Mucopolysaccharidosis IVA: Diagnosis, Treatment, and Management

 ,
,  ,
,  , and
, and 
Abstract
:1. Introduction
2. Diagnosis
2.1. Clinical Diagnosis and Phenotype
2.2. GALNS Enzyme and Genetic Diagnosis
2.3. Radiographic Diagnosis
2.4. Biochemical Diagnosis
2.4.1. GAG Level in Urine
2.4.2. KS Level
2.4.3. C6S Level
2.4.4. Enzyme Assay
3. Treatment and Management
3.1. ERT
3.2. HSCT
3.3. Orthopedic Surgery
3.4. Management of Tracheal Obstruction
3.5. Future Therapies
3.5.1. Substrate Degradation Enzyme Therapy (SDET)
3.5.2. Gene Therapy
3.5.3. Nanomedicine
3.5.4. Pharmacological Chaperone Therapy
4. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| AAV | Adeno-associated virus |
| ADL | Activity of daily living |
| BBB | Blood-brain barrier |
| BMD | Bone mineral density |
| BMT | Bone marrow transplantation |
| C6S | Chondroitin-6-sulfate |
| CBA | Chicken β-actin |
| CSF | Cerebrospinal fluid |
| CT | Computed tomography |
| D8 | Aspartic acid octapeptide |
| DBS | Dried blood spot |
| DMB | Dimethylmethylene blue |
| ECM | Extracellular matrix |
| ELISA | Enzyme-linked immunosorbent assay |
| EMA | European Medicines Agency |
| ERT | Enzyme replacement therapy |
| FDA | Food and Drug Administration |
| FEV1 | Forced expiratory volume in 1s |
| FVC | Forced vital capacity |
| GAG | Glycosaminoglycans |
| GALNS | N-acetylgalactosamine 6-sulfate sulfatase |
| GVHD | Graft versus host disease |
| HS | Heparan sulfate |
| HSCT | Hematopoietic stem cell transplantation |
| KS | Keratan sulfate |
| LC-MS/MS | Liquid chromatography tandem mass spectrometry |
| MPS | Mucopolysaccharidosis |
| MRI | Magnetic resonance imaging |
| NLC | Nanostructured lipid carriers |
| SDET | Substrate degradation enzyme therapy |
References
- Neufeld, E.F.; Muenzer, J. The mucopolysaccharidoses. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3421–3452. [Google Scholar]
- Khan, S.; Alméciga-Díaz, C.J.; Sawamoto, K.; Mackenzie, W.G.; Theroux, M.C.; Pizarro, C.; Mason, R.W.; Orii, T.; Tomatsu, S. Mucopolysaccharidosis IVA and glycosaminoglycans. Mol. Genet. Metab. 2017, 120, 78–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peracha, H.; Sawamoto, K.; Averill, L.; Kecskemethy, H.; Theroux, M.; Thacker, M.; Nagao, K.; Pizarro, C.; Mackenzie, W.; Kobayashi, H.; et al. Diagnosis and prognosis of Mucopolysaccharidosis IVA. Mol. Genet. Metab. 2018, 125, 18–37. [Google Scholar] [CrossRef] [PubMed]
- Sawamoto, K.; Alméciga-Díaz, C.J.; Mason, R.W.; Orii, T.; Tomatsu, S. Mucopolysaccharidosis type IVA: Clinical features, biochemistry, diagnosis, genetics, and treatment. Mucopolysaccharidoses Update; Tomatsu, S., Ed.; Nova Science Publishers: New York, NY, USA, 2018; pp. 235–272. [Google Scholar]
- Tomatsu, S.; Averill, L.W.; Sawamoto, K.; Mackenzie, W.G.; Bober, M.B.; Pizarro, C.; Goff, C.J.; Xie, L.; Orii, T.; Theroux, M. Obstructive airway in Morquio A syndrome, the past, the present and the future. Mol. Genet. Metab. 2016, 117, 150–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melbouci, M.; Mason, R.W.; Suzuki, Y.; Fukao, T.; Orii, T.; Tomatsu, S. Growth impairment in mucopolysaccharidoses. Mol. Genet. Metab. 2018, 124, 1–10. [Google Scholar] [CrossRef]
- Brailsford, J.F. Chondro-osteo-dystrophy, roentgenopgraphic & clinical features of a child with dislocation of vertebrae. Am. J. Surg. 1929, 7, 404–410. [Google Scholar]
- Morquio, L. Sur une forme de dystrophie osseuse familial. Arch. Méd. Enfants Paris. 1929, 32, 129–135. [Google Scholar]
- Khan, S.A.; Peracha, H.; Ballhausen, D.; Wiesbauer, A.; Rohrbach, M.; Gautschi, M.; Mason, R.W.; Giugliani, R.; Suzuki, Y.; Orii, K.E.; et al. Epidemiology of mucopolysaccharidoses. Mol. Genet. Metab. 2017, 121, 227–240. [Google Scholar] [CrossRef]
- Nelson, J.; Crowhurst, J.; Carey, B.; Greed, L. Incidence of the mucopolysaccharidoses in Western Australia. Hum. Genet. 2003, 123, 310–313. [Google Scholar] [CrossRef]
- Montaño, A.M.; Tomatsu, S.; Gottesman, G.S.; Smith, M.; Orii, T. International Morquio A Registry: Clinical manifestation and natural course of Morquio A disease. J. Inherit. Metab. Dis. 2007, 30, 165–174. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montaño, A.M.; Oikawa, H.; Smith, M.; Barrera, L.; Chinen, Y.; Thacker, M.M.; Mackenzie, W.G.; Suzuki, Y.; Orii, T. Mucopolysaccharidosis type IVA (Morquio A disease): Clinical review and current treatment. Curr. Pharm. Biotechnol. 2011, 12, 931–945. [Google Scholar] [CrossRef]
- Tomatsu, S.; Mackenzie, W.G.; Theroux, M.C.; Mason, R.W.; Thacker, M.M.; Shaffer, T.H.; Montaño, A.M.; Rowan, D.; Sly, W.; Alméciga-Díaz, C.J.; et al. Current and emerging treatments and surgical interventions for Morquio A Syndrome: A review. Res. Rep. Endocr. Disord. 2012, 2012, 65–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomatsu, S.; Sawamoto, K.; Shimada, T.; Bober, M.B.; Kubaski, F.; Yasuda, E.; Mason, R.W.; Khan, S.; Alméciga-Díaz, C.J.; Barrera, L.A.; et al. Enzyme replacement therapy for treating mucopolysaccharidosis type IVA (Morquio A syndrome): Effect and limitations. Expert Opin. Orphan. Drugs 2015, 3, 1279–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomatsu, S.; Sawamoto, K.; Alméciga-Díaz, C.J.; Shimada, T.; Bober, M.B.; Chinen, Y.; Yabe, H.; Montaño, A.M.; Giugliani, R.; Kubaski, F.; et al. Impact of enzyme replacement therapy and hematopoietic stem cell transplantation in patients with Morquio A syndrome. Drug Des. Dev. Ther. 2015, 9, 1937–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawamoto, K.; Suzuki, Y.; Mackenzie, W.G.; Theroux, M.C.; Pizarro, C.; Yabe, H.; Orii, K.E.; Mason, R.W.; Orii, T.; Tomatsu, S. Current therapies for Morquio A syndrome and their clinical outcomes. Expert Opin. Orphan Drugs 2016, 4, 941–951. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Musson, D.G.; Schweighardt, B.; Tompkins, T.; Jesaitis, L.; Shaywitz, A.J.; Yang, K.; O’Neill, C.A. Pharmacokinetic and pharmacodynamic evaluation of elosulfase alfa, an enzyme replacement therapy in patients with Morquio A syndrome. Clin. Pharmacokinet. 2014, 53, 1137–1147. [Google Scholar] [CrossRef] [Green Version]
