AGAP2: Modulating TGFβ1-Signaling in the Regulation of Liver Fibrosis

 , and
, and 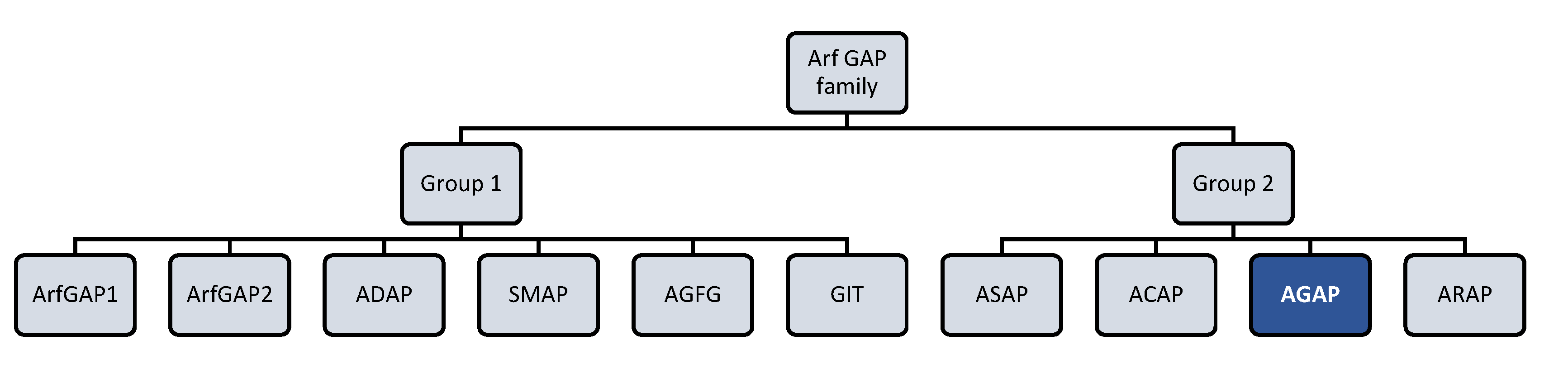{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. AGAP2: A Unique Member of the Arf GAP Family
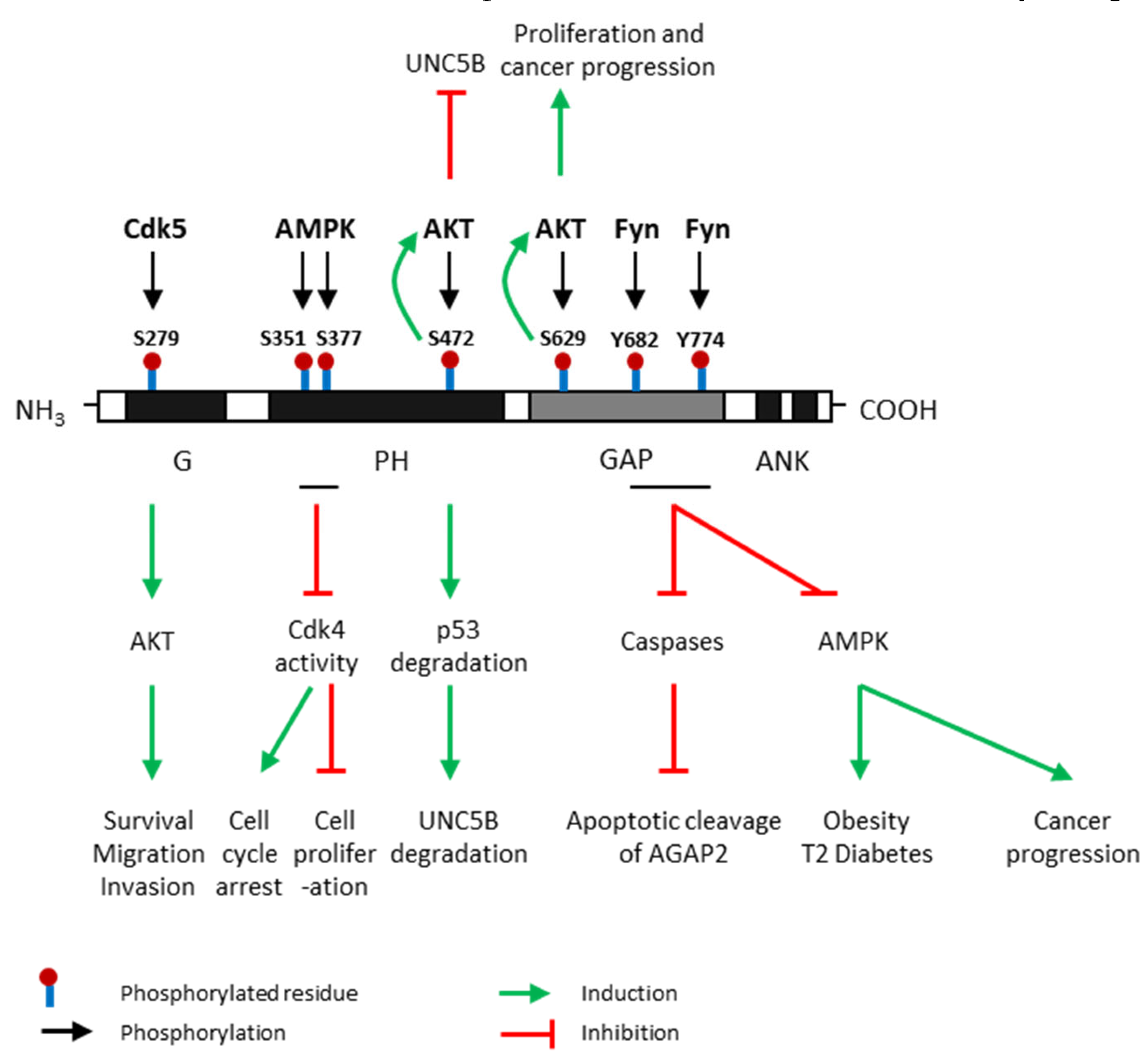
2. Two Human AGAP2 Isoforms: PIKE-L and AGAP2
3. Regulation of AGAP2 Expression and Activity
4. TGFβ and AGAP2 in Liver Fibrosis
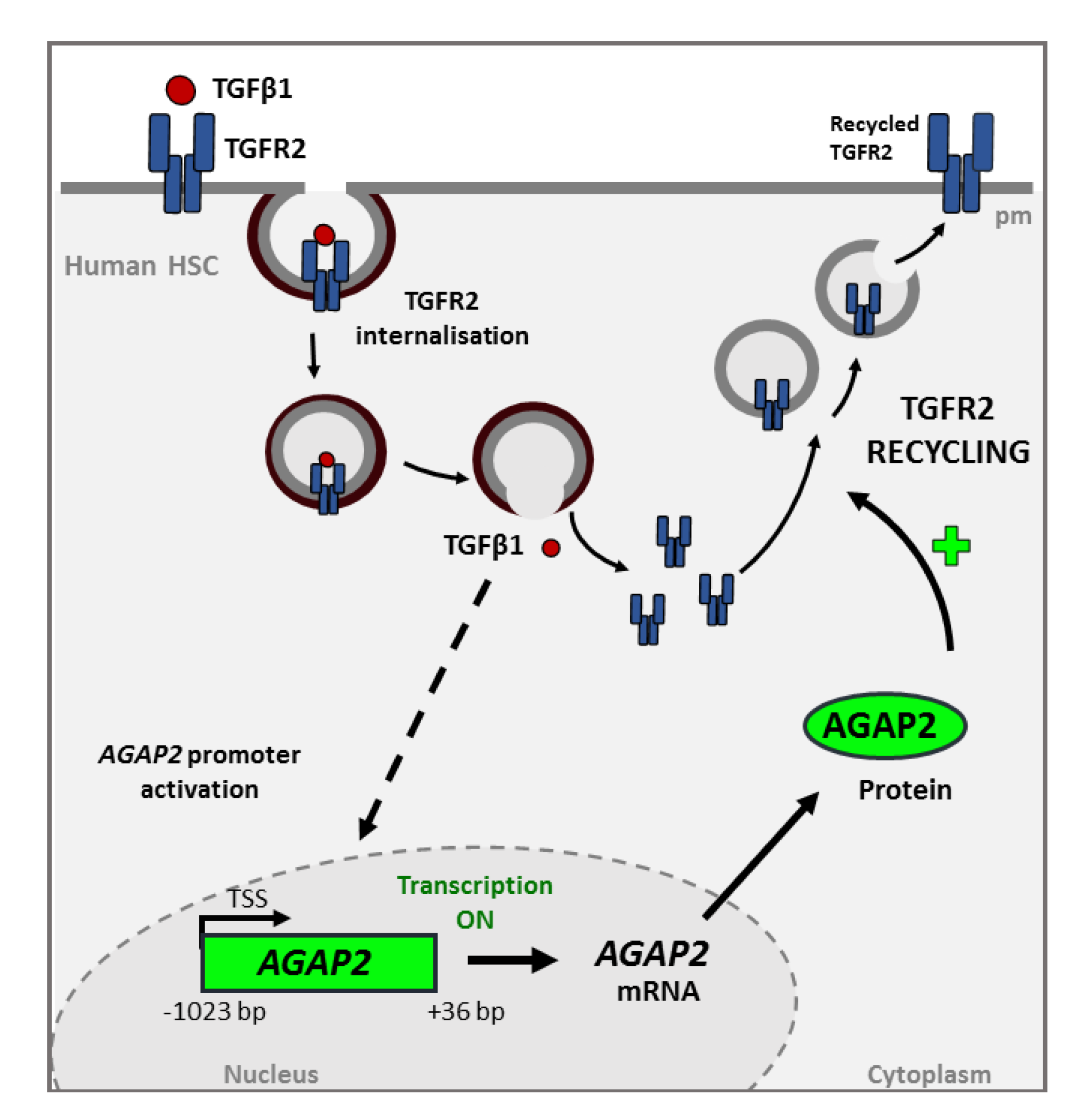
4.1. AGAP2 in TGFβ Receptor Trafficking
4.2. TGFβ1 and AGAP2 in HSC Proliferation and Migration
4.3. AGAP2 in TGFβ1-Induced Fibrogenesis on HSC
4.4. TGFβ1 Induces AGAP2 in HSC
5. Conclusions and Prospects
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| α-SMA | α-Smooth muscle actin |
| AGAP2 | Arf GAP with GTP-binding protein-like domain, Ankyrin repeat and PH domain 2 |
| AKT | AKT serine/threonine kinase, also known as ‘protein kinase B’ |
| AMPK | AMP-activated protein kinase |
| ANK | Ankyrin |
| AP | Adaptor protein |
| Arf | ADP-ribosylation factor |
| Arf GAP | ADP-ribosylation factor GTPase activating proteins |
| ATRA | all-trans retinoic acid |
| β2-AR | β2-adrenergic receptor |
| Cdk | Cyclin-dependent kinase |
| Cnt-g1 | Centauring-gamma 1 |
| DR5 | Direct repeat (DR) of two motif (G/AGTTCA) separated by 5 bp |
| ECM | Extracellular matrix |
| EGF | Epithelial growth factor |
| ERK | Extracellular-signal-regulated kinase |
| FAK | Focal adhesion kinase |
| GAPs | GTPase-activating proteins |
| GEFs | Guanine nucleotide exchange factors |
| GLUT4 | Glucose transporter type 4 |
| GSV | GLUT4-storage vesicles (GSV) |
| HSC | Hepatic stellate cells |
| MMP | Metalloproteinase |
| NK | Natural killer cells |
| NSL | Nuclear signal localization |
| PCAF | Acetyl transferase P300/CBP-associated factor |
| p38 MAP | p38 mitogen-activated protein kinase |
| PDGF | Platelet-derived growth factor 2 |
| PH | Pleckstrin homology |
| PI3K | Phosphoinositide 3-kinase |
| PIKE-L | PI 3-Kinase Enhancer longer isoform L |
| PRD | Proline-rich domain |
| Rab | Ras-associated binding protein |
| RACK1 | Receptor of activated protein kinase C1 |
| RARE | Retinoic acid response element |
| SARA | Smad anchor for receptor activation |
| SP1 | Specificity protein 1 |
| TAA | Thioacetamide |
| TIMP | Tissue inhibitor of metallopeptidases |
| TGFR | Transforming growth factor β receptor |
| Treg | T regulatory cells |
| UNC5B | Uncoordinated-5 netrin receptor B |
References
- Donaldson, J.G.; Jackson, C.L. ARF family G proteins and their regulators: Roles in membrane transport, development and disease. Nat. Rev. Mol. Cell Biol. 2011, 12, 362–375. [Google Scholar] [CrossRef] [PubMed]
