Structural Analysis and Design of Chionodracine-Derived Peptides Using Circular Dichroism and Molecular Dynamics Simulations

 , ,
, ,  and
and
Abstract
:
1. Introduction
2. Results
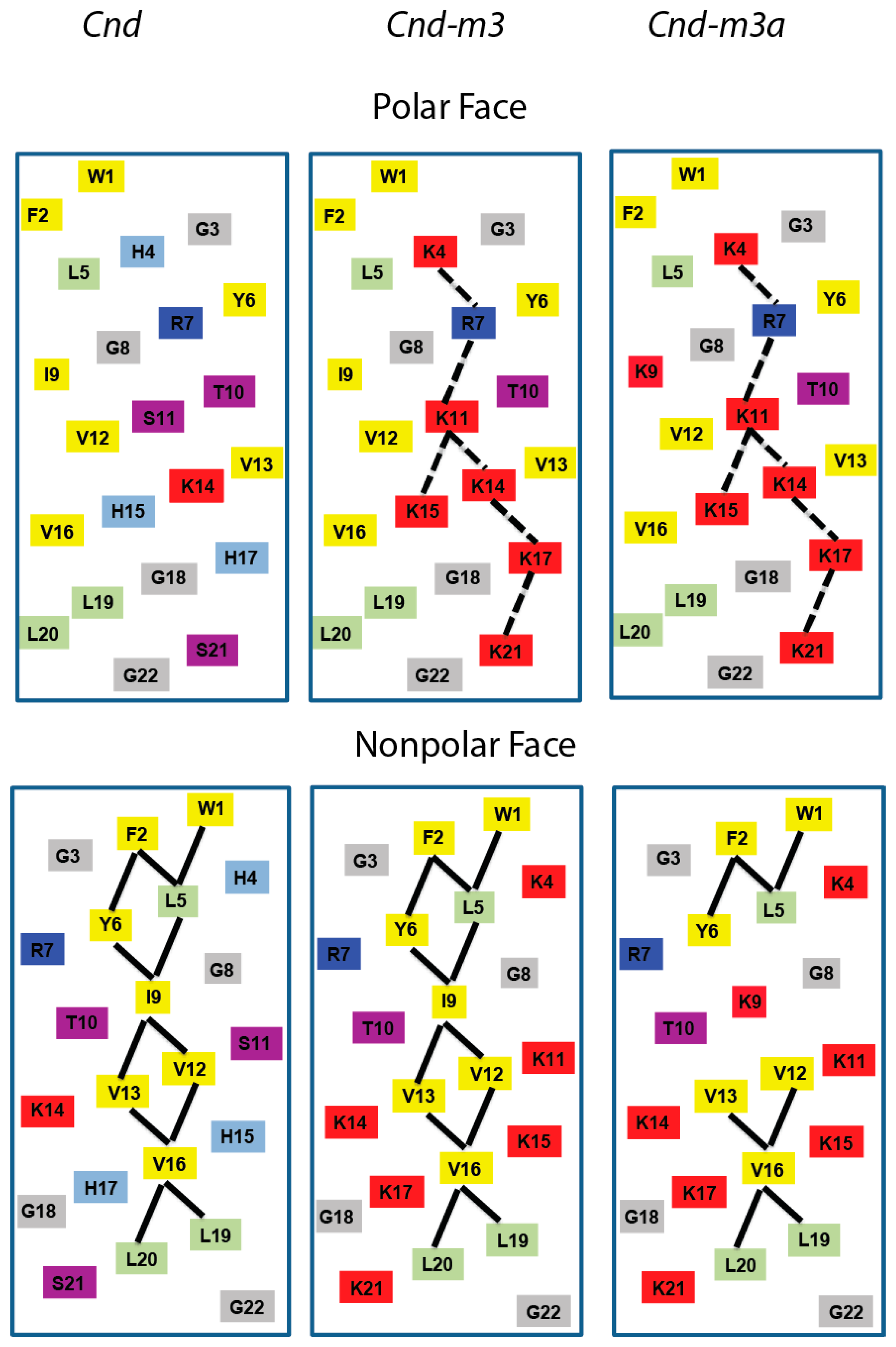
2.1. Peptide Design and Physico-Chemical Properties
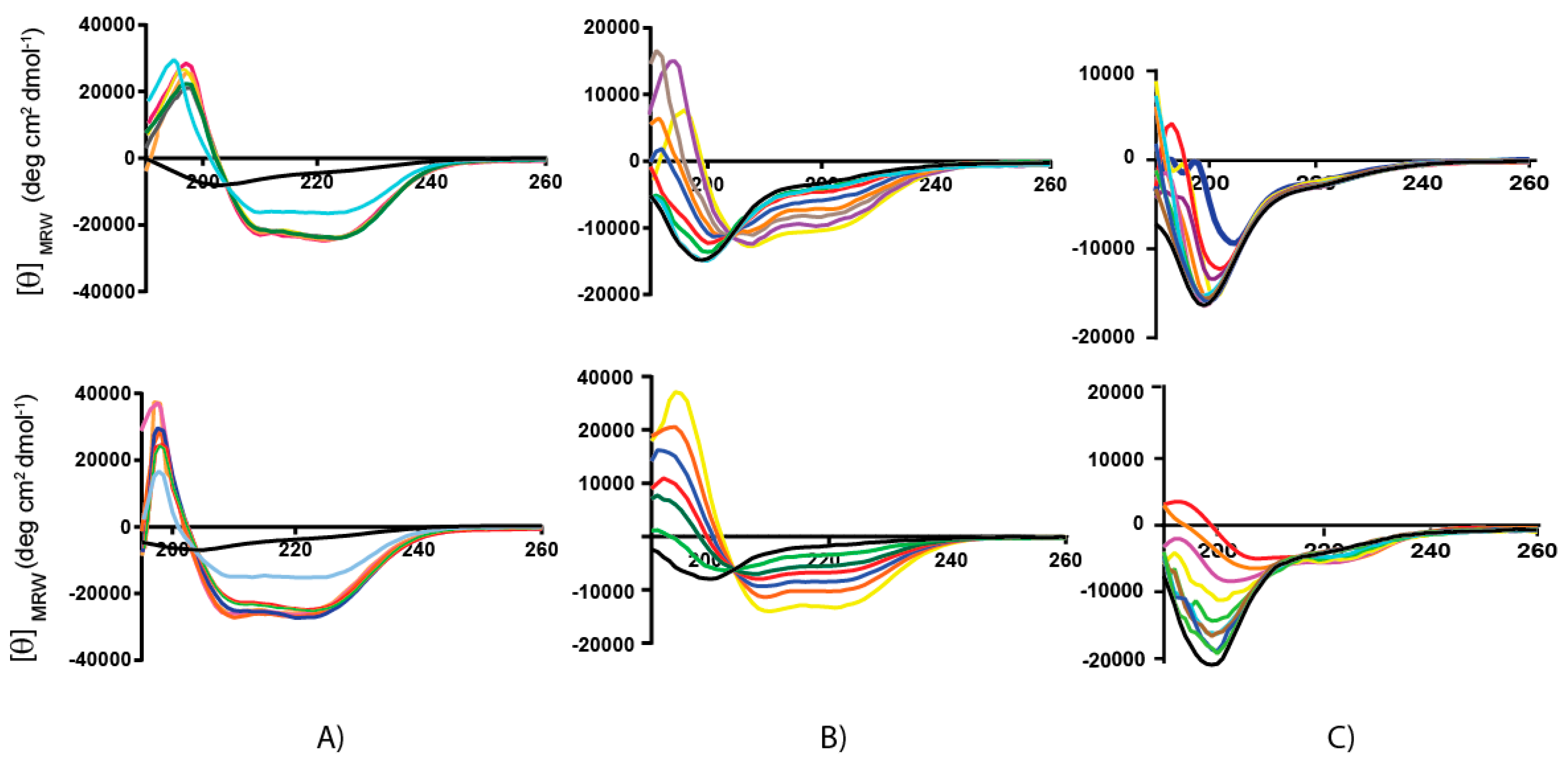
2.2. Secondary Structure Analysis by Circular Dichroism
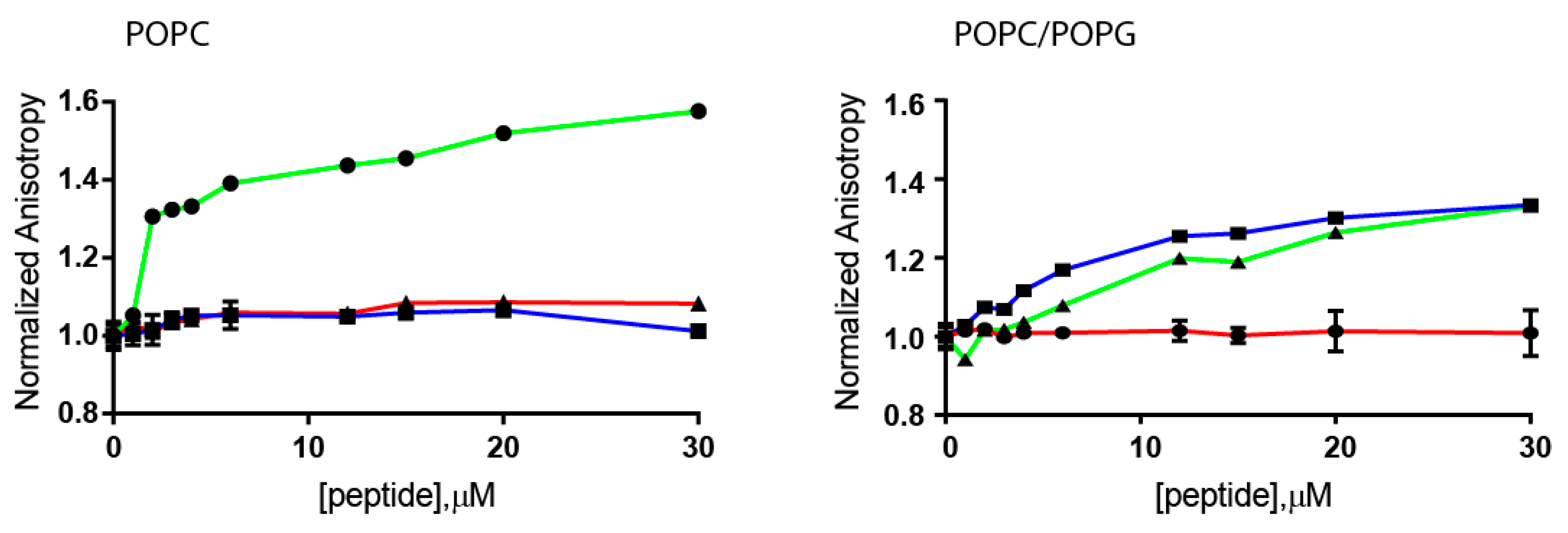
2.3. Steady-State Anisotropy
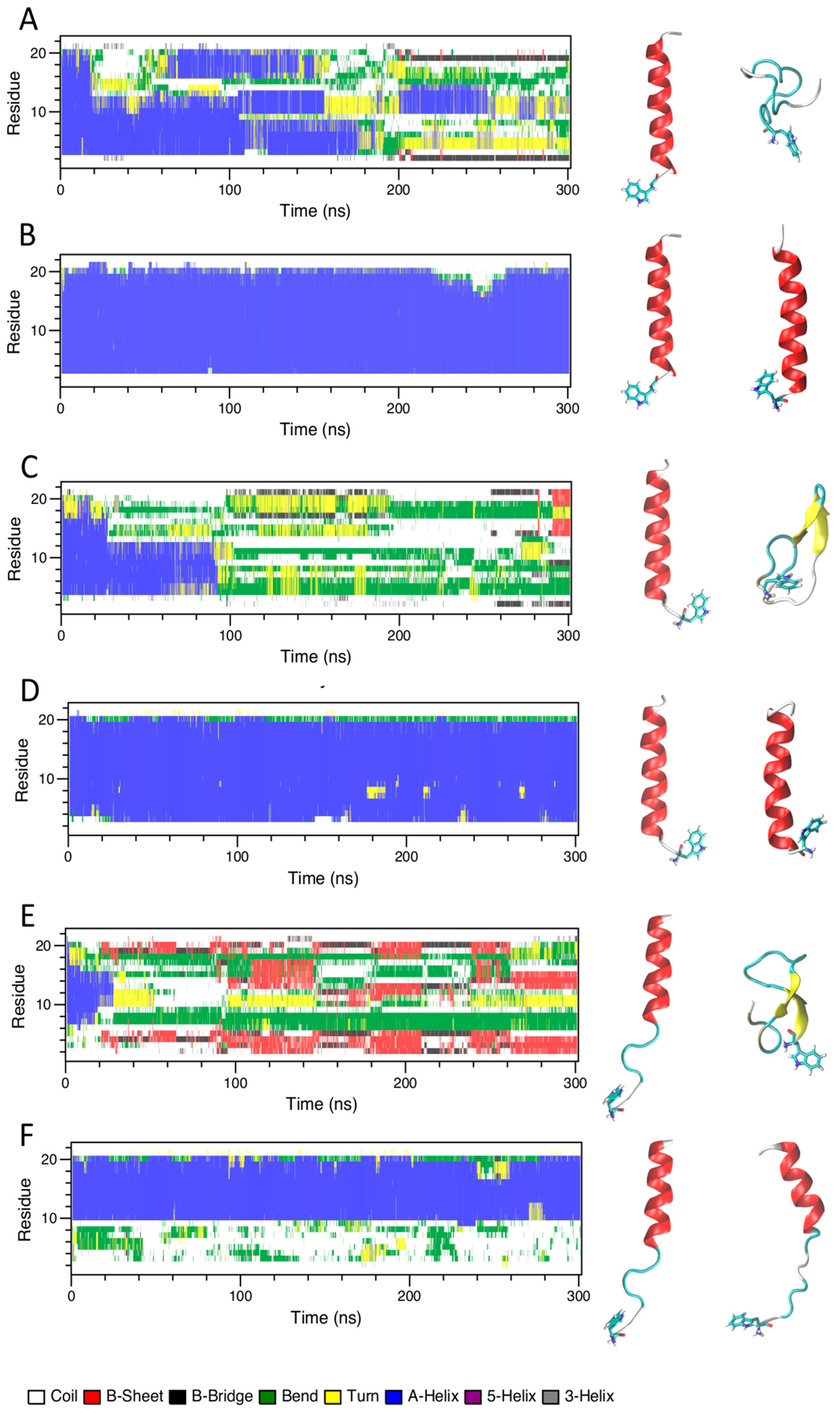
2.4. Molecular Dynamics Simulation
2.4.1. Water and TFE/Water Solution
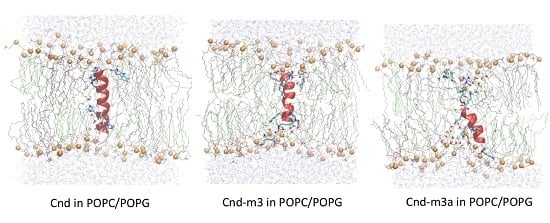
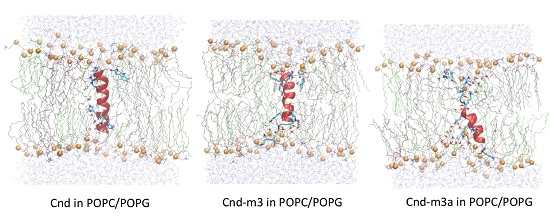
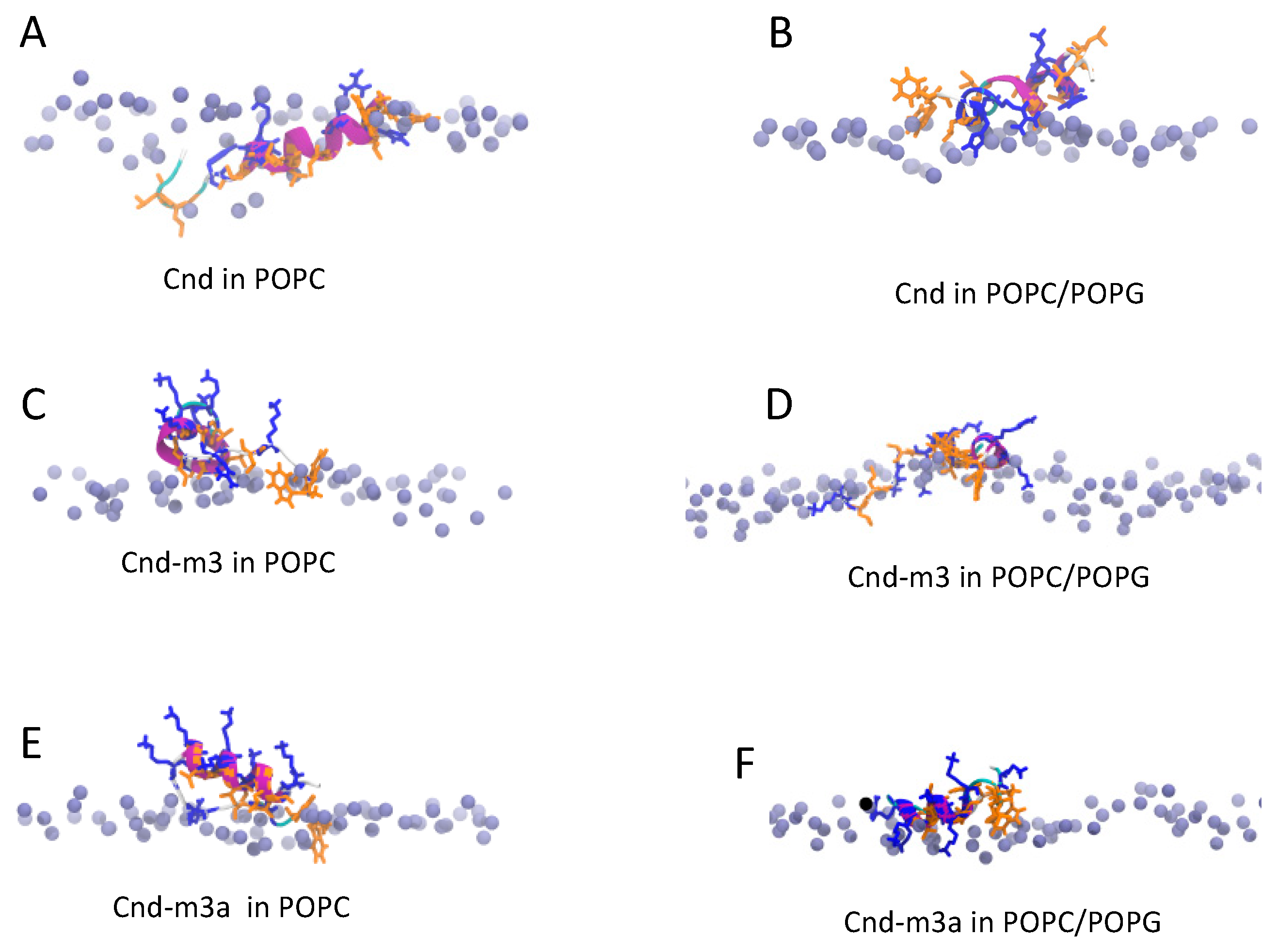
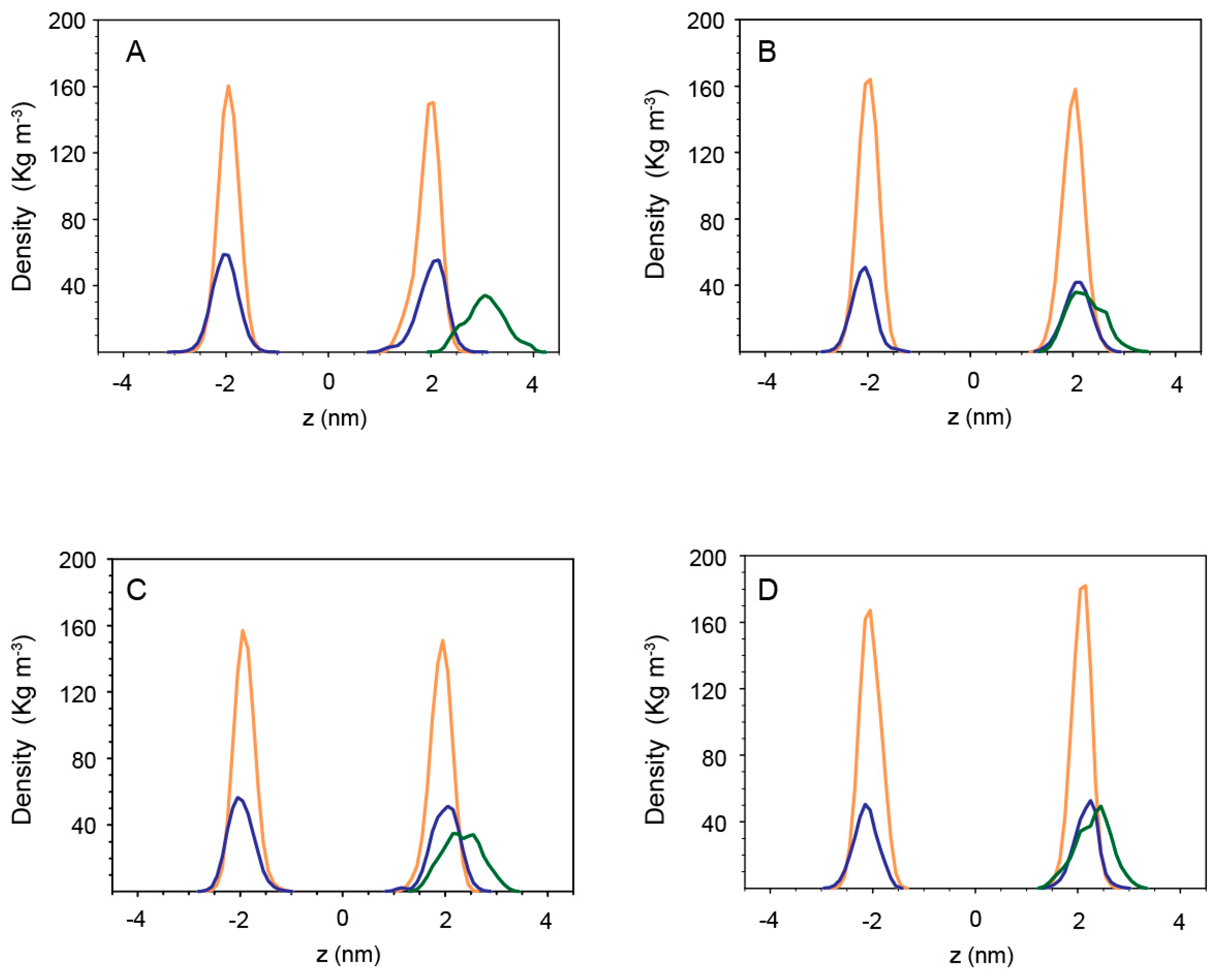
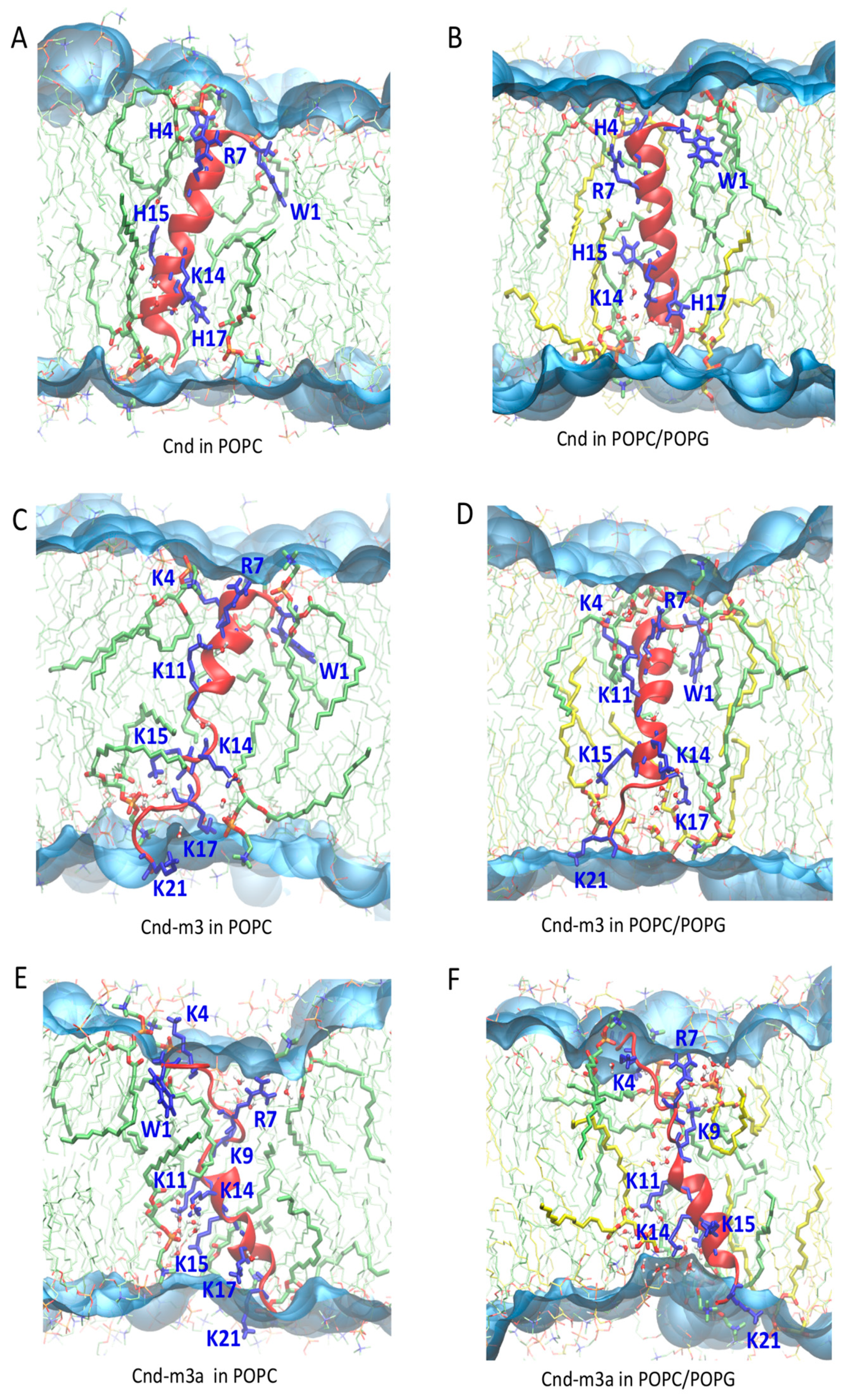
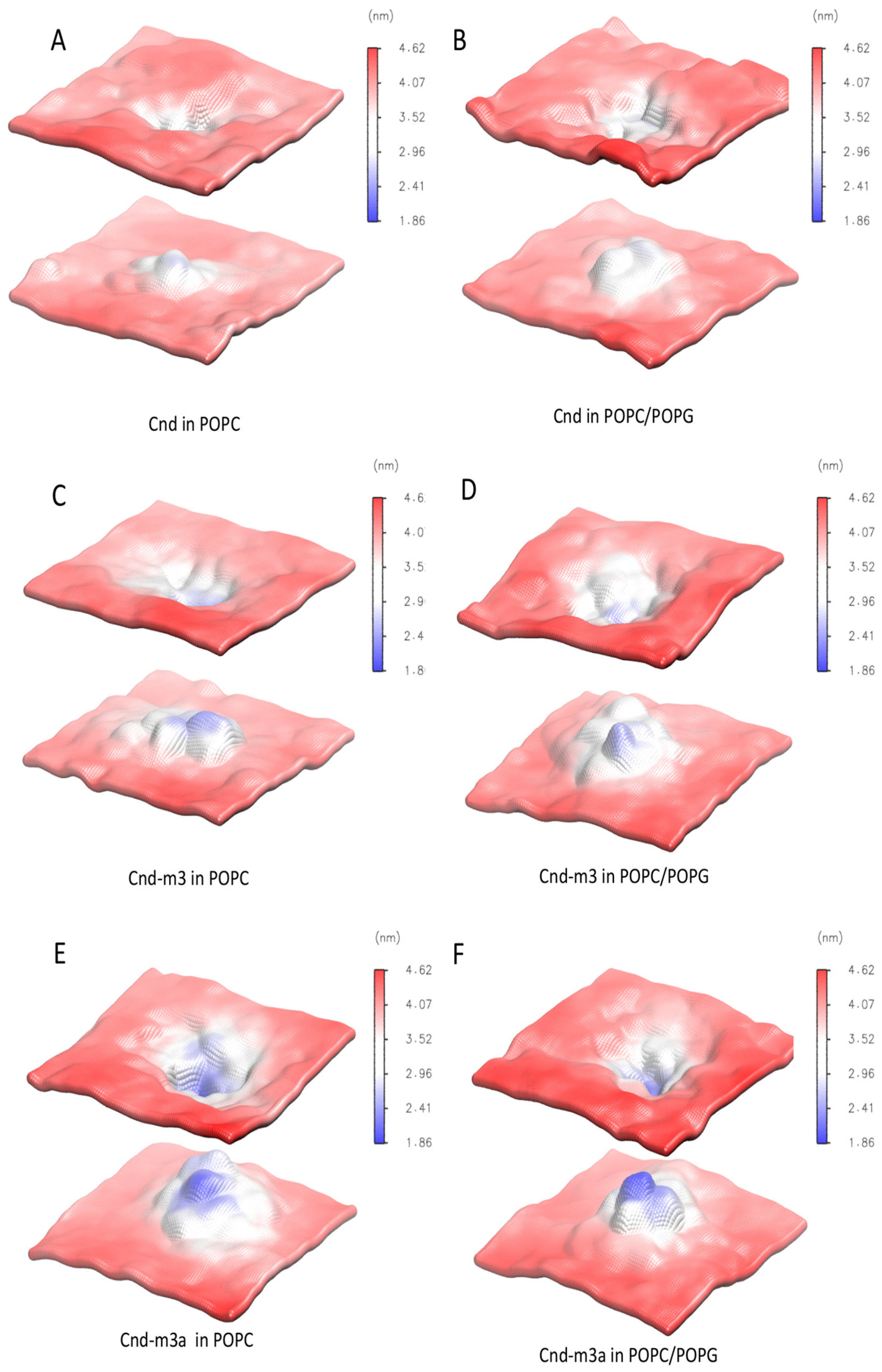
2.4.2. Peptides with Lipid Bilayers
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Preparation of Lipid Vesicles
4.3. Circular Dichroism Spectroscopy
4.4. Steady-State Anisotropy Measurements
4.5. Computational Methods
4.5.1. Preparation of MD Simulations
- (1)
- in water: the peptides were immersed in a cubic box (5.0 nm) and solvated with at list 4000 water molecules;
- (2)
- in a mixture of TFE/water (30% v/v): the peptides were immersed in a cubic box (5.0 nm) and solvated with about 270 molecules of TFE and 2650 water molecules;
- (3)
- with the peptide absorbed on lipid bilayer surface of POPC: the peptide was positioned above the pre-equilibrated bilayer surface of POPC with the helical axis parallel to the lipid surface with a distance of 0.5 nm between the center of mass of peptide and the phosphate atoms of lipids. A rectangular box of 6.5 × 6.5 × 8.0 nm3 was used with at list 5140 water molecules;
- (4)
- with the peptide embedded in POPC lipid bilayer: the peptide was embedded in a pre-equilibrated lipid bilayer of POPC with the helical axis parallel to the normal of lipid bilayer in a rectangular box of 6.5 × 6.5 × 8.0 nm3 with at list 5140 water molecules;
- (5)