- Tomatsu, S.; Montaño, A.M.; Ohashi, A.; Gutierrez, M.A.; Oikawa, H.; Oguma, T.; Dung, V.C.; Nishioka, T.; Orii, T.; Sly, W.S. Enzyme replacement therapy in a murine model of Morquio A syndrome. Hum. Mol Genet. 2008, 17, 815–824. [Google Scholar] [CrossRef] [Green Version]
- Pizarro, C.; Davies, R.R.; Theroux, M.; Spurrier, E.A.; Averill, L.W.; Tomatsu, S. Surgical Reconstruction for Severe Tracheal Obstruction in Morquio A Syndrome. Ann. Thorac. Surg. 2016, 102, e329–e331. [Google Scholar] [CrossRef] [Green Version]
- Doherty, C.; Stapleton, M.; Piechnik, M.; Mason, R.W.; Mackenzie, W.G.; Yamaguchi, S.; Kobayashi, H.; Suzuki, Y.; Tomatsu, S. Effect of enzyme replacement therapy on the growth of patients with Morquio A. J. Hum. Genet. 2019, 64, 625–635. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montaño, A.M.; Dung, V.C.; Ohashi, A.; Oikawa, H.; Oguma, T.; Orii, T.; Barrera, L.; Sly, W.S. Enhancement of drug delivery: Enzyme-replacement therapy for murine Morquio A syndrome. Mol. Ther. 2010, 18, 1094–1102. [Google Scholar] [CrossRef]
- Chinen, Y.; Higa, T.; Tomatsu, S.; Suzuki, Y.; Orii, T.; Hyakuna, N. Long-term therapeutic efficacy of allogenic bone marrow transplantation in a patient with mucopolysaccharidosis IVA. Mol. Genet. Metab. Rep. 2014, 1, 31–41. [Google Scholar] [CrossRef]
- Yabe, H.; Tanaka, A.; Chinen, Y.; Kato, S.; Sawamoto, K.; Yasuda, E.; Shintaku, H.; Suzuki, Y.; Orii, T.; Tomatsu, S. Hematopoietic stem cell transplantation for Morquio A syndrome. Mol. Genet. Metab. 2016, 117, 84–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Luan, Z.; Jiang, H.; Fang, J.; Qin, M.; Lee, V.; Chen, J. Allogeneic hematopoietic stem cell transplantation in thirty-four pediatric cases of mucopolysaccharidosis: A ten-year report from the China Childrens Transplant Group. Biol. Blood Marrow Transplant. 2016, 22, 2104–2108. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Khan, S.; Stapleton, M.; Wang, J.; Chen, J.; Wynn, R.; Yabe, H.; Chinen, Y.; Boelens, J.J.; Mason, R.W.; et al. Hematopoietic Stem Cell Transplantation for Mucopolysaccharidoses: Past, Present, and Future. Biol. Blood Marrow Transplant. 2019, 25, e226–e246. [Google Scholar] [CrossRef] [PubMed]
- Pshezhetsky, A.V.; Potier, M. Association of N-acetylgalactosamine-6-sulfate sulfatase with the multienzyme lysosomal complex of beta-galactosidase, cathepsin A., and neuraminidase. Possible implication for intralysosomal catabolism of keratan sulfate. J. Biol. Chem. 1996, 271, 28359–28365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regier, D.S.; Oetgen, M.; Tanpaiboon, P. Mucopolysaccharidosis Type IVA. In GeneReviews®; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Tummolo, A.; Gabrielli, O.; Gaeta, A.; Masciopinto, M.; Zampini, L.; Pavone, L.M.; Di Natale, P.; Papadia, F. Bisphosphonate Treatment in a Patient Affected by MPS IVA with Osteoporotic Phenotype. Case Rep. Med. 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhlebach, M.S.; Wooten, W.; Muenzer, J. Respiratory Manifestations in Mucopolysaccharidoses. Paediatr. Respir. Rev. 2011, 12, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Hendriksz, C.J.; Harmatz, P.; Beck, M.; Jones, S.; Wood, T.; Lachman, R.; Gravance, C.G.; Orii, T.; Tomatsu, S. Review of clinical presentation and diagnosis of mucopolysaccharidosis IVA. Mol. Genet. Metab. 2013, 110, 54–64. [Google Scholar] [CrossRef] [Green Version]
- Masue, M.; Sukegawa, K.; Orii, T.; Hashimoto, T. N-acetylgalactosamine-6-sulfate sulfatase in human placenta: Purification and characteristics. J. Biochem. 1991, 110, 965–970. [Google Scholar] [CrossRef]
- Nakashima, Y.; Tomatsu, S.; Hori, T.; Fukuda, S.; Sukegawa, K.; Kondo, N.; Suzuki, Y.; Shimozawa, N.; Orii, T. Mucopolysaccharidosis IV A: Molecular cloning of the human N-acetylgalactosamine-6-sulfatase gene (GALNS) and analysis of the 5′-flanking region. Genomics 1994, 20, 99–104. [Google Scholar] [CrossRef]
- Tomatsu, S.; Fukuda, S.; Masue, M.; Sukegawa, K.; Fukao, T.; Yamagishi, A.; Hori, T.; Iwata, H.; Ogawa, T.; Nakashima, Y.; et al. Morquio disease: Isolation, characterization and expression of full-length cDNA for human N-acetylgalactosamine-6-sulfate sulfatase. Biochem. Biophys. Res. Commun. 1991, 181, 677–683. [Google Scholar] [CrossRef]
- Pshezhetsky, A.V.; Ashmarina, M. Lysosomal multienzyme complex: Biochemistry, genetics, and molecular pathophysiology. Prog. Nucleic Acid Res. Mol. Biol. 2001, 69, 81–114. [Google Scholar] [PubMed]
- Hiraiwa, M. Cathepsin A/protective protein: An unusual lysosomal multifunctional protein. Cell Mol. Life Sci. 1999, 56, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Morrone, A.; Caciotti, A.; Atwood, R.; Davidson, K.; Du, C.; Francis-Lyon, P.; Harmatz, P.; Mealiffe, M.; Mooney, S.; Oron, T.R.; et al. Morquio A syndrome-associated mutations: A review of alterations in the GALNS gene and a new locus-specific database. Hum. Mutat. 2014, 35, 1271–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomatsu, S.; Montaño, A.M.; Nishioka, T.; Gutierrez, M.A.; Peña, O.M.; Tranda Firescu, G.G.; Lopez, P.; Yamaguchi, S.; Noguchi, A.; Orii, T. Mutation and polymorphism spectrum of the GALNS gene in mucopolysaccharidosis IVA (Morquio A). Hum. Mutat. 2005, 26, 500–512. [Google Scholar] [CrossRef]
- Available online: http://www.hgmd.cf.ac.uk/ac/gene.php?gene=GALNS (accessed on 19 February 2020).