- East, M.P.; Kahn, R.A. Models for the functions of Arf GAPs. Semin. Cell Dev. Biol. 2011, 22, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cukierman, E.; Huber, I.; Rotman, M.; Cassel, D. The ARF1 GTPase-activating protein: Zinc finger motif and Golgi complex localization. Science 1995, 270, 1999–2002. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Randazzo, P.A. Arf GAPs and their interacting proteins. Traffic 2007, 8, 1465–1475. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Dara, L.; Li, T.W.; Zheng, Y.; Yang, H.; Tomasi, M.L.; Tomasi, I.; Giordano, P.; Mato, J.M.; Lu, S.C. MAT2B-GIT1 interplay activates MEK1/ERK 1 and 2 to induce growth in human liver and colon cancer. Hepatology 2013, 57, 2299–2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, S.Y.; Jang, S.W.; Ko, J. Regulation of ADP-ribosylation factor 4 expression by small leucine zipper protein and involvement in breast cancer cell migration. Cancer Lett. 2012, 314, 185–197. [Google Scholar] [CrossRef]
- Hashimoto, S.; Onodera, Y.; Hashimoto, A.; Tanaka, M.; Hamaguchi, M.; Yamada, A.; Sabe, H. Requirement for Arf6 in breast cancer invasive activities. Proc. Natl. Acad. Sci. USA 2004, 101, 6647–6652. [Google Scholar] [CrossRef] [Green Version]
- Schlienger, S.; Campbell, S.; Pasquin, S.; Gaboury, L.; Claing, A. ADP-ribosylation factor 1 expression regulates epithelial-mesenchymal transition and predicts poor clinical outcome in triple-negative breast cancer. Oncotarget 2016, 7, 15811–15827. [Google Scholar] [CrossRef] [Green Version]
- Bidkhori, G.; Narimani, Z.; Hosseini Ashtiani, S.; Moeini, A.; Nowzari-Dalini, A.; Masoudi-Nejad, A. Reconstruction of an integrated genome-scale co-expression network reveals key modules involved in lung adenocarcinoma. PLoS ONE 2013, 8, e67552. [Google Scholar] [CrossRef]
- Chi, J.H.; Panner, A.; Cachola, K.; Crane, C.A.; Murray, J.; Pieper, R.O.; James, C.D.; Parsa, A.T. Increased expression of the glioma-associated antigen ARF4L after loss of the tumor suppressor PTEN. Laboratory investigation. J. Neurosurg. 2008, 108, 299–303. [Google Scholar] [CrossRef]
- Davis, J.E.; Xie, X.; Guo, J.; Huang, W.; Chu, W.M.; Huang, S.; Teng, Y.; Wu, G. ARF1 promotes prostate tumorigenesis via targeting oncogenic MAPK signaling. Oncotarget 2016, 7, 39834–39845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Shi, B.; Jarzynka, M.J.; Yiin, J.J.; D’Souza-Schorey, C.; Cheng, S.Y. ADP-ribosylation factor 6 regulates glioma cell invasion through the IQ-domain GTPase-activating protein 1-Rac1-mediated pathway. Cancer Res. 2009, 69, 794–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahn, R.A.; Bruford, E.; Inoue, H.; Logsdon, J.M.J.r.; Nie, Z.; Premont, R.T.; Randazzo, P.A.; Satake, M.; Theibert, A.B.; Zapp, M.L.; et al. Consensus nomenclature for the human ArfGAP domain-containing proteins. J. Cell Biol. 2008, 182, 1039–1044. [Google Scholar] [CrossRef] [PubMed]
- Spang, A.; Shiba, Y.; Randazzo, P.A. Arf GAPs: Gatekeepers of vesicle generation. FEBS Lett. 2010, 584, 2646–2651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, K.; Hurt, K.J.; Wu, F.Y.; Fang, M.; Luo, H.R.; Hong, J.J.; Blackshaw, S.; Ferris, C.D.; Snyder, S.H. Pike. A nuclear gtpase that enhances PI3kinase activity and is regulated by protein 4.1N. Cell 2000, 103, 919–930. [Google Scholar] [CrossRef] [Green Version]