- with the peptide absorbed on lipid bilayer surface of POPC:POPG: the peptide was positioned above the pre-equilibrated bilayer surface of POPC:POPG with the helical axis parallel to the lipid surface and a distance of 0.5 nm between the center of mass of peptide and the phosphate atoms of lipids. A rectangular box of 6.2 × 6.2 × 8.0 nm3 was used with at list 4300 water molecule;
- (6)
- with the peptide embedded in POPC/POPG lipid bilayer: the peptide was embedded in a pre-equilibrated lipid bilayer of POPC/POPG (70:30) with the helical axis parallel to the normal of lipid bilayer in a rectangular box of 6.2 × 6.2 × 8.0 nm3 with at list 4300 water molecules.
4.5.2. The MD Systems Setup
4.5.3. The MD Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AMP | Antimicrobial peptide |
| CD | Circular dichroism |
| Cnd | Chionodracine |
| DPH | 1,6-Diphenyl-1,3,5-hexatriene |
| EDTA | Ethylenediaminetetraacetic acid |
| LUV | Large Unilamellar Vesicles |
| MD | Molecular Dynamics |
| MDR | Multidrug resistant |
| MIC | Minimum inhibitory concentration |
| MLVS | Multilamellar vesicles |
| OD | Optical Density |
| PB | Phosphate buffer |
| POPC | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine |
| POPG | 1-palmitoyl-2-oleoyl-sn-glycero-3-(1′-rac-glycerol) sodium salt |
| TFE | 2,2,2-Trifluoroethanol |
References
- Cooper, M.A.; Shlaes, D. Fix the antibiotics pipeline. Nature 2011, 472, 32. [Google Scholar] [CrossRef]
- Administration, F.A.D. CFR Annual Print Title 21 food and Drugs. 2018. Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm (accessed on 19 July 2019).
- Baindara, P.; Mandal, S.M. Antimicrobial Peptides and Vaccine Development to Control Multi-drug Resistant Bacteria. Protein Pept. Lett. 2019, 26, 324–331. [Google Scholar] [CrossRef]
- Alvarez, A.; Fernandez, L.; Gutierrez, D.; Iglesias, B.; Rodriguez, A.; Garcia, P. Methicillin-resistant Staphylococcus aureus (MRSA) in hospitals: Latest trends and treatments based on bacteriophages. J. Clin. Microbiol. 2019. [Google Scholar] [CrossRef]
- Ghosh, C.; Sarkar, P.; Issa, R.; Haldar, J. Alternatives to Conventional Antibiotics in the Era of Antimicrobial Resistance. Trends Microbiol. 2019, 27, 323–338. [Google Scholar] [CrossRef]