- Jezela-Stanek, A.; Różdżyńska-Świątkowska, A.; Kulpanovich, A.; Ciara, E.; Marucha, J.; Tylki-Szymańska, A. Novel data on growth phenotype and causative genotypes in 29 patients with Morquio (Morquio-Brailsford) syndrome from Central-Eastern Europe. J. Appl. Genet. 2019, 60, 163–174. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, S.; Tomatsu, S.; Masue, M.; Sukegawa, K.; Iwata, H.; Ogawa, T.; Nakashima, Y.; Hori, T.; Yamagishi, A.; Hanyu, Y.; et al. Mucopolysaccharidosis type IVA. N-acetylgalactosamine-6-sulfate sulfatase exonic point mutations in classical Morquio and mild cases. J. Clin. Investig. 1992, 90, 1049–1053. [Google Scholar] [CrossRef] [Green Version]
- Hori, T.; Tomatsu, S.; Nakashima, Y.; Uchiyama, A.; Fukuda, S.; Sukegawa, K.; Shimozawa, N.; Suzuki, Y.; Kondo, N.; Horiuchi, T.; et al. Mucopolysaccharidosis type IVA: Common double deletion in the N-acetylgalactosamine-6-sulfatase gene (GALNS). Genomics 1995, 26, 535–542. [Google Scholar] [CrossRef]
- Kato, Z.; Fukuda, S.; Tomatsu, S.; Vega, H.; Yasunaga, T.; Yamagishi, A.; Yamada, N.; Valencia, A.; Barrera, L.A.; Sukegawa, K.; et al. A novel common missense mutation G301C in the N-acetylgalactosamine-6-sulfate sulfatase gene in mucopolysaccharidosis IVA. Hum. Genet. 1997, 101, 97–101. [Google Scholar] [CrossRef]
- Tomatsu, S.; Filocamo, M.; Orii, K.O.; Sly, W.S.; Gutierrez, M.A.; Nishioka, T.; Yamada, N.; Valencia, A.; Barrera, L.A.; Sukegawa, K.; et al. Mucopolysaccharidosis IVA (Morquio A): Identification of novel common mutations in the N-acetylgalactosamine-6-sulfate sulfatase (GALNS) gene in Italian patients. Hum. Mutat. 2004, 24, 187–188. [Google Scholar] [CrossRef]
- Tomatsu, S.; Fukuda, S.; Cooper, A.; Wraith, J.E.; Ferreira, P.; Di Natale, P.; Tortora, P.; Fujimoto, A.; Kato, Z.; Yamada, N.; et al. Fourteen novel mucopolysaccharidosis IVA producing mutations in GALNS gene. Hum. Mutat. 1997, 10, 368–375. [Google Scholar] [CrossRef]
- Tomatsu, S.; Fukuda, S.; Cooper, A.; Wraith, J.E.; Rezvi, G.M.; Yamagishi, A.; Yamada, N.; Kato, Z.; Isogai, K.; Sukegawa, K.; et al. Mucopolysaccharidosis IVA: Identification of a common missense mutation I113F in the N-Acetylgalactosamine-6-sulfate sulfatase gene. Am. J. Hum. Genet. 1995, 57, 556–563. [Google Scholar] [PubMed]
- Tomatsu, S.; Fukuda, S.; Yamagishi, A.; Cooper, A.; Wraith, J.F.; Hori, T.; Kato, Z.; Yamada, N.; Isogai, K.; Sukegawa, K.; et al. Mucopolysaccharidosis IVA: Four new exonic mutations in patients with N-acetylgalactosamine-6-sulfate sulfatase deficiency. Am. J. Hum. Genet. 1996, 58, 950–962. [Google Scholar] [PubMed]
- Yamada, N.; Fukuda, S.; Tomatsu, S.; Muller, V.; Hopwood, J.J.; Nelson, J.; Kato, Z.; Yamagishi, A.; Sukegawa, K.; Kondo, N.; et al. Molecular heterogeneity in mucopolysaccharidosis IVA in Australia and Northern Ireland: Nine novel mutations including T312S, a common allele that confers a mild phenotype. Hum. Mutat. 1998, 11, 202–208. [Google Scholar] [CrossRef]
- Tamarozzi, E.R.; Torrieri, E.; Semighini, E.P.; Giuliatti, S. In silico analysis of mutations occurring in the protein N-acetylgalactosamine-6-sulfatase (GALNS) and causing mucopolysaccharidosis IVA. Genet. Mol. Res. GMR. 2014, 13, 10025–10034. [Google Scholar] [CrossRef] [Green Version]
- Leong, H.Y.; Abdul Azize, N.A.; Chew, H.B.; Keng, W.T.; Thong, M.K.; Mohd Khalid, M.K.N.; Hung, L.C.; Mohamed Zainudin, N.; Ramlee, A.; Md Haniffa, M.A.; et al. Clinical, biochemical and genetic profiles of patients with mucopolysaccharidosis type IVA (Morquio A syndrome) in Malaysia: The first national natural history cohort study. Orphanet. J. Rare Dis. 2019, 14, 143. [Google Scholar] [CrossRef]
- Lavery, C.; Hendriksz, C. Mortality in patients with morquio syndrome a. JIMD Rep. 2015, 15, 59–66. [Google Scholar]
- Eggli, K.D.; Dorst, J.P. The mucopolysaccharidoses and related conditions. Semin. Roentgenol. 1986, 21, 275–294. [Google Scholar] [CrossRef]
- Dhawale, A.A.; Thacker, M.M.; Belthur, M.V.; Rogers, K.; Bober, M.B.; Mackenzie, W.G. The Lower Extremity in Morquio Syndrome. J. Pediatr. Orthop. 2012, 32, 534–540. [Google Scholar] [CrossRef]
- White, K.K.; Jester, A.; Bache, C.E.; Harmatz, P.R.; Shediac, R.; Thacker, M.M.; Mackenzie, W.G. Orthopedic management of the extremities in patients with Morquio A syndrome. J. Child. Orthop. 2014, 8, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Whitley, C.B.; Ridnour, M.D.; Draper, K.A.; Dutton, C.M.; Neglia, J.P. Diagnostic test for mucopolysaccharidosis. I. Direct method for quantifying excessive urinary glycosaminoglycan excretion. Clin. Chem. 1989, 35, 374–379. [Google Scholar] [CrossRef]
- Piraud, M.; Boyer, S.; Mathieu, M.; Maire, I. Diagnosis of mucopolysaccharidoses in a clinically selected population by urinary glycosaminoglycan analysis: A study of 2,000 urine samples. Clin. Chim. Acta. Int. J. Clin. Chem. 1993, 221, 171–181. [Google Scholar] [CrossRef]