- Xia, C.; Ma, W.; Stafford, L.J.; Liu, C.; Gong, L.; Martin, J.F.; Liu, M. GGAPs, a new family of bifunctional GTP-binding and GTPase-activating proteins. Mol. Cell. Biol. 2003, 23, 2476–2488. [Google Scholar] [CrossRef] [Green Version]
- Casalou, C.; Faustino, A.; Barral, D.C. Arf proteins in cancer cell migration. Small GTPases 2016, 7, 270–282. [Google Scholar] [CrossRef]
- Nagase, T.; Seki, N.; Ishikawa, K.; Tanaka, A.; Nomura, N. Prediction of the coding sequences of unidentified human genes. V. The coding sequences of 40 new genes (KIAA0161-KIAA0200) deduced by analysis of cDNA clones from human cell line KG-1. DNA Res. 1996, 3, 17–24. [Google Scholar] [CrossRef]
- Elkahloun, A.G.; Krizman, D.B.; Wang, Z.; Hofmann, T.A.; Roe, B.; Meltzer, P.S. Transcript mapping in a 46-kb sequenced region at the core of 12q13.3 amplification in human cancers. Genomics 1997, 42, 295–301. [Google Scholar] [CrossRef] [Green Version]
- Ahn, J.Y.; Rong, R.; Kroll, T.G.; Van Meir, E.G.; Snyder, S.H.; Ye, K. PIKE (phosphatidylinositol 3-kinase enhancer)-A GTPase stimulates Akt activity and mediates cellular invasion. J. Biol. Chem. 2004, 279, 16441–16451. [Google Scholar] [CrossRef] [Green Version]
- Campa, F.; Yoon, H.Y.; Ha, V.L.; Szentpetery, Z.; Balla, T.; Randazzo, P.A. A PH domain in the Arf GTPase-activating protein (GAP) ARAP1 binds phosphatidylinositol 3,4,5-trisphosphate and regulates Arf GAP activity independently of recruitment to the plasma membranes. J. Biol. Chem. 2009, 284, 28069–28083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Liu, Z.; Ye, K. Phosphoinositol lipids bind to phosphatidylinositol 3 (PI3)-kinase enhancer GTPase and mediate its stimulatory effect on PI3-kinase and Akt signalings. Proc. Natl. Acad. Sci. USA 2005, 102, 16853–16858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Q.; Ye, K. The roles of PIKE in tumorigenesis. Acta Pharmacol. Sin. 2013, 34, 991–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Wang, M.; Marcora, E.M.; Zhang, B.; Goate, A.M. Promoter DNA hypermethylation—Implications for Alzheimer’s disease. Neurosci. Lett. 2019, 711, 134403. [Google Scholar] [CrossRef]
- Doush, Y.; Surani, A.A.; Navarro-Corcuera, A.; McArdle, S.; Billett, E.E.; Montiel-Duarte, C. SP1 and RARalpha regulate AGAP2 expression in cancer. Sci. Rep. 2019, 9, 390. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Tian, B.; Gearing, M.; Hunter, S.; Ye, K.; Mao, Z. Cdk5-mediated regulation of the PIKE-A-Akt pathway and glioblastoma cell invasion. Proc. Natl. Acad. Sci. USA 2008, 105, 7570–7575. [Google Scholar] [CrossRef] [Green Version]
- He, K.; Jang, S.W.; Joshi, J.; Yoo, M.H.; Ye, K. Akt-phosphorylated PIKE-A inhibits UNC5B-induced apoptosis in cancer cell lines in a p53-dependent manner. Mol. Biol. Cell 2011, 22, 1943–1954. [Google Scholar] [CrossRef]
