The Novel Small-molecule Annexin-A1 Mimetic, Compound 17b, Elicits Vasoprotective Actions in Streptozotocin-induced Diabetic Mice

 , , ,
, , ,
Abstract
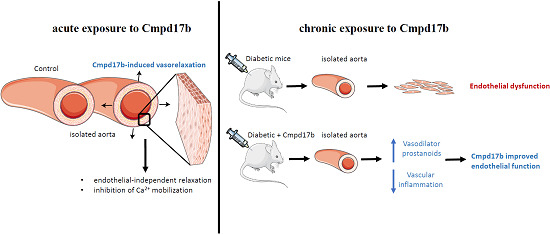
:
1. Introduction
2. Results
2.1. Localization of FPR1 and FPR2 in the Aorta
2.2. Cmpd17b But not Cmpd43 Is a Vasodilator in the Aorta
2.3. Mechanisms of Cmpd17b-Induced Relaxation
2.4. Systemic Characteristics of STZ-Induced Diabetes
2.5. Investigating Mechanisms of Cmpd17b Action in Diabetes
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Isolation of Aorta
4.3. Immunohistochemistry
4.4. Quantitative Real-Time PCR
4.5. Assessment of Vascular Reactivity
4.6. Induction of Type 1 Diabetic Mice
4.7. Tissue Collection from Diabetic and Non-Diabetic Mice for Vascular Analysis
4.8. Vascular Reactivity of Aorta from Diabetic Mice
4.9. Quantitative PCR from Diabetic Mice
4.10. Reagents
4.11. Statistical Analysis
4.12. Chemical Compounds
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACh | Acetylcholine |
| Akt | Protein kinase B |
| Apamin | Small conductance calcium-activated potassium channel inhibitor |
| ANX-A | Annexin A1, endogenous ligand for FPR |
| cAMP | Cyclic adenosine monophosphate |
| CB | Citrate buffer |
| Cmpd17b | Compound 17b, agonist of FPR1/2 |
| Cmpd43 | Compound 43, agonist of FPR1/2 |
| COX 1 | Cyclooxygenase-1 |
| COX 2 | Cyclooxygenase-2 |
| Cq | Quantification cycle |
| DMSO | Dimethyl sulfoxide |
| EDH | Endothelium-derived hyperpolarization |
| Emax | Maximum contraction |
| ERK1/2 | Extracellular signal-regulated kinases 1/2 |
| FPR | Formyl peptide receptor family |
| FPR1 | Formyl peptide receptor 1 |
| FPR2 | Formyl peptide receptor 2 |
| Glibenclamide | ATP-sensitive potassium (KATP) channel blocker |
| HbA1c | Glycated haemoglobin A1c |
| IKca | Intermediate conductance calcium-activated potassium channel |
| KCaB | Combination of inhibitors (Apamin and TRAM34) |
| Indo | Indomethacin |
| L-NAME | nitric oxide synthase (NOS) inhibitor, L-NG-Nitroarginine methyl ester |
| LxA4 | Lipoxin 4 |
| NGS | Normal goat serum |
| Nifedipine | Calcium channel blocker |
| NO | Nitric oxide |
| NOS | Nitric oxide synthase |
| ODQ | Soluble guanylate cyclase inhibitor, 1H-(1,2,4)oxadiazolo[4,3-a]quinoxalin-1-one |
| PBS | Phosphate buffered saline |
| pEC50 | Sensitivity |
| PGI2 | Prostacyclin |
| PSS | Physiological saline solution |
| PTGIR | Prostaglandin I2 receptor |
| PTGIS | Prostacyclin synthase |
| Rn18s | 18s Ribosomal RNA |
| Rmax | Maximum relaxation |
| SKca | Small conductance calcium-activated potassium channel |
| SNP | Sodium nitroprusside |
| STZ | Streptozotocin |
| TRAM34 | Intermediate conductance calcium-activated potassium channel inhibitor, 1-[(2-Chlorophenyl)diphenylmethyl]-1H-pyrazole |
| U46619 | Thromboxane A2 mimetic |
References
- Ye, R.D.; Boulay, F.; Wang, J.M.; Dahlgren, C.; Gerard, C.; Parmentier, M.; Serhan, C.N.; Murphy, P.M. International Union of Basic and Clinical Pharmacology. LXXIII. Nomenclature for the formyl peptide receptor (FPR) family. Pharm. Rev. 2009, 61, 119–161. [Google Scholar] [CrossRef] [PubMed]