- Tomatsu, S.; Okamura, K.; Taketani, T.; Orii, K.O.; Nishioka, T.; Gutierrez, M.A.; Velez-Castrillon, S.; Fachel, A.A.; Grubb, J.H.; Cooper, A.; et al. Development and testing of new screening method for keratan sulfate in mucopolysaccharidosis IVA. Pediatr Res. 2004, 55, 592–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, G.; Claridge, P.; Jenkinson, L.; Green, A. Quantitation of urinary glycosaminoglycans using dimethylene blue as a screening technique for the diagnosis of mucopolysaccharidoses: An evaluation. Ann. Clin. Biochem. 2007, 44, 360–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oguma, T.; Tomatsu, S.; Montano, A.M.; Okazaki, O. Analytical method for the determination of disaccharides derived from keratan, heparan, and dermatan sulfates in human serum and plasma by high-performance liquid chromatography/turbo ionspray ionization tandem mass spectrometry. Anal. Biochem. 2007, 368, 79–86. [Google Scholar] [CrossRef]
- Oguma, T.; Tomatsu, S.; Okazaki, O. Analytical method for determination of disaccharides derived from keratan sulfates in human serum and plasma by high-performance liquid chromatography/turbo-ionspray ionization tandem mass spectrometry. Biomed. Chromatogr. 2007, 21, 356–362. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montaño, A.M.; Oguma, T.; Dung, V.C.; Oikawa, H.; de Carvalho, T.G.; Gutiérrez, M.L.; Yamaguchi, S.; Suzuki, Y.; Fukushi, M.; et al. Validation of keratan sulfate level in mucopolysaccharidosis type IVA by liquid chromatography-tandem mass spectrometry. J. Inherit. Metab. Dis. 2010, 33, S35–S42. [Google Scholar] [CrossRef]
- Hintze, J.P.; Tomatsu, S.; Fujii, T.; Montaño, A.M.; Yamaguchi, S.; Suzuki, Y.; Fukushi, M.; Ishimaru, T.; Orii, T. Comparison of liquid chromatography-tandem mass spectrometry and sandwich ELISA for determination of keratan sulfate in plasma and urine. Biomark Insights. 2011, 6, 69–78. [Google Scholar] [CrossRef]
- Martell, L.A.; Cunico, R.L.; Ohh, J.; Fulkerson, W.; Furneaux, R.; Foehr, E.D. Validation of an LC-MS/MS assay for detecting relevant disaccharides from keratan sulfate as a biomarker for Morquio A syndrome. Bioanalysis 2011, 3, 1855–1866. [Google Scholar] [CrossRef]
- Tomatsu, S.; Shimada, T.; Mason, R.W.; Kelly, J.; LaMarr, W.A.; Yasuda, E.; Shibata, Y.; Futatsumori, H.; Montaño, A.M.; Yamaguchi, S.; et al. Assay for Glycosaminoglycans by Tandem Mass Spectrometry and its Applications. J. Anal. Bioanal. Tech. 2014, 2014, 006. [Google Scholar]
- Tomatsu, S.; Shimada, T.; Mason, R.W.; Montaño, A.M.; Kelly, J.; LaMarr, W.A.; Kubaski, F.; Giugliani, R.; Guha, A.; Yasuda, E.; et al. Establishment of glycosaminoglycan assays for mucopolysaccharidoses. Metabolites 2014, 4, 655–679. [Google Scholar] [CrossRef] [Green Version]
- Shimada, T.; Tomatsu, S.; Mason, R.W.; Yasuda, E.; Mackenzie, W.G.; Hossain, J.; Shibata, Y.; Montaño, A.M.; Kubaski, F.; Giugliani, R.; et al. Di-sulfated Keratan Sulfate as a Novel Biomarker for Mucopolysaccharidosis II, IVA, and IVB. JIMD Rep. 2015, 21, 1–13. [Google Scholar] [PubMed] [Green Version]
- Kubaski, F.; Mason, R.W.; Nakatomi, A.; Shintaku, H.; Xie, L.; van Vlies, N.N.; Church, H.; Giugliani, R.; Kobayashi, H.; Yamaguchi, S.; et al. Newborn screening for mucopolysaccharidoses: A pilot study of measurement of glycosaminoglycans by tandem mass spectrometry. J. Inherit. Metab. Dis. 2017, 40, 151–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.Y.; Lee, C.L.; Lo, Y.T.; Tu, R.Y.; Chang, Y.H.; Chang, C.Y.; Chiu, P.C.; Chang, T.M.; Tsai, W.H.; Niu, D.M.; et al. An At-Risk Population Screening Program for Mucopolysaccharidoses by Measuring Urinary Glycosaminoglycans in Taiwan. Diagnostics (Basel). 2019, 9, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.Y.; Lee, C.L.; Lo, Y.T.; Wang, T.J.; Huang, S.F.; Chen, T.L.; Wang, Y.S.; Niu, D.M.; Chuang, C.K.; Lin, S.P. The relationships between urinary glycosaminoglycan levels and phenotypes of mucopolysaccharidoses. Mol. Genet. Genomic Med. 2018, 6, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Stapleton, M.; Kubaski, F.; Mason, R.W.; Shintaku, H.; Kobayashi, H.; Yamaguchi, S.; Taketani, T.; Suzuki, Y.; Orii, K.; Orii, T.; et al. Newborn screening for Mucopolysaccharidoses: Measurement of glycosaminoglycans by LC-MS/MS. Mol. Genet. Metab. Rep 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Martell, L.; Lau, K.; Mei, M.; Burnett, V.; Decker, C.; Foehr, E.D. Biomarker analysis of Morquio syndrome: Identification of disease state and drug responsive markers. Orphanet. J. Rare Dis. 2011, 6, 84. [Google Scholar] [CrossRef] [Green Version]
- Fujitsuka, H.; Sawamoto, K.; Peracha, H.; Mason, R.W.; Mackenzie, W.; Kobayashi, H.; Yamaguchi, S.; Suzuki, Y.; Orii, K.; Orii, T.; et al. Biomarkers in patients with mucopolysaccharidosis type II and IV. Mol. Genet. Metab. Rep. 2019, 19, 100455. [Google Scholar] [CrossRef]
- Algahim, M.F.; Almassi, G.H. Current and emerging management options for patients with Morquio A syndrome. Ther. Clin. Risk Manag. 2013, 9, 45–53. [Google Scholar]
- Mourão, P.A. Distribution of chondroitin 4-sulfate and chondroitin 6-sulfate in human articular and growth cartilage. Arthritis Rheum. 1988, 31, 1028–1033. [Google Scholar] [CrossRef]
- Yasuda, E.; Fushimi, K.; Suzuki, Y.; Shimizu, K.; Takami, T.; Zustin, J.; Patel, P.; Ruhnke, K.; Shimada, T.; Boyce, B.; et al. Pathogenesis of Morquio A syndrome: An autopsied case reveals systemic storage disorder. Mol. Genet. Metab. 2013, 109, 301–311. [Google Scholar] [CrossRef]