- Cai, Y.; Wang, J.; Li, R.; Ayala, G.; Ittmann, M.; Liu, M. GGAP2/PIKE-a directly activates both the Akt and nuclear factor-kappaB pathways and promotes prostate cancer progression. Cancer Res. 2009, 69, 819–827. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Feng, Y.; Ye, K. Src-family tyrosine kinase fyn phosphorylates phosphatidylinositol 3-kinase enhancer-activating Akt, preventing its apoptotic cleavage and promoting cell survival. Cell Death Differ. 2007, 14, 368–377. [Google Scholar] [CrossRef]
- Zhang, S.; Sheng, H.; Zhang, X.; Qi, Q.; Chan, C.B.; Li, L.; Shan, C.; Ye, K. Cellular energy stress induces AMPK-mediated regulation of glioblastoma cell proliferation by PIKE-A phosphorylation. Cell Death Dis. 2019, 10, 222. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y.D.; Jensen, A.R.; Salgia, R.; Posadas, E.M. Fyn: A novel molecular target in cancer. Cancer 2010, 116, 1629–1637. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Ye, K. Pike tyrosine phosphorylation regulates its apoptotic cleavage during programmed cell death. Adv. Enzym. Regul. 2006, 46, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caja, L.; Dituri, F.; Mancarella, S.; Caballero-Diaz, D.; Moustakas, A.; Giannelli, G.; Fabregat, I. TGF-beta and the Tissue Microenvironment: Relevance in Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 1294. [Google Scholar] [CrossRef] [Green Version]
- Novo, E.; di Bonzo, L.V.; Cannito, S.; Colombatto, S.; Parola, M. Hepatic myofibroblasts: A heterogeneous population of multifunctional cells in liver fibrogenesis. Int. J. Biochem. Cell Biol. 2009, 41, 2089–2093. [Google Scholar] [CrossRef]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef]
- Dooley, S.; ten Dijke, P. TGF-beta in progression of liver disease. Cell Tissue Res. 2012, 347, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Ignotz, R.A.; Massague, J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J. Biol. Chem. 1986, 261, 4337–4345. [Google Scholar]
- Heino, J.; Ignotz, R.A.; Hemler, M.E.; Crouse, C.; Massague, J. Regulation of cell adhesion receptors by transforming growth factor-beta. Concomitant regulation of integrins that share a common beta 1 subunit. J. Biol. Chem. 1989, 264, 380–388. [Google Scholar]
- Edwards, D.R.; Murphy, G.; Reynolds, J.J.; Whitham, S.E.; Docherty, A.J.; Angel, P.; Heath, J.K. Transforming growth factor beta modulates the expression of collagenase and metalloproteinase inhibitor. EMBO J. 1987, 6, 1899–1904. [Google Scholar] [CrossRef]