- He, H.Q.; Ye, R.D. The formyl peptide receptors: Diversity of ligands and mechanism for recognition. Molecules 2017, 22, 455. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Yang, Y.H.; May, L.; Gao, X.; Stewart, A.G.; Tu, Y.; Woodman, O.L.; Ritchie, R.H. Cardioprotective potential of annexin-A1 mimetics in myocardial infarction. Pharm. Ther. 2015, 148, 47–65. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Buxton, K.D.; Pepe, S.; Cao, A.H.; Venardos, K.; Love, J.E.; Kaye, D.M.; Yang, Y.H.; Morand, E.F.; Ritchie, R.H. Reperfusion-induced myocardial dysfunction is prevented by endogenous annexin-A1 and its N-terminal-derived peptide Ac-ANX-A1(2-26). Br. J. Pharmacol. 2013, 168, 238–252. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, R.H.; Sun, X.; Bilszta, J.L.; Gulluyan, L.M.; Dusting, G.J. Cardioprotective actions of an N-terminal fragment of annexin-1 in rat myocardium in vitro. Eur. J. Pharm. 2003, 461, 171–179. [Google Scholar] [CrossRef]
- Ritchie, R.H.; Gordon, J.M.; Woodman, O.L.; Cao, A.H.; Dusting, G.J. Annexin-1 peptide Anx-1(2-26) protects adult rat cardiac myocytes from cellular injury induced by simulated ischaemia. Br. J. Pharmacol. 2005, 145, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Maderna, P.; Cottell, D.C.; Toivonen, T.; Dufton, N.; Dalli, J.; Perretti, M.; Godson, C. FPR2/ALX receptor expression and internalization are critical for lipoxin A4 and annexin-derived peptide-stimulated phagocytosis. FASEB J. 2010, 24, 4240–4249. [Google Scholar] [CrossRef] [Green Version]
- Vital, S.A.; Becker, F.; Holloway, P.M.; Russell, J.; Perretti, M.; Granger, D.N.; Gavins, F.N. Formyl-Peptide Receptor 2/3/Lipoxin A4 Receptor Regulates Neutrophil-Platelet Aggregation and Attenuates Cerebral Inflammation: Impact for Therapy in Cardiovascular Disease. Circulation 2016, 133, 2169–2179. [Google Scholar] [CrossRef] [Green Version]
- Smith, H.K.; Gil, C.D.; Oliani, S.M.; Gavins, F.N. Targeting formyl peptide receptor 2 reduces leukocyte-endothelial interactions in a murine model of stroke. FASEB J. 2015, 29, 2161–2171. [Google Scholar] [CrossRef] [Green Version]
- Peshavariya, H.M.; Taylor, C.J.; Goh, C.; Liu, G.S.; Jiang, F.; Chan, E.C.; Dusting, G.J. Annexin peptide Ac2-26 suppresses TNFalpha-induced inflammatory responses via inhibition of Rac1-dependent NADPH oxidase in human endothelial cells. PLoS ONE 2013, 8, e60790. [Google Scholar] [CrossRef] [Green Version]
- Flower, R.J.; Blackwell, G.J. Anti-inflammatory steroids induce biosynthesis of a phospholipase A2 inhibitor which prevents prostaglandin generation. Nature 1979, 278, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Jelinic, M.; Kahlberg, N.; Leo, C.H.; Ng, H.H.; Rosli, S.; Deo, M.; Li, M.; Finlayson, S.; Walsh, J.; Parry, L.J.; et al. Annexin-A1 deficiency exacerabates pathological remodelling of the mesenteric vasculature in insulin-resistance but not insulin-deficiency. Br. J. Pharm. 2019. [Google Scholar] [CrossRef]