- Shimada, T.; Tomatsu, S.; Yasuda, E.; Mason, R.W.; Mackenzie, W.G.; Shibata, Y.; Kubaski, F.; Giugliani, R.; Yamaguchi, S.; Suzuki, Y.; et al. Chondroitin 6-Sulfate as a Novel Biomarker for Mucopolysaccharidosis IVA and VII. JIMD Rep. 2014, 16, 15–24. [Google Scholar] [PubMed] [Green Version]
- Duffey, T.A.; Khaliq, T.; Scott, C.R.; Turecek, F.; Gelb, M.H. Design and synthesis of substrates for newborn screening of Maroteaux-Lamy and Morquio A syndromes. Bioorg. Med. Chem. Lett. 2010, 20, 5994–5996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camelier, M.V.; Burin, M.G.; De Mari, J.; Vieira, T.A.; Marasca, G.; Giugliani, R. Practical and reliable enzyme test for the detection of mucopolysaccharidosis IVA (Morquio Syndrome type A) in dried blood samples. Clin. Chim. Acta Int. J. Clin. Chem. 2011, 412, 1805–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleijer, W.J.; Geilen, G.C.; Garritsen, V.; Huijmans, J.G.; Los, F.J.; Voznyi, Y.V.; van Diggelen, O.P. First-trimester diagnosis of Morquio disease type A. Prenat. Diagn. 2000, 20, 183–185. [Google Scholar] [CrossRef]
- Zhao, H.; Van Diggelen, O.P.; Thoomes, R.; Huijmans, J.; Young, E.; Mazurczak, T.; Kleijer, W.J. Prenatal diagnosis of Morquio disease type A using a simple fluorometric enzyme assay. Prenat. Diagn. 1990, 10, 85–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aboul Nasr, A.; Fateen, E. Prenatal diagnosis of mucopolysaccharidoses (MPS): The first Egyptian experience. Bratisl. Lek. Listy. 2004, 105, 310–314. [Google Scholar]
- Shams, S.; Barazandeh Tehrani, M.; Civallero, G.; Minookherad, K.; Giugliani, R.; Setoodeh, A.; Haghi Ashtiani, M.T. Diagnosing Mucopolysaccharidosis type IV a by the fluorometric assay of N-Acetylgalactosamine-6-sulfate sulfatase activity. J. Diabetes Metab. Disord. 2017, 16, 37. [Google Scholar] [CrossRef] [Green Version]
- Chuang, C.K.; Lin, H.Y.; Wang, T.J.; Huang, S.F.; Lin, S.P. Bio-Plex immunoassay measuring the quantity of lysosomal N-acetylgalactosamine-6-sulfatase protein in dried blood spots for the screening of mucopolysaccharidosis IVA in newborn: A pilot study. BMJ Open. 2017, 7, 014410. [Google Scholar] [CrossRef]
- Tomatsu, S.; Fujii, T.; Fukushi, M.; Oguma, T.; Shimada, T.; Maeda, M.; Kida, K.; Shibata, Y.; Futatsumori, H.; Montaño, A.M.; et al. Newborn screening and diagnosis of mucopolysaccharidoses. Mol. Genet. Metab. 2013, 110, 42–53. [Google Scholar] [CrossRef] [Green Version]
- Colón, C.; Alvarez, J.V.; Castaño, C.; Gutierrez-Solana, L.G.; Marquez, A.M.; O’Callaghan, M.; Sánchez-Valverde, F.; Yeste, C.; Couce, M.-L. A selective screening program for the early detection of mucopolysaccharidosis: Results of the FIND project – a 2-year follow-up study. Medicine 2017, 96, 6887. [Google Scholar] [CrossRef]
- Hendriksz, C.J.; Burton, B.; Fleming, T.R.; Harmatz, P.; Hughes, D.; Jones, S.A.; Lin, S.P.; Mengel, E.; Scarpa, M.; Valayannopoulos, V.; et al. Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): A phase 3 randomised placebo-controlled study. J. Inherit. Metab. Dis. 2014, 37, 979–990. [Google Scholar] [CrossRef] [Green Version]
- Hendriksz, C.J.; Giugliani, R.; Harmatz, P.; Mengel, E.; Guffon, N.; Valayannopoulos, V.; Parini, R.; Hughes, D.; Pastores, G.M.; Lau, H.A.; et al. Multi-domain impact of elosulfase alfa in Morquio A syndrome in the pivotal phase III trial. Mol. Genet. Metab. 2015, 114, 178–185. [Google Scholar] [CrossRef]
- Do Cao, J.; Wiedemann, A.; Quinaux, T.; Battaglia-Hsu, S.F.; Mainard, L.; Froissart, R.; Bonnemains, C.; Ragot, S.; Leheup, B.; Journeau, P. 30 months follow-up of an early enzyme replacement therapy in a severe Morquio A patient: About one case. Mol. Genet. Metab. Rep. 2016, 9, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Harmatz, P.R.; Mengel, E.; Geberhiwot, T.; Muschol, N.; Hendriksz, C.J.; Burton, B.K.; Jameson, E.; Berger, K.I.; Jester, A.; Treadwell, M.; et al. Impact of elosulfase alfa in patients with morquio A syndrome who have limited ambulation: An open_ label, phase 2 study. Am. J. Med. Genet. A 2017, 173, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.; Giugliani, R.; Guffon, N.; Jones, S.A.; Mengel, K.E.; Parini, R.; Matousek, R.; Hawley, S.M.; Quartel, A. Clinical outcomes in a subpopulation of adults with Morquio A syndrome: Results from a long-term extension study of elosulfase alfa. Orphanet. J. Rare Dis. 2017, 12, 98. [Google Scholar] [CrossRef] [PubMed]
- Pintos-Morell, G.; Blasco-Alonso, J.; Couce, M.L.; Gutiérrez-Solana, L.G.; Guillén-Navarro, E.; O’Callaghan, M.; Del Toro, M. Elosulfase alfa for mucopolysaccharidosis type IVA: Real-world experience in 7 patients from the Spanish Morquio-A early access program. Mol. Genet. Metab. Rep. 2018, 15, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Chuang, C.K.; Ke, Y.Y.; Hsu, C.C.; Chiu, P.C.; Niu, D.M.; Tsai, F.J.; Hwu, W.L.; Lin, J.L.; Lin, S.P. Long-term effects of enzyme replacement therapy for Taiwanese patients with mucopolysaccharidosis IVA. Pediatr. Neonatol. 2019, 60, 342–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.A.; Mason, R.W.; Giugliani, R.; Orii, K.; Fukao, T.; Suzuki, Y.; Yamaguchi, S.; Kobayashi, H.; Orii, T.; Tomatsu, S. Glycosaminoglycans analysis in blood and urine of patients with mucopolysaccharidosis. Mol. Genet. Metab. 