- Boman, H.G. Antibacterial peptides: Basic facts and emerging concepts. J. Intern. Med. 2003, 254, 197–215. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Hancock, R.E.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nature Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef]
- Diamond, G.; Beckloff, N.; Weinberg, A.; Kisich, K.O. The roles of antimicrobial peptides in innate host defense. Curr. Pharm. Des. 2009, 15, 2377–2392. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.K.; Song, J.W.; Gong, F.; Li, S.B.; Chang, H.Y.; Xie, H.M.; Gao, H.W.; Tan, Y.X.; Ji, S.P. Design of an alpha-helical antimicrobial peptide with improved cell-selective and potent anti-biofilm activity. Sci. Rep. 2016, 6, 27394. [Google Scholar] [CrossRef] [Green Version]
- White, S.H.; Wimley, W.C. Hydrophobic interactions of peptides with membrane interfaces. Biochim. Biophys. Acta 1998, 1376, 339–352. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Mant, C.T.; Farmer, S.W.; Hancock, R.E.; Vasil, M.L.; Hodges, R.S. Rational design of alpha-helical antimicrobial peptides with enhanced activities and specificity/therapeutic index. J. Biol. Chem. 2005, 280, 12316–12329. [Google Scholar] [CrossRef] [Green Version]
- Zelezetsky, I.; Tossi, A. Alpha-helical antimicrobial peptides--using a sequence template to guide structure-activity relationship studies. Biochim. Biophys. Acta 2006, 1758, 1436–1449. [Google Scholar] [CrossRef] [Green Version]
- Bartels, E.J.H.; Dekker, D.; Amiche, M. Dermaseptins, Multifunctional Antimicrobial Peptides: A Review of Their Pharmacology, Effectivity, Mechanism of Action, and Possible Future Directions. Front. Pharmacol. 2019, 10, 1421. [Google Scholar] [CrossRef] [Green Version]
- Latendorf, T.; Gerstel, U.; Wu, Z.; Bartels, J.; Becker, A.; Tholey, A.; Schroder, J.M. Cationic Intrinsically Disordered Antimicrobial Peptides (CIDAMPs) Represent a New Paradigm of Innate Defense with a Potential for Novel Anti-Infectives. Sci. Rep. 2019, 9, 3331. [Google Scholar] [CrossRef] [Green Version]
- Romero, S.M.; Cardillo, A.B.; Martinez Ceron, M.C.; Camperi, S.A.; Giudicessi, S.L. Temporins: An Approach of Potential Pharmaceutic Candidates. Surg. Infect. (Larchmt) 2019. [Google Scholar] [CrossRef]