- Cilibrizzi, A.; Quinn, M.T.; Kirpotina, L.N.; Schepetkin, I.A.; Holderness, J.; Ye, R.D.; Rabiet, M.J.; Biancalani, C.; Cesari, N.; Graziano, A.; et al. 6-methyl-2,4-disubstituted pyridazin-3(2H)-ones: A novel class of small-molecule agonists for formyl peptide receptors. J. Med. Chem. 2009, 52, 5044–5057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burli, R.W.; Xu, H.; Zou, X.; Muller, K.; Golden, J.; Frohn, M.; Adlam, M.; Plant, M.H.; Wong, M.; McElvain, M.; et al. Potent hFPRL1 (ALXR) agonists as potential anti-inflammatory agents. Bioorganic Med. Chem. Lett. 2006, 16, 3713–3718. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.X.; May, L.T.; Li, R.; Cao, N.; Rosli, S.; Deo, M.; Alexander, A.E.; Horlock, D.; Bourke, J.E.; Yang, Y.H.; et al. Small-molecule-biased formyl peptide receptor agonist compound 17b protects against myocardial ischaemia-reperfusion injury in mice. Nat. Commun. 2017, 8, 14232. [Google Scholar] [CrossRef] [PubMed]
- Jelinic, M.; Marshall, S.A.; Leo, C.H.; Parry, L.J.; Tare, M. From pregnancy to cardiovascular disease: Lessons from relaxin-deficient animals to understand relaxin actions in the vascular system. Microcirculation 2019, 26, e12464. [Google Scholar] [CrossRef]
- Jelinic, M.; Marshall, S.A.; Stewart, D.; Unemori, E.; Parry, L.J.; Leo, C.H. The peptide hormone relaxin: From bench to bedside. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 314, R753–R760. [Google Scholar] [CrossRef]
- Leo, C.H.; Jelinic, M.; Ng, H.H.; Marshall, S.A.; Novak, J.; Tare, M.; Conrad, K.P.; Parry, L.J. Vascular actions of relaxin: Nitric oxide and beyond. Br. J. Pharm. 2017, 174, 1002–1014. [Google Scholar] [CrossRef] [Green Version]
- Berkestedt, I.; Nelson, A.; Bodelsson, M. Endogenous antimicrobial peptide LL-37 induces human vasodilatation. Br. J. Anaesth. 2008, 100, 803–809. [Google Scholar] [CrossRef] [Green Version]
- von der Weid, P.Y.; Hollenberg, M.D.; Fiorucci, S.; Wallace, J.L. Aspirin-triggered, cyclooxygenase-2-dependent lipoxin synthesis modulates vascular tone. Circulation 2004, 110, 1320–1325. [Google Scholar] [CrossRef] [Green Version]
- Horewicz, V.V.; Crestani, S.; de Sordi, R.; Rezende, E.; Assreuy, J. FPR2/ALX activation reverses LPS-induced vascular hyporeactivity in aorta and increases survival in a pneumosepsis model. Eur. J. Pharm. 2015, 746, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Wenceslau, C.F.; McCarthy, C.G.; Szasz, T.; Webb, R.C. Lipoxin A4 mediates aortic contraction via RHOA/RHO kinase, endothelial dysfunction and reactive oxygen species. J. Vasc. Res. 2014, 51, 407–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenceslau, C.F.; McCarthy, C.G.; Webb, R.C. Formyl Peptide Receptor Activation Elicits Endothelial Cell Contraction and Vascular Leakage. Front. Pharm. 2016, 7, 297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgescu, A. Vascular dysfunction in diabetes: The endothelial progenitor cells as new therapeutic strategy. World J. Diabetes 2011, 2, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Leo, C.H.; Jelinic, M.; Ng, H.H.; Tare, M.; Parry, L.J. Serelaxin: A novel therapeutic for vascular diseases. Trends Pharm. Sci. 2016, 37, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Loader, J.; Montero, D.; Lorenzen, C.; Watts, R.; Meziat, C.; Reboul, C.; Stewart, S.; Walther, G. Acute hyperglycemia impairs vascular function in healthy and cardiometabolic diseased subjects: Systematic review and meta-analysis. Arter. Thromb. Vasc. Biol. 2015, 35, 2060–2072. [Google Scholar] [CrossRef] [Green Version]