2018, 125, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Schweighardt, B.; Tompkins, T.; Lau, K.; Jesaitis, L.; Qi, Y.; Musson, D.G.; Farmer, P.; Haller, C.; Shaywitz, A.J.; Yang, K.; et al. Immunogenicity of Elosulfase Alfa, an Enzyme Replacement Therapy in Patients with Morquio A Syndrome: Results From MOR-004, a Phase III Trial. Clin. Ther. 2015, 37, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Long, B.; Tompkins, T.; Decker, C.; Jesaitis, L.; Khan, S.; Slasor, P.; Harmatz, P.; O’Neill, C.A.; Schweighardt, B. Long-term Immunogenicity of Elosulfase Alfa in the Treatment of Morquio A Syndrome: Results From MOR-005, a Phase III Extension Study. Clin. Ther. 2017, 39, 118–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenth, J.J.; Thompson, G.; Fullwood, C.; Wilkinson, S.; Jones, S.; Bruce, I.A. The characterisation of pulmonary function in patients with mucopolysaccharidoses IVA: A longitudinal analysis. Mol. Genet. Metab. Rep. 2019, 20, 100487. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, E.; Suzuki, Y.; Shimada, T.; Sawamoto, K.; Mackenzie, W.G.; Theroux, M.C.; Pizarro, C.; Xie, L.; Miller, F.; Rahman, T.; et al. Activity of daily living for Morquio A syndrome. Mol. Genet. Metab. 2016, 118, 111–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theroux, M.C.; Nerker, T.; Ditro, C.; Mackenzie, W.G. Anesthetic care and perioperative complications of children with Morquio syndrome. Paediatr. Anaesth. 2012, 22, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.M.; Kendall, B.E.; Crockard, H.A.; Ransford, A. The odontoid process in Morquio-Brailsford’s disease. The effects of occipitocervical fusion. J. Bone Joint Surg. Br. 1991, 73, 851–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, D.G.; Chadderton, R.D.; Cowie, R.A.; Wraith, J.E.; Jenkins, J.P. MRI of the brain and craniocervical junction in Morquio’s disease. Neuroradiology 1997, 39, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Ransford, A.O.; Crockard, H.A.; Stevens, J.M.; Modaghegh, S. Occipito-atlanto-axial fusion in morquio-brailsford syndrome. A ten-year experience. J. Bone Joint Surg. Br. 1996, 78, 307–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipson, S.J. Dysplasia of the odontoid process in Morquio’s syndrome causing quadriparesis. J. Bone Joint Surg. Am. 1977, 59, 340–344. [Google Scholar] [CrossRef]
- Dede, O.; Thacker, M.M.; Rogers, K.J.; Oto, M.; Belthur, M.V.; Baratela, W.; Mackenzie, W.G. Upper cervical fusion in children with Morquio syndrome: Intermediate to long-term results. J. Bone Joint Surg. Am. 2013, 95, 1228–1234. [Google Scholar] [CrossRef]
- Mikles, M.; Stanton, R.P. A review of Morquio syndrome. Am. J. Orthop. 1997, 26, 533–540. [Google Scholar]
- Tomatsu, S.; Yasuda, E.; Patel, P.; Ruhnke, K.; Shimada, T.; Mackenzie, W.G.; Mason, R.; Thacker, M.M.; Theroux, M.; Montaño, A.M.; et al. Morquio A syndrome: Diagnosis and current and future therapies. Pediatr. Endocrinol. Rev. 2014, 12, 141–151. [Google Scholar]
- Hendriksz, C.J.; Berger, K.I.; Giugliani, R.; Harmatz, P.; Kampmann, C.; Mackenzie, W.G.; Raiman, J.; Villarreal, M.S.; Savarirayan, R. International guidelines for the management and treatment of Morquio A syndrome. Am. J. Med. Genet. A 2015, 167, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Sumner, J.; Tomatsu, S.; Stapleton, M.; Davies, R.R.; Pizarro, C. Management of tracheal obstruction in MPS. In Mucopolysaccharidoses Update; Tomatsu, S., Ed.; Nova Science Publishers: New York, NY, USA, 2018; pp. 689–710. [Google Scholar]
- Bank, R.A.; Groener, J.E.; van Gemund, J.J.; Maaswinkel, P.D.; Hoeben, K.A.; Schut, H.A.; Everts, V. Deficiency in N-acetylgalactosamine-6-sulfate sulfatase results in collagen perturbations in cartilage of Morquio syndrome A patients. Mol. Genet. Metab. 2009, 97, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Simonaro, C.M.; Ge, Y.; Eliyahu, E.; He, X.; Jepsen, K.J.; Schuchman, E.H. Involvement of the Toll-like receptor 4 pathway and use of TNF-alpha antagonists for treatment of the mucopolysaccharidoses. Proc. Natl. Acad. Sci. USA 2010, 107, 222–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zustin, J. Morquio disease: The role of cartilage canals in the pathogenesis of chondrogenic dwarfism. Med. Hypotheses 2010, 75, 642–644. [Google Scholar] [CrossRef]
- Frohbergh, M.; Ge, Y.; Meng, F.; Karabul, N.; Solyom, A.; Lai, A.; Iatridis, J.; Schuchman, E.H.; Simonaro, C.M. Dose responsive effects of subcutaneous pentosan polysulfate injection in mucopolysaccharidosis type VI rats and comparison to oral treatment. PLoS ONE 2014, 9, e100882. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, K.; Suzuki, K.; Imai, K.; Mochizuki, H.; Morikawa, K.; Kyogashima, M.; Kimata, K.; Watanabe, H. Purification, characterization, and molecular cloning of a novel keratan sulfate hydrolase, endo-beta-N-acetylglucosaminidase, from Bacillus circulans. J. Biol. Chem. 2003, 278, 25766–25772. [Google Scholar] [CrossRef] [Green Version]
- Sawamoto, K.; Tomatsu, S. Development of Substrate Degradation Enzyme Therapy for Mucopolysaccharidosis IVA Murine Model. Int. J. Mol. Sci. 2019, 20, 4139. [Google Scholar] [CrossRef] [Green Version]