- Jenssen, H.; Hamill, P.; Hancock, R.E. Peptide antimicrobial agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar] [CrossRef] [Green Version]
- Silva, P.M.; Goncalves, S.; Santos, N.C. Defensins: Antifungal lessons from eukaryotes. Front. Microbiol. 2014, 5, 97. [Google Scholar] [CrossRef] [Green Version]
- Wimley, W.C.; Hristova, K. Antimicrobial peptides: Successes, challenges and unanswered questions. J. Membr. Biol. 2011, 239, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef]
- Porcelli, F.; Ramamoorthy, A.; Barany, G.; Veglia, G. On the role of NMR spectroscopy for characterization of antimicrobial peptides. Methods Mol. Biol. 2013, 1063, 159–180. [Google Scholar]
- Shai, Y. Mode of action of membrane active antimicrobial peptides. Biopolymers 2002, 66, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Aisenbrey, C.; Marquette, A.; Bechinger, B. The Mechanisms of Action of Cationic Antimicrobial Peptides Refined by Novel Concepts from Biophysical Investigations. Adv. Exp. Med. Biol. 2019, 1117, 33–64. [Google Scholar] [PubMed] [Green Version]
- Huang, H.W. Action of antimicrobial peptides: Two-state model. Biochemistry 2000, 39, 8347–8352. [Google Scholar] [CrossRef]
- Wimley, W.C. Describing the mechanism of antimicrobial peptide action with the interfacial activity model. ACS Chem. Biol. 2010, 5, 905–917. [Google Scholar] [CrossRef] [Green Version]
- Milne, D.J.; Fernandez-Montero, A.; Gundappa, M.K.; Wang, T.; Acosta, F.; Torrecillas, S.; Montero, D.; Zou, J.; Sweetman, J.; Secombes, C.J. An insight into piscidins: The discovery, modulation and bioactivity of greater amberjack, Seriola dumerili, piscidin. Mol. Immunol. 2019, 114, 378–388. [Google Scholar] [CrossRef]
- Noga, E.J.; Silphaduang, U. Piscidins: A novel family of peptide antibiotics from fish. Drug News Perspect. 2003, 16, 87–92. [Google Scholar] [CrossRef]