- Leo, C.H.; Joshi, A.; Hart, J.L.; Woodman, O.L. Endothelium-dependent nitroxyl-mediated relaxation is resistant to superoxide anion scavenging and preserved in diabetic rat aorta. Pharm. Res. 2012, 66, 383–391. [Google Scholar] [CrossRef]
- Leo, C.H.; Joshi, A.; Woodman, O.L. Short term type 1 diabetes alters the mechanism of endothelium-dependent relaxation in the rat carotid artery. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H502–H511. [Google Scholar] [CrossRef] [Green Version]
- Ng, H.H.; Leo, C.H.; Parry, L.J. Serelaxin (recombinant human relaxin-2) prevents high glucose-induced endothelial dysfunction by ameliorating prostacyclin production in the mouse aorta. Pharm. Res. 2016, 107, 220–228. [Google Scholar] [CrossRef]
- Guzik, T.J.; Mussa, S.; Gastaldi, D.; Sadowski, J.; Ratnatunga, C.; Pillai, R.; Channon, K.M. Mechanisms of increased vascular superoxide production in human diabetes mellitus: Role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation 2002, 105, 1656–1662. [Google Scholar] [CrossRef] [Green Version]
- Skyrme-Jones, R.A.; O’Brien, R.C.; Luo, M.; Meredith, I.T. Endothelial vasodilator function is related to low-density lipoprotein particle size and low-density lipoprotein vitamin E content in type 1 diabetes. J. Am. Coll Cardiol. 2000, 35, 292–299. [Google Scholar] [CrossRef] [Green Version]
- Tabit, C.E.; Chung, W.B.; Hamburg, N.M.; Vita, J.A. Endothelial dysfunction in diabetes mellitus: Molecular mechanisms and clinical implications. Rev. Endocr. Metab. Disord. 2010, 11, 61–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leo, C.H.; Woodman, O.L. Flavonols in the Prevention of Diabetes-induced Vascular Dysfunction. J. Cardiovasc. Pharm. 2015, 65, 532–544. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.H.; Leo, C.H.; Parry, L.J.; Ritchie, R.H. Relaxin as a Therapeutic Target for the Cardiovascular Complications of Diabetes. Front. Pharm. 2018, 9, 501. [Google Scholar] [CrossRef]
- Feuerstein, G.; Siren, A.L. Mesenteric vascular responses to i.v. administration of lipoxin A4 and lipoxin B4 in the conscious rat. FEBS Lett. 1988, 232, 51–55. [Google Scholar] [CrossRef] [Green Version]
- Nelson, M.T.; Quayle, J.M. Physiological roles and properties of potassium channels in arterial smooth muscle. Am. J. Physiol. 1995, 268, C799–C822. [Google Scholar] [CrossRef]
- Thorneloe, K.S.; Nelson, M.T. Ion channels in smooth muscle: Regulators of intracellular calcium and contractility. Can. J. Physiol. Pharm. 2005, 83, 215–242. [Google Scholar] [CrossRef]
- Hawkins, T.E.; Merrifield, C.J.; Moss, S.E. Calcium signaling and annexins. Cell Biochem. Biophys. 2000, 33, 275–296. [Google Scholar] [CrossRef]
- Gollasch, M.; Nelson, M.T. Voltage-dependent Ca2+ channels in arterial smooth muscle cells. Kidney Blood Press Res. 1997, 20, 355–371. [Google Scholar] [CrossRef]