- Islam, M.S.; Kusakabe, M.; Horiguchi, K.; Iino, S.; Nakamura, T.; Iwanaga, K.; Hashimoto, H.; Matsumoto, S.; Murata, T.; Hori, M.; et al. PDGF and TGF-β promote tenascin-C expression in subepithelial myofibroblasts and contribute to intestinal mucosal protection in mice. Br. J. Pharmacol. 2014, 171, 375–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neubauer, K.; Krüger, M.; Quondamatteo, F.; Knittel, T.; Saile, B.; Ramadori, G. Transforming growth factor-beta1 stimulates the synthesis of basement membrane proteins laminin, Collagen Type IV and Entactin in Rat Liver Sinusoidal Endothelial Cells. J. Hepatol. 1999, 31, 692–702. [Google Scholar] [CrossRef]
- Okabe, H.; Furukawa, Y.; Kato, T.; Hasegawa, S.; Yamaoka, Y.; Nakamura, Y. Isolation of development and differentiation enhancing factor-like 1 (DDEFL1) as a drug target for hepatocellular carcinomas. Int. J. Oncol. 2004, 24, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Santibanez, J.F.; Guerrero, J.; Quintanilla, M.; Fabra, A.; Martinez, J. Transforming growth factor-beta1 modulates matrix metalloproteinase-9 production through the Ras/MAPK signaling pathway in transformed keratinocytes. Biochem. Biophys. Res. Commun. 2002, 296, 267–273. [Google Scholar] [CrossRef]
- Porther, N.; Barbieri, M.A. The role of endocytic Rab GTPases in regulation of growth factor signaling and the migration and invasion of tumor cells. Small GTPases 2015, 6, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Kardassis, D.; Murphy, C.; Fotsis, T.; Moustakas, A.; Stournaras, C. Control of transforming growth factor beta signal transduction by small GTPases. FEBS J. 2009, 276, 2947–2965. [Google Scholar] [CrossRef]
- Martinez-Salgado, C.; Rodriguez-Pena, A.B.; Lopez-Novoa, J.M. Involvement of small Ras GTPases and their effectors in chronic renal disease. Cell. Mol. Life Sci. 2008, 65, 477–492. [Google Scholar] [CrossRef]
- Nomikou, E.; Livitsanou, M.; Stournaras, C.; Kardassis, D. Transcriptional and post-transcriptional regulation of the genes encoding the small GTPases RhoA, RhoB, and RhoC: Implications for the pathogenesis of human diseases. Cell. Mol. Life Sci. 2018, 75, 2111–2124. [Google Scholar] [CrossRef]
- Ren, H.; Li, Y.; Chen, Y.; Wang, L. Endostatin attenuates PDGF-BB- or TGF-beta1-induced HSCs activation via suppressing RhoA/ROCK1 signal pathways. Drug Des. Dev. Ther. 2019, 13, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Bottner, M.; Krieglstein, K.; Unsicker, K. The transforming growth factor-betas: Structure, signaling, and roles in nervous system development and functions. J. Neurochem. 2000, 75, 2227–2240. [Google Scholar] [CrossRef]
- Vander Ark, A.; Cao, J.; Li, X. TGF-β receptors: In and beyond TGF-β Signaling. Cell Signal. 2018, 52, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-beta and the TGF-beta Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.K.; Sheppard, D.; Chapman, H.A. TGF-beta1 Signaling and Tissue Fibrosis. Cold Spring Harb. Perspect. Biol. 2018, 10, a022293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massague, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [Green Version]