- Buonocore, F.; Randelli, E.; Casani, D.; Picchietti, S.; Belardinelli, M.C.; de Pascale, D.; De Santi, C.; Scapigliati, G. A piscidin-like antimicrobial peptide from the icefish Chionodraco hamatus (Perciformes: Channichthyidae) molecular characterization, localization and bactericidal activity. Fish Shellfish Immunol. 2012, 33, 1183–1191. [Google Scholar] [CrossRef]
- Olivieri, C.; Buonocore, F.; Picchietti, S.; Taddei, A.R.; Bernini, C.; Scapigliati, G.; Dicke, A.A.; Vostrikov, V.V.; Veglia, G.; Porcelli, F. Structure and membrane interactions of chionodracine, a piscidin-like antimicrobial peptide from the icefish Chionodraco hamatus. Biochim. Biophys. Acta 2015, 1848, 1285–1293. [Google Scholar] [CrossRef] [Green Version]
- Olivieri, C.; Bugli, F.; Menchinelli, G.; Veglia, G.; Buonocore, F.; Scapigliati, G.; Stocchi, V.; Ceccacci, F.; Papi, M.; Sanguinetti, M.; et al. Design and characterization of chionodracine-derived antimicrobial peptides with enhanced activity against drug-resistant human pathogens. RSC Adv. 2018, 8, 41331–41346. [Google Scholar] [CrossRef] [Green Version]
- Buonocore, F.; Picchietti, S.; Porcelli, F.; Della Pelle, G.; Olivieri, C.; Poerio, E.; Bugli, F.; Menchinelli, G.; Sanguinetti, M.; Bresciani, A.; et al. Fish-derived antimicrobial peptides: Activity of a chionodracine mutant against bacterial models and human bacterial pathogens. Dev. Comp. Immunol. 2019, 96, 9–17. [Google Scholar] [CrossRef]
- Dathe, M.; Nikolenko, H.; Meyer, J.; Beyermann, M.; Bienert, M. Optimization of the antimicrobial activity of magainin peptides by modification of charge. FEBS Lett. 2001, 501, 146–150. [Google Scholar] [CrossRef] [Green Version]
- Sudha, T.S.; Vijayakumar, E.K.; Balaram, P. Circular dichroism studies of helical oligopeptides. Can 3(10) and alpha-helical conformations be chiroptically distinguished? Int. J. Pept. Protein Res. 1983, 22, 464–468. [Google Scholar] [CrossRef]
- Manning, M.C.; Woody, R.W. Theoretical CD studies of polypeptide helices: Examination of important electronic and geometric factors. Biopolymers 1991, 31, 569–586. [Google Scholar] [CrossRef]
- Andersen, N.H.; Liu, Z.; Prickett, K.S. Efforts toward deriving the CD spectrum of a 3(10) helix in aqueous medium. FEBS Lett. 1996, 399, 47–52. [Google Scholar] [CrossRef] [Green Version]
