–1.91 | 0.625 | |||||||||||
| PG § | 135 | 28 | 0.83 | 0.36–1.90 | 0.652 | |||||||||||
| Risk of Bone Events | Risk of Severe Thrombocytopenia | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Univariate Analysis | Multivariable Analysis | Univariate Analysis | Multivariable Analysis | |||||||||||||
| n | Events | HR | 95%CI | p | HR | 95%CI | p | n | Events | HR | 95%CI | p | HR | 95%CI | p | |
| PG§ | 190 | 76 | 1.29 | 0.73–2.27 | 0.381 | 1.27 | 0.70–2.30 | 0.427 | 197 | 47 | 0.89 | 0.42–1.91 | 0.769 | 1.19 | 0.55–2.59 | 0.658 |
| Age at GD diagnosis | 190 | 76 | 1.01 | 0.99–1.02 | 0.476 | 1.01 | 0.99–1.02 | 0.503 | 197 | 47 | 1.01 | 0.99–1.03 | 0.209 | 1.01 | 0.99–1.03 | 0.296 |
| Male sex | 190 | 76 | 0.95 | 0.59–1.51 | 0.818 | 1.25 | 0.76–2.06 | 0.378 | 197 | 47 | 1.13 | 0.64–2.00 | 0.678 | 1.03 | 0.58–1.85 | 0.915 |
| Splenectomy during follow-up § | 190 | 76 | 2.37 | 1.49–3.77 | <0.001 | 2.63 | 1.60–4.33 | <0.001 | 197 | 47 | 0.41 | 0.18–0.90 | 0.026 | 0.34 | 0.15–0.76 | 0.009 |
| Treatment during follow-up *§ | 190 | 76 | 0.98 | 0.59–1.65 | 0.948 | 1.14 | 0.65–1.99 | 0.64 | 197 | 47 | 0.33 | 0.14–0.82 | 0.016 | 0.26 | 0.10–0.68 | 0.006 |
| Risk of Bone Events | Risk of Severe Thrombocytopenia | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Univariate Analysis | Multivariable Analysis | Univariate Analysis | Multivariable Analysis | |||||||||||||
| n | Events | HR | 95%CI | p | HR | 95%CI | p | n | Events | HR | 95%CI | p | HR | 95%CI | p | |
| MG§ | 150 | 61 | 1.40 | 0.69–2.86 | 0.353 | 1.28 | 0.60–2.75 | 0.525 | 151 | 36 | 0.58 | 0.14–2.42 | 0.453 | 0.55 | 0.12–2.47 | 0.439 |
| Age at GD diagnosis | 150 | 61 | 1.01 | 1.00–1.03 | 0.097 | 1.01 | 0.99–1.03 | 0.326 | 151 | 36 | 1 | 0.98–1.02 | 0.917 | 1.01 | 0.98–1.03 | 0.666 |
| Male sex | 150 | 61 | 1.04 | 0.62–1.73 | 0.713 | 1.28 | 0.73–2.23 | 0.525 | 151 | 36 | 0.96 | 0.50–1.86 | 0.906 | 0.81 | 0.41–1.60 | 0.548 |
| Splenectomy during follow-up § | 150 | 61 | 2.30 | 1.37–3.87 | 0.002 | 2.60 | 1.49–4.55 | 0.001 | 151 | 36 | 0.53 | 0.22–1.25 | 0.148 | 0.46 | 0.19–1.11 | 0.085 |
| Treatment during follow-up *§ | 150 | 61 | 1.22 | 0.70–2.14 | 0.479 | 1.44 | 0.81–2.57 | 0.218 | 151 | 36 | 0.6 | 0.24–1.46 | 0.261 | 0.56 | 0.23–1.38 | 0.207 |
| Patient | Sex | Age at GD Diagnosis (Years) | PG (g/L) | Age at PG Diagnosis (Years) | MG (type) | Age at First GM (years) | Malignant Hemopathy | Age at Hemopathy | Age at GD Treatment | Status |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 61 | 21.7 | 68 | - | - | AITL | 83 | 79 | Deceased |
| 2 | F | 29 | 25 | 47 | - | - | B-NHL | 57 | 45 | Deceased |
| 3 | F | 24 | 16.1 | 49 | IgM λ | 48 | MALT lymphoma | 47 | 51 | Alive |
| 4 | F | 56 | 22.1 | 61 | IgG κ | 65 | Lymphocytic lymphoma | 61 | Not treated | Alive |
| 5 | M | 42 | - | - | IgG λ | 42 | B-NHL | 54 | NA | Deceased |
| 6 | F | 62 | - | - | IgG λ | 75 | MM IgG λ | 75 | 75 | Deceased |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, Y.; Stirnemann, J.; Lautredoux, F.; Cador, B.; Bengherbia, M.; Yousfi, K.; Hamroun, D.; Astudillo, L.; Billette de Villemeur, T.; Brassier, A.; et al. Immunoglobulin Abnormalities in Gaucher Disease: an Analysis of 278 Patients Included in the French Gaucher Disease Registry. Int. J. Mol. Sci. 2020, 21, 1247. https://doi.org/10.3390/ijms21041247
Nguyen Y, Stirnemann J, Lautredoux F, Cador B, Bengherbia M, Yousfi K, Hamroun D, Astudillo L, Billette de Villemeur T, Brassier A, et al. Immunoglobulin Abnormalities in Gaucher Disease: an Analysis of 278 Patients Included in the French Gaucher Disease Registry. International Journal of Molecular Sciences. 2020; 21(4):1247. https://doi.org/10.3390/ijms21041247
Chicago/Turabian StyleNguyen, Yann, Jérôme Stirnemann, Florent Lautredoux, Bérengère Cador, Monia Bengherbia, Karima Yousfi, Dalil Hamroun, Leonardo Astudillo, Thierry Billette de Villemeur, Anaïs Brassier, and et al. 2020. "Immunoglobulin Abnormalities in Gaucher Disease: an Analysis of 278 Patients Included in the French Gaucher Disease Registry" International Journal of Molecular Sciences 21, no. 4: 1247. https://doi.org/10.3390/ijms21041247
APA StyleNguyen, Y., Stirnemann, J., Lautredoux, F., Cador, B., Bengherbia, M., Yousfi, K., Hamroun, D., Astudillo, L., Billette de Villemeur, T., Brassier, A., Camou, F., Dalbies, F., Dobbelaere, D., Gaches, F., Leguy-Seguin, V., Masseau, A., Pers, Y. -M., Pichard, S., Serratrice, C., ... on behalf of the French Evaluation of Gaucher Disease Treatment Committee. (2020). Immunoglobulin Abnormalities in Gaucher Disease: an Analysis of 278 Patients Included in the French Gaucher Disease Registry. International Journal of Molecular Sciences, 21(4), 1247. https://doi.org/10.3390/ijms21041247