- Brennan, E.P.; Mohan, M.; McClelland, A.; de Gaetano, M.; Tikellis, C.; Marai, M.; Crean, D.; Dai, A.; Beuscart, O.; Derouiche, S.; et al. Lipoxins Protect Against Inflammation in Diabetes-Associated Atherosclerosis. Diabetes 2018, 67, 2657–2667. [Google Scholar] [CrossRef] [Green Version]
- Ng, H.H.; Leo, C.H.; O’Sullivan, K.; Alexander, S.A.; Davies, M.J.; Schiesser, C.H.; Parry, L.J. 1,4-Anhydro-4-seleno-d-talitol (SeTal) protects endothelial function in the mouse aorta by scavenging superoxide radicals under conditions of acute oxidative stress. Biochem. Pharm. 2017, 128, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Kahlberg, N.; Qin, C.X.; Anthonisz, J.; Jap, E.; Ng, H.H.; Jelinic, M.; Parry, L.J.; Kemp-Harper, B.K.; Ritchie, R.H.; Leo, C.H. Adverse vascular remodelling is more sensitive than endothelial dysfunction to hyperglycaemia in diabetic rat mesenteric arteries. Pharm. Res. 2016, 111, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Leo, C.H.; Hart, J.L.; Woodman, O.L. Impairment of both nitric oxide-mediated and EDHF-type relaxation in small mesenteric arteries from rats with streptozotocin-induced diabetes. Br J Pharm. 2011, 162, 365–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leo, C.H.; Hart, J.L.; Woodman, O.L. 3’,4’-dihydroxyflavonol restores endothelium dependent relaxation in small mesenteric artery from rats with type 1 and type 2 diabetes. Eur. J. Pharm. 2011, 659, 193–198. [Google Scholar] [CrossRef]
- Leo, C.H.; Hart, J.L.; Woodman, O.L. 3’,4’-dihydroxyflavonol reduces superoxide and improves nitric oxide function in diabetic rat mesenteric arteries. PLoS ONE 2011, 6, e20813. [Google Scholar] [CrossRef] [Green Version]
- Ng, H.H.; Leo, C.H.; Prakoso, D.; Qin, C.; Ritchie, R.H.; Parry, L.J. Serelaxin treatment reverses vascular dysfunction and left ventricular hypertrophy in a mouse model of Type 1 diabetes. Sci. Rep. 2017, 7, 39604. [Google Scholar] [CrossRef] [Green Version]
- Purvis, G.S.D.; Chiazza, F.; Chen, J.; Azevedo-Loiola, R.; Martin, L.; Kusters, D.H.M.; Reutelingsperger, C.; Fountoulakis, N.; Gnudi, L.; Yaqoob, M.M.; et al. Annexin A1 attenuates microvascular complications through restoration of Akt signalling in a murine model of type 1 diabetes. Diabetologia 2018, 61, 482–495. [Google Scholar] [CrossRef] [Green Version]
- Qin, C.X.; Anthonisz, J.; Leo, C.H.; Kahlberg, N.; Velagic, A.; Li, M.; Jap, E.; Woodman, O.L.; Parry, L.J.; Horowitz, J.D.; et al. NO• resistance, induced in the myocardium by diabetes is circumvented by the NO redox sibling, nitroxyl. Antioxid. Redox Signal. 2019. [Google Scholar] [CrossRef]
- Leo, C.H.; Ng, H.H.; Marshall, S.A.; Jelinic, M.; Rupasinghe, T.; Qin, C.; Roessner, U.; Ritchie, R.H.; Tare, M.; Parry, L.J. Relaxin reduces endothelium-derived vasoconstriction in hypertension: Revealing new therapeutic insights. Br. J. Pharm. 2019. [Google Scholar] [CrossRef]
- Lu, Q.Y.; Jin, Y.; Mao, J.T.; Zhang, Z.F.; Heber, D.; Dubinett, S.M.; Rao, J. Green tea inhibits cycolooxygenase-2 in non-small cell lung cancer cells through the induction of Annexin-1. Biochem. Biophys. Res. Commun. 2012, 427, 725–730. [Google Scholar] [CrossRef] [Green Version]