- Duan, D.; Derynck, R. Transforming growth factor-beta (TGF-beta)-induced up-regulation of TGF-beta receptors at the cell surface amplifies the TGF-beta response. J. Biol. Chem. 2019, 294, 8490–8504. [Google Scholar] [CrossRef]
- Zwaagstra, J.C.; El-Alfy, M.; O’Connor-McCourt, M.D. Transforming growth factor (TGF)-beta 1 internalization: Modulation by ligand interaction with TGF-beta receptors types I and II and a mechanism that is distinct from clathrin-mediated endocytosis. J. Biol. Chem. 2001, 276, 27237–27245. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Chen, Y.G. Regulation of TGF-beta receptor activity. Cell Biosci. 2012, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Rozés-Salvador, V.; Siri, S.O.; Musri, M.M.; Conde, C. New Player in Endosomal Trafficking: Differential Roles of Smad Anchor for Receptor Activation (SARA) Protein. Mol. Cell. Biol. 2018, 38, e00446–18. [Google Scholar] [CrossRef] [Green Version]
- Navarro-Corcuera, A.; Lopez-Zabalza, M.J.; Martinez-Irujo, J.J.; Alvarez-Sola, G.; Avila, M.A.; Iraburu, M.J.; Ansorena, E.; Montiel-Duarte, C. Role of AGAP2 in the profibrogenic effects induced by TGFbeta in LX-2 hepatic stellate cells. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 673–685. [Google Scholar] [CrossRef]
- Mitchell, H.; Choudhury, A.; Pagano, R.E.; Leof, E.B. Ligand-dependent and -independent transforming growth factor-beta receptor recycling regulated by clathrin-mediated endocytosis and Rab11. Mol. Biol. Cell 2004, 15, 4166–4178. [Google Scholar] [CrossRef]
- Nie, Z.; Fei, J.; Premont, R.T.; Randazzo, P.A. The Arf GAPs AGAP1 and AGAP2 distinguish between the adaptor protein complexes AP-1 and AP-3. J. Cell Sci. 2005, 118, 3555–3566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiba, Y.; Romer, W.; Mardones, G.A.; Burgos, P.V.; Lamaze, C.; Johannes, L. AGAP2 regulates retrograde transport between early endosomes and the TGN. J. Cell Sci. 2010, 123, 2381–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobilka, B.K. Structural insights into adrenergic receptor function and pharmacology. Trends Pharmacol. Sci. 2011, 32, 213–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefkowitz, R.J.; Rajagopal, K.; Whalen, E.J. New roles for beta-arrestins in cell signaling: Not just for seven-transmembrane receptors. Mol. Cell 2006, 24, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhao, Y.; Ma, X.; Zhu, Y.; Patel, J.; Nie, Z. The Arf GAP AGAP2 interacts with beta-arrestin2 and regulates beta2-adrenergic receptor recycling and ERK activation. Biochem. J. 2013, 452, 411–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Li, J.; Xiang, X.; Guo, L.; Tu, K.; Liu, Q.; Shah, V.H.; Kang, N. PDGF receptor-alpha promotes TGF-beta signaling in hepatic stellate cells via transcriptional and posttranscriptional regulation of TGF-beta receptors. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G749–G759. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Zeisberg, M.; Mosterman, B.; Sudhakar, A.; Yerramalla, U.; Holthaus, K.; Xu, L.; Eng, F.; Afdhal, N.; Kalluri, R. Liver fibrosis: Insights into migration of hepatic stellate cells in response to extracellular matrix and growth factors. Gastroenterology 2003, 124, 147–159. [Google Scholar] [CrossRef]