- Heldermon, C.D.; Qin, E.Y.; Ohlemiller, K.K.; Herzog, E.D.; Brown, J.R.; Vogler, C.; Hou, W.; Orrock, J.L.; Crawford, B.E.; Sands, M.S. Disease correction by combined neonatal intracranial AAV and systemic lentiviral gene therapy in Sanfilippo Syndrome type B mice. Gene Ther. 2013, 20, 913–921. [Google Scholar] [CrossRef]
- Di Natale, P.; Di Domenico, C.; Gargiulo, N.; Castaldo, S.; Gonzalez, Y.; Reyero, E.; Mithbaokar, P.; De Felice, M.; Follenzi, A.; Naldini, L.; et al. Treatment of the mouse model of mucopolysaccharidosis type IIIB with lentiviral-NAGLU vector. Biochem. J. 2005, 388, 639–646. [Google Scholar] [CrossRef] [Green Version]
- Baldo, G.; Wozniak, D.F.; Ohlemiller, K.K.; Zhang, Y.; Giugliani, R.; Ponder, K.P. Retroviral-vector-mediated gene therapy to mucopolysaccharidosis I mice improves sensorimotor impairments and other behavioral deficits. J. Inherit. Metab. Dis. 2013, 36, 499–512. [Google Scholar] [CrossRef] [Green Version]
- Tordo, J.; O’Leary, C.; Antunes, A.S.L.M.; Palomar, N.; Aldrin-Kirk, P.; Basche, M.; Bennett, A.; D’Souza, Z.; Gleitz, H.; Godwin, A.; et al. A novel adeno-associated virus capsid with enhanced neurotropism corrects a lysosomal transmembrane enzyme deficiency. Brain 2018, 141, 2014–2031. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Zaraspe, K.; Murakami, N.; Meadows, A.S.; Pineda, R.J.; McCarty, D.M.; Muenzer, J. Targeting Root Cause by Systemic scAAV9-hIDS Gene Delivery: Functional Correction and Reversal of Severe MPS II in Mice. Mol. Ther. Methods Clin. Dev. 2018, 10, 327–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zincarelli, C.; Soltys, S.; Rengo, G.; Rabinowitz, J.E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 2008, 16, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Alméciga-Díaz, C.J.; Montaño, A.M.; Tomatsu, S.; Barrera, L.A. Adeno-associated virus gene transfer in Morquio A disease-effect of promoters and sulfatase-modifying factor 1. FEBS J. 2010, 277, 3608–3619. [Google Scholar] [CrossRef] [PubMed]
- Alméciga-Díaz, C.J.; Rueda-Paramo, M.A.; Espejo, A.J.; Echeverri, O.Y.; Montaño, A.; Tomatsu, S.; Barrera, L.A. Effect of elongation factor 1alpha promoter and SUMF1 over in vitro expression of N-acetylgalactosamine-6-sulfate sulfatase. Mol. Biol. Rep. 2009, 36, 1863–1870. [Google Scholar] [CrossRef] [PubMed]
- Toietta, G.; Severini, G.M.; Traversari, C.; Tomatsu, S.; Sukegawa, K.; Fukuda, S.; Kondo, N.; Tortora, P.; Bordignon, C. Various cells retrovirally transduced with N-acetylgalactosoamine-6-sulfate sulfatase correct Morquio skin fibroblasts in vitro. Hum. Gene Ther. 2001, 12, 2007–2016. [Google Scholar] [CrossRef]
- Alméciga-Díaz, C.J.; Montaño, A.M.; Barrera, L.A.; Tomatsu, S. Tailoring the AAV2 capsid vector for bone-targeting. Pediatr. Res. 2018, 84, 545–551. [Google Scholar] [CrossRef] [Green Version]
- Gutiérrez, M.A.; García-Vallejo, F.; Tomatsu, S.; Cerón, F.; Alméciga-Díaz, C.J.; Domínguez, M.C.; Barrera, L.A. Construction of an adenoassociated, viral derived, expression vector to correct the genetic defect in Morquio A disease. Biomedica 2008, 28, 448–459. [Google Scholar]
- Mingozzi, F.; High, K.A. Immune responses to AAV vectors: Overcoming barriers to successful gene therapy. Blood 2013, 122, 23–36. [Google Scholar] [CrossRef]
- Vandamme, C.; Adjali, O.; Mingozzi, F. Unraveling the Complex Story of Immune Responses to AAV Vectors Trial After Trial. Hum. Gene Ther. 2017, 28, 1061–1074. [Google Scholar] [CrossRef]
- Selot, R.; Arumugam, S.; Mary, B.; Cheemadan, S.; Jayandharan, G.R. Optimized AAV rh.10 Vectors That Partially Evade Neutralizing Antibodies during Hepatic Gene Transfer. Front. Pharmacol. 2017, 8, 441. [Google Scholar] [CrossRef] [PubMed]
- Colella, P.; Sellier, P.; Costa Verdera, H.; Puzzo, F.; van Wittenberghe, L.; Guerchet, N.; Daniele, N.; Gjata, B.; Marmier, S.; Charles, S.; et al. AAV Gene Transfer with Tandem Promoter Design Prevents Anti-transgene Immunity and Provides Persistent Efficacy in Neonate Pompe Mice. Mol. Ther./Methods Clin. Dev. 2018, 12, 85–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manno, C.S.; Pierce, G.F.; Arruda, V.R.; Glader, B.; Ragni, M.; Rasko, J.J.; Ozelo, M.C.; Hoots, K.; Blatt, P.; Konkle, B.; et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006, 12, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Meliani, A.; Boisgerault, F.; Fitzpatrick, Z.; Marmier, S.; Leborgne, C.; Collaud, F.; Simon Sola, M.; Charles, S.; Ronzitti, G.; Vignaud, A.; et al. Enhanced liver gene transfer and evasion of preexisting humoral immunity with exosome-enveloped AAV vectors. Blood Adv. 2017, 1, 2019–2031. [Google Scholar] [CrossRef] [Green Version]
- Maguire, C.A.; Balaj, L.; Sivaraman, S.; Crommentuijn, M.H.; Ericsson, M.; Mincheva-Nilsson, L.; Baranov, V.; Gianni, D.; Tannous, B.A.; Sena-Esteves, M.; et al. Microvesicle-associated AAV vector as a novel gene delivery system. Mol. Ther. 2012, 20, 960–971. [Google Scholar] [CrossRef] [Green Version]
- REGENXBIO Inc. REGENXBIO Announces Interim Data from Phase I/II Trial of RGX-121 for the Treatment of Mucopolysaccharidosis type II (MPS II). PR Newswire. Available online: https://www.prnewswire.com/news-releases/regenxbio-announces-interim-data-from-phase-iii-trial-of-rgx-121-for-the-treatment-of-mucopolysaccharidosis-type-ii-mps-ii-300976867.html (accessed on 18 December 2019).