- Gluais, P.; Lonchampt, M.; Morrow, J.D.; Vanhoutte, P.M.; Feletou, M. Acetylcholine-induced endothelium-dependent contractions in the SHR aorta: The Janus face of prostacyclin. Br. J. Pharm. 2005, 146, 834–845. [Google Scholar] [CrossRef] [Green Version]
- Qin, C.X.; Rosli, S.; Deo, M.; Cao, N.; Walsh, J.; Tate, M.; Alexander, A.E.; Donner, D.; Horlock, D.; Li, R.; et al. Cardioprotective actions of the annexin-A1 N-terminal peptide, Ac2-26, against myocardial infarction. Front. Pharm. 2019, 10, 269. [Google Scholar] [CrossRef] [PubMed]
- Jelinic, M.; Leo, C.H.; Post Uiterweer, E.D.; Sandow, S.L.; Gooi, J.H.; Wlodek, M.E.; Conrad, K.P.; Parkington, H.; Tare, M.; Parry, L.J. Localization of relaxin receptors in arteries and veins, and region-specific increases in compliance and bradykinin-mediated relaxation after in vivo serelaxin treatment. FASEB J. 2014, 28, 275–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leo, C.H.; Jelinic, M.; Ng, H.H.; Tare, M.; Parry, L.J. Time-dependent activation of prostacyclin and nitric oxide pathways during continuous i.v. infusion of serelaxin (recombinant human H2 relaxin). Br. J. Pharm. 2016, 173, 1005–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leo, C.H.; Jelinic, M.; Parkington, H.C.; Tare, M.; Parry, L.J. Acute intravenous injection of serelaxin (recombinant human relaxin-2) causes rapid and sustained bradykinin-mediated vasorelaxation. J. Am. Heart Assoc. 2014, 3, e000493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, S.A.; Leo, C.H.; Girling, J.E.; Tare, M.; Beard, S.; Hannan, N.J.; Parry, L.J. Relaxin treatment reduces angiotensin II-induced vasoconstriction in pregnancy and protects against endothelial dysfunction. Biol. Reprod. 2017, 96, 895–906. [Google Scholar] [CrossRef] [Green Version]
- Marshall, S.A.; Leo, C.H.; Senadheera, S.N.; Girling, J.E.; Tare, M.; Parry, L.J. Relaxin deficiency attenuates pregnancy-induced adaptation of the mesenteric artery to angiotensin II in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R847–R857. [Google Scholar] [CrossRef] [Green Version]
- Ng, H.H.; Jelinic, M.; Parry, L.J.; Leo, C.H. Increased superoxide production and altered nitric oxide-mediated relaxation in the aorta of young but not old male relaxin-deficient mice. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H285–H296. [Google Scholar] [CrossRef] [Green Version]
- Leo, C.H.; Fernando, D.T.; Tran, L.; Ng, H.H.; Marshall, S.A.; Parry, L.J. Serelaxin Treatment Reduces Oxidative Stress and Increases Aldehyde Dehydrogenase-2 to Attenuate Nitrate Tolerance. Front. Pharm. 2017, 8, 141. [Google Scholar] [CrossRef] [Green Version]
- Marshall, S.A.; O’Sullivan, K.; Ng, H.H.; Bathgate, R.A.D.; Parry, L.J.; Hossain, M.A.; Leo, C.H. B7-33 replicates the vasoprotective functions of human relaxin-2 (serelaxin). Eur. J. Pharm. 2017, 807, 190–197. [Google Scholar] [CrossRef]