- Shah, R.; Reyes-Gordillo, K.; Arellanes-Robledo, J.; Lechuga, C.G.; Hernandez-Nazara, Z.; Cotty, A.; Rojkind, M.; Lakshman, M.R. TGF-beta1 up-regulates the expression of PDGF-beta receptor mRNA and induces a delayed PI3K-, AKT-, and p70(S6K) -dependent proliferative response in activated hepatic stellate cells. Alcohol. Clin. Exp. Res. 2013, 37, 1838–1848. [Google Scholar] [CrossRef]
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell migration: Integrating signals from front to back. Science 2003, 302, 1704–1709. [Google Scholar] [CrossRef] [Green Version]
- Lauffenburger, D.A.; Horwitz, A.F. Cell migration: A physically integrated molecular process. Cell 1996, 84, 359–369. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Guan, J.L. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv. Drug Deliv Rev. 2011, 63, 610–615. [Google Scholar] [CrossRef] [Green Version]
- Bissell, D.M. Hepatic fibrosis as wound repair: A progress report. J. Gastroenterol. 1998, 33, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.D.; Borgman, C.A.; Cobb, B.S.; Vines, R.R.; Reynolds, A.B.; Parsons, J.T. pp125FAK a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc. Natl. Acad. Sci. USA 1992, 89, 5192–5196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atherton, P.; Stutchbury, B.; Jethwa, D.; Ballestrem, C. Mechanosensitive components of integrin adhesions: Role of vinculin. Exp. Cell Res. 2016, 343, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Huttenlocher, A.; Horwitz, A.R. Integrins in cell migration. Cold Spring Harb. Perspect. Biol. 2011, 3, a005074. [Google Scholar] [CrossRef] [PubMed]
- Gardel, M.L.; Schneider, I.C.; Aratyn-Schaus, Y.; Waterman, C.M. Mechanical integration of actin and adhesion dynamics in cell migration. Annu. Rev. Cell Dev. Biol. 2010, 26, 315–333. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Loijens, J.C.; Martin, K.H.; Karginov, A.V.; Parsons, J.T. The association of ASAP1, an ADP ribosylation factor-GTPase activating protein, with focal adhesion kinase contributes to the process of focal adhesion assembly. Mol. Biol. Cell 2002, 13, 2147–2156. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Wu, Y.; Kim, J.I.; Wang, Z.; Daaka, Y.; Nie, Z. Arf GTPase-activating protein AGAP2 regulates focal adhesion kinase activity and focal adhesion remodeling. J. Biol. Chem. 2009, 284, 13489–13496. [Google Scholar] [CrossRef] [Green Version]
- Dwane, S.; Durack, E.; O’Connor, R.; Kiely, P.A. RACK1 promotes neurite outgrowth by scaffolding AGAP2 to FAK. Cell Signal. 2014, 26, 9–18. [Google Scholar] [CrossRef]