- Sani, M.A.; Lee, T.H.; Aguilar, M.I.; Separovic, F. Proline-15 creates an amphipathic wedge in maculatin 1.1 peptides that drives lipid membrane disruption. Biochim. Biophys. Acta 2015, 1848, 2277–2289. [Google Scholar] [CrossRef] [Green Version]
- Plasek, J.; Jarolim, P. Interaction of the fluorescent probe 1,6-diphenyl-1,3,5-hexatriene with biomembranes. Gen. Physiol. Biophys. 1987, 6, 425–437. [Google Scholar]
- Strandberg, E.; Killian, J.A. Snorkeling of lysine side chains in transmembrane helices: How easy can it get? FEBS Lett. 2003, 544, 69–73. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wang, G. APD: The Antimicrobial Peptide Database. Nucleic Acids Res. 2004, 32, D590–D592. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Vasil, A.I.; Gera, L.; Vasil, M.L.; Hodges, R.S. Rational design of alpha-helical antimicrobial peptides to target Gram-negative pathogens, Acinetobacter baumannii and Pseudomonas aeruginosa: Utilization of charge, ‘specificity determinants’, total hydrophobicity, hydrophobe type and location as design parameters to improve the therapeutic ratio. Chem. Biol. Drug Des. 2011, 77, 225–240. [Google Scholar]
- Chen, Y.; Guarnieri, M.T.; Vasil, A.I.; Vasil, M.L.; Mant, C.T.; Hodges, R.S. Role of peptide hydrophobicity in the mechanism of action of alpha-helical antimicrobial peptides. Antimicrob. Agents Chemother. 2007, 51, 1398–1406. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Vidal, M.; White, S.H.; Ladokhin, A.S. Membrane partitioning: “classical” and “nonclassical” hydrophobic effects. J. Membr. Biol. 2011, 239, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Luo, P.; Baldwin, R.L. Mechanism of helix induction by trifluoroethanol: A framework for extrapolating the helix-forming properties of peptides from trifluoroethanol/water mixtures back to water. Biochemistry 1997, 36, 8413–8421. [Google Scholar] [CrossRef]
- Suzuki, M.; Miura, T. Effect of amyloid beta-peptide on the fluidity of phosphatidylcholine membranes: Uses and limitations of diphenylhexatriene fluorescence anisotropy. Biochim. Biophys. Acta 2015, 1848, 753–759. [Google Scholar] [CrossRef] [Green Version]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy; Springer: New York, NY, USA, 2009. [Google Scholar]