- Huynh, K.; McMullen, J.R.; Julius, T.L.; Tan, J.W.; Love, J.E.; Cemerlang, N.; Kiriazis, H.; Du, X.J.; Ritchie, R.H. Cardiac-specific IGF-1 receptor transgenic expression protects against cardiac fibrosis and diastolic dysfunction in a mouse model of diabetic cardiomyopathy. Diabetes 2010, 59, 1512–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agonist | Sample Size | Cmpd17b | |
| n | pEC50 | Rmax | |
| Control (DMSO) | 12 | 5.17 ± 0.06 | 69 ± 5 |
| L-NAME | 8 | 5.08 ± 0.04 | 72 ± 2 |
| Indo | 4 | 5.09 ± 0.15 | 74 ± 4 |
| Indo+L-NAME | 4 | 5.17 ± 0.10 | 67 ± 8 |
| Indo+L-NAME+KCaB | 4 | 5.27 ± 0.06 | 75 ± 8 |
| Endothelium intact | 6 | 5.07 ± 0.03 | 62 ± 5 |
| Endothelium denuded | 6 | 5.06 ± 0.03 | 55 ± 10 |
| Control (DMSO) | 6 | 5.09 ± 0.05 | 60 ± 2 |
| 50mM K+ | 4 | 5.13 ± 0.05 | 73 ± 8 |
| ODQ | 5 | 5.14 ± 0.09 | 71 ± 2 |
| Glibenclamide | 6 | 5.20 ± 0.18 | 52 ± 3 |
| Agonist | CaCl2 | ||
| n | pEC50 | Emax | |
| Control (DMSO) | 3 | 2.34 ± 0.23 | 111 ± 10 |
| Cmpd17b | 3 | ND | 0 * |
| Nifedipine | 3 | ND | 0 * |
| Drug | Sample Size | Control | Sample Size | Diabetic | Sample Size | Diabetic +Cmpd17b | |||
|---|---|---|---|---|---|---|---|---|---|
| ACh | n | pEC50 | Rmax | n | pEC50 | Rmax | n | pEC50 | Rmax |
| Control | 6 | 7.09 ± 0.07 | 92 ± 1 | 9 | 6.70 ± 0.06 * | 88 ± 2 | 7 | 7.11 ± 0.11 # | 90 ± 2 |
| Indo | 7 | 6.97 ± 0.12 | 91 ± 1 | 7 | 6.82 ± 0.12 | 88 ± 3 | 7 | 6.68 ± 0.11^ | 86 ± 2 |
| Indo+L-NAME | 7 | ND | 9 ± 4^ | 9 | ND | 13 ± 6^ | 8 | ND | 11 ± 8 ^ |
| SNP | n | pEC50 | Rmax | n | pEC50 | Rmax | n | pEC50 | Rmax |
| Control | 7 | 7.55 ± 0.11 | 95 ± 1 | 7 | 7.80 ± 0.12 | 96 ± 1 | 7 | 7.58 ± 0.08 | 94 ± 1 |
| Cmpd17b | n | pEC50 | Rmax | n | pEC50 | Rmax | n | pEC50 | Rmax |
| Control | 6 | 5.12 ± 0.13 | 70 ± 5 | 4 | 5.14 ± 0.13 | 67 ± 5 | ND | ND | ND |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marshall, S.A.; Qin, C.X.; Jelinic, M.; O’Sullivan, K.; Deo, M.; Walsh, J.; Li, M.; Parry, L.J.; Ritchie, R.H.; Leo, C.H. The Novel Small-molecule Annexin-A1 Mimetic, Compound 17b, Elicits Vasoprotective Actions in Streptozotocin-induced Diabetic Mice. Int. J. Mol. Sci. 2020, 21, 1384. https://doi.org/10.3390/ijms21041384
Marshall SA, Qin CX, Jelinic M, O’Sullivan K, Deo M, Walsh J, Li M, Parry LJ, Ritchie RH, Leo CH. The Novel Small-molecule Annexin-A1 Mimetic, Compound 17b, Elicits Vasoprotective Actions in Streptozotocin-induced Diabetic Mice. International Journal of Molecular Sciences. 2020; 21(4):1384. https://doi.org/10.3390/ijms21041384
Chicago/Turabian StyleMarshall, Sarah A, Cheng Xue Qin, Maria Jelinic, Kelly O’Sullivan, Minh Deo, Jesse Walsh, Mandy Li, Laura J Parry, Rebecca H. Ritchie, and Chen Huei Leo. 2020. "The Novel Small-molecule Annexin-A1 Mimetic, Compound 17b, Elicits Vasoprotective Actions in Streptozotocin-induced Diabetic Mice" International Journal of Molecular Sciences 21, no. 4: 1384. https://doi.org/10.3390/ijms21041384
APA StyleMarshall, S. A., Qin, C. X., Jelinic, M., O’Sullivan, K., Deo, M., Walsh, J., Li, M., Parry, L. J., Ritchie, R. H., & Leo, C. H. (2020). The Novel Small-molecule Annexin-A1 Mimetic, Compound 17b, Elicits Vasoprotective Actions in Streptozotocin-induced Diabetic Mice. International Journal of Molecular Sciences, 21(4), 1384. https://doi.org/10.3390/ijms21041384