- Zhao, X.K.; Yu, L.; Cheng, M.L.; Che, P.; Lu, Y.Y.; Zhang, Q.; Mu, M.; Li, H.; Zhu, L.L.; Zhu, J.J.; et al. Focal Adhesion Kinase Regulates Hepatic Stellate Cell Activation and Liver Fibrosis. Sci. Rep. 2017, 7, 4032. [Google Scholar] [CrossRef]
- Varadaraj, A.; Jenkins, L.M.; Singh, P.; Chanda, A.; Snider, J.; Lee, N.Y.; Amsalem-Zafran, A.R.; Ehrlich, M.; Henis, Y.I.; Mythreye, K. TGF-beta triggers rapid fibrillogenesis via a novel TbetaRII-dependent fibronectin-trafficking mechanism. Mol. Biol. Cell 2017, 28, 1195–1207. [Google Scholar] [CrossRef] [PubMed]
- Rippe, R.A.; Almounajed, G.; Brenner, D.A. Sp1 binding activity increases in activated Ito cells. Hepatology 1995, 22, 241–251. [Google Scholar] [PubMed]
- Chen, H.; Zhou, Y.; Chen, K.Q.; An, G.; Ji, S.Y.; Chen, Q.K. Anti-fibrotic effects via regulation of transcription factor Sp1 on hepatic stellate cells. Cell. Physiol. Biochem. 2012, 29, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Y.; Ju, D.; Zhang, D.W.; Li, H.; Kong, L.M.; Guo, Y.; Li, C.; Wang, X.L.; Chen, Z.N.; Bian, H. Activation of TGF-beta1-CD147 positive feedback loop in hepatic stellate cells promotes liver fibrosis. Sci. Rep. 2015, 5, 16552. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Wang, Y.; Wang, L.; Yao, B.; Sun, L.; Liu, R.; Chen, T.; Niu, Y.; Tu, K.; Liu, Q. Long non-coding RNA AGAP2-AS1, functioning as a competitive endogenous RNA, upregulates ANXA11 expression by sponging miR-16-5p and promotes proliferation and metastasis in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2019, 38, 194. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.G.; Su, X.; Su, G.; Scotton, C.J.; Camerer, E.; Laurent, G.J.; Davis, G.E.; Chambers, R.C.; Matthay, M.A.; Sheppard, D. Ligation of protease-activated receptor 1 enhances alpha(v)beta6 integrin-dependent TGF-beta activation and promotes acute lung injury. J. Clin. Investig. 2006, 116, 1606–1614. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Navarro-Corcuera, A.; Ansorena, E.; Montiel-Duarte, C.; Iraburu, M.J. AGAP2: Modulating TGFβ1-Signaling in the Regulation of Liver Fibrosis. Int. J. Mol. Sci. 2020, 21, 1400. https://doi.org/10.3390/ijms21041400
Navarro-Corcuera A, Ansorena E, Montiel-Duarte C, Iraburu MJ. AGAP2: Modulating TGFβ1-Signaling in the Regulation of Liver Fibrosis. International Journal of Molecular Sciences. 2020; 21(4):1400. https://doi.org/10.3390/ijms21041400
Chicago/Turabian StyleNavarro-Corcuera, Amaia, Eduardo Ansorena, Cristina Montiel-Duarte, and María J. Iraburu. 2020. "AGAP2: Modulating TGFβ1-Signaling in the Regulation of Liver Fibrosis" International Journal of Molecular Sciences 21, no. 4: 1400. https://doi.org/10.3390/ijms21041400
APA StyleNavarro-Corcuera, A., Ansorena, E., Montiel-Duarte, C., & Iraburu, M. J. (2020). AGAP2: Modulating TGFβ1-Signaling in the Regulation of Liver Fibrosis. International Journal of Molecular Sciences, 21(4), 1400. https://doi.org/10.3390/ijms21041400