- do Canto, A.; Robalo, J.R.; Santos, P.D.; Carvalho, A.J.P.; Ramalho, J.P.P.; Loura, L.M.S. Diphenylhexatriene membrane probes DPH and TMA-DPH: A comparative molecular dynamics simulation study. Biochim. Biophys. Acta 2016, 1858, 2647–2661. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Martinez, L.; Andrade, R.; Birgin, E.G.; Martinez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Javanainen, M. Universal Method for Embedding Proteins into Complex Lipid Bilayers for Molecular Dynamics Simulations. J. Chem. Theory Comput. 2014, 10, 2577–2582. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef]
- Kukol, A. Lipid Models for United-Atom Molecular Dynamics Simulations of Proteins. J. Chem. Theory. Comput. 2009, 5, 615–626. [Google Scholar] [CrossRef] [Green Version]
- Poger, D.; Van Gunsteren, W.F.; Mark, A.E. A new force field for simulating phosphatidylcholine bilayers. J. Comput. Chem. 2010, 31, 1117–1125. [Google Scholar] [CrossRef]
- Poger, D.; Mark, A.E. On the Validation of Molecular Dynamics Simulations of Saturated and cis-Monounsaturated Phosphatidylcholine Lipid Bilayers: A Comparison with Experiment. J. Chem. Theory Comput. 2010, 6, 325–336. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Hermans, J. Interaction Models for Water in Relation to Protein Hydration. In Intermolecular Forces; Reidel Publishing Company: Dordrecht, The Netherlands, 1981; pp. 331–342. [Google Scholar]
- Fioroni, M.; Burger, K.; Mark, A.E.; Roccatano, D. A new 2,2,2-Trifluoroethanol model for molecular dynamics simulations. J. Phys. Chem. B 2000, 104, 12347–12354. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comp. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Hess, B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2007, 4, 116–122. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. SETTLE: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comp. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 14101–14107. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; Di Nola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Gapsys, V.; de Groot, B.L.; Briones, R. Computational analysis of local membrane properties. J. Comput. Aided Mol. Des. 2013, 27, 845–858. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence | Net Charge | pI | (μH) |
|---|---|---|---|---|
| Cnd | WFGHLYRGITSVVKHVHGLLSG | +2 | 9.99 | 0.574 |
| Cnd-m3 | WFGKLYRGITKVVKKVKGLLKG | +7 | 10.58 | 0.684 |
| Cnd-m3a | WFGKLYRGKTKVVKKVKGLLKG | +8 | 10.82 | 0.564 |
| POPC | POPC/POPG (70:30) | POPC vs. POPC/POPG | |||
|---|---|---|---|---|---|
| θ222nm | % helicity | θ222nm | % helicity | ||
| Cnd | −24,900 | 62.8 | −25,500 | 64.4 | 0.98 |
| Cnd-m3 | −12,300 | 30.6 | −31,000 | 78.5 | 0.40 |
| Cnd-m3a | −5000 | 11.9 | −5100 | 14.0 | 0.98 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borocci, S.; Della Pelle, G.; Ceccacci, F.; Olivieri, C.; Buonocore, F.; Porcelli, F. Structural Analysis and Design of Chionodracine-Derived Peptides Using Circular Dichroism and Molecular Dynamics Simulations. Int. J. Mol. Sci. 2020, 21, 1401. https://doi.org/10.3390/ijms21041401
Borocci S, Della Pelle G, Ceccacci F, Olivieri C, Buonocore F, Porcelli F. Structural Analysis and Design of Chionodracine-Derived Peptides Using Circular Dichroism and Molecular Dynamics Simulations. International Journal of Molecular Sciences. 2020; 21(4):1401. https://doi.org/10.3390/ijms21041401
Chicago/Turabian StyleBorocci, Stefano, Giulia Della Pelle, Francesca Ceccacci, Cristina Olivieri, Francesco Buonocore, and Fernando Porcelli. 2020. "Structural Analysis and Design of Chionodracine-Derived Peptides Using Circular Dichroism and Molecular Dynamics Simulations" International Journal of Molecular Sciences 21, no. 4: 1401. https://doi.org/10.3390/ijms21041401
APA StyleBorocci, S., Della Pelle, G., Ceccacci, F., Olivieri, C., Buonocore, F., & Porcelli, F. (2020). Structural Analysis and Design of Chionodracine-Derived Peptides Using Circular Dichroism and Molecular Dynamics Simulations. International Journal of Molecular Sciences, 21(4), 1401. https://doi.org/10.3390/ijms21041401