- Abeona Therapeutics. Abeona Therapeutics Announces Positive Interim Data from the ABO-102 Phase 1/2 Gene Therapy Clinical Trial in MPS IIIA. Globe Newswire. Available online: https://www.globenewswire.com/news-release/2019/07/25/1888073/0/en/Abeona-Therapeutics-Announces-Positive-Interim-Data-from-the-ABO-102-Phase-1-2-Gene-Therapy-Clinical-Trial-in-MPS-IIIA.html (accessed on 25 July 2019).
- Hocquemiller, M.; Hemsley, K.M.; Douglass, M.L. AAVrh10 vector corrects disease pathology in MPS IIIA mice and achieves widespread distribution of sulfamidase in the brain of large animals. Mol. Ther. Methods Clin. Dev. 2019, 17, 174–187. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04201405?term=gene+therapy&cond=MPS&draw=1&rank=9 (accessed on 19 February 2020).
- Ellison, S.M.; Liao, A.; Wood, S.; Taylor, J.; Youshani, A.S.; Rowlston, S.; Parker, H.; Armant, M.; Biffi, A.; Chan, L.; et al. Pre-clinical Safety and Efficacy of Lentiviral Vector-Mediated Ex Vivo Stem Cell Gene Therapy for the Treatment of Mucopolysaccharidosis IIIA. Mol. Ther. Methods Clin. Dev. 2019, 13, 399–413. [Google Scholar] [CrossRef] [Green Version]
- Lee, L.R.; Peacock, L.; Lisowski, L.; Little, D.G.; Munns, C.F.; Schindeler, A. Targeting Adeno-Associated Virus Vectors for Local Delivery to Fractures and Systemic Delivery to the Skeleton. Mol. Ther. Methods Clin. Dev. 2019, 15, 101–111. [Google Scholar] [CrossRef] [Green Version]
- Kreuter, J. Nanoparticles—A historical perspective. Int. J. Pharm. 2007, 331, 1–10. [Google Scholar] [CrossRef]
- Bourdenx, M.; Daniel, J.; Genin, E.; Soria, F.N.; Blanchard-Desce, M.; Bezard, E.; Dehay, B. Nanoparticles restore lysosomal acidification defects: Implications for Parkinson and other lysosomal-related diseases. Autophagy 2016, 12, 472–483. [Google Scholar] [CrossRef] [Green Version]
- Mayer, F.Q.; Adorne, M.D.; Bender, E.A.; de Carvalho, T.G.; Dilda, A.C.; Beck, R.C.; Guterres, S.S.; Giugliani, R.; Matte, U.; Pohlmann, A.R. Laronidase-functionalized multiple-wall lipid-core nanocapsules: Promising formulation for a more effective treatment of mucopolysaccharidosis type I. Pharm. Res. 2015, 32, 941–954. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.; de Carvalho, T.G.; Bidone, J.; Schuh, R.S.; Matte, U.; Teixeira, H.F. Factors influencing transfection efficiency of pIDUA/nanoemulsion complexes in a mucopolysaccharidosis type I murine model. Int. J. Nanomedicine. 2017, 15, 2061–2067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrera, I.; Abasolo, I.; Corchero, J.L.; Elizondo, E.; Gil, P.R.; Moreno, E.; Faraudo, J.; Sala, S.; Bueno, D.; González-Mira, E.; et al. α-Galactosidase-A Loaded-Nanoliposomes with Enhanced Enzymatic Activity and Intracellular Penetration. Adv. Healthc. Mater. 2016, 6, 829–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Álvarez, J.V.; Herrero Filgueira, C.; González, A.F.; Colón Mejeras, C.; Beiras Iglesias, A.; Tomatsu, S.; Blanco Méndez, J.; LuzardoÁlvarez, A.; Couce, M.L.; Otero Espinar, F.J. Enzyme-loaded gel core nanostructured lipid carriers to improve treatment of lysosomal storage diseases: Formulation and in vitro cellular studies of elosulfase alfa- loaded systems. Pharmaceutics 2019, 11, 11. [Google Scholar]
- Salvalaio, M.; Rigon, L.; Belletti, D.; D’Avanzo, F.; Pederzoli, F.; Ruozi, B.; Marin, O.; Vandelli, M.A.; Forni, F.; Scarpa, M.; et al. Targeted Polymeric Nanoparticles for Brain Delivery of High Molecular Weight Molecules in Lysosomal Storage Disorders. PLoS ONE 2016, 26, 0156452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigon, L.; Salvalai, M.; Pederzoli, F.; Legnini, E.; Duskey, J.T.; D’Avanzo, F.; De Filippis, C.; Ruozi, B.; Marin, O.; Vandelli, M.A.; et al. Targeting Brain Disease in MPSII: Preclinical Evaluation of IDS-Loaded PLGA Nanoparticles. Int. J. Mol. Sci. 2019, 28, 2014. [Google Scholar] [CrossRef] [Green Version]
- Donida, B.; Tauffner, B.; Raabe, M.; Immich, M.F.; de Farias, M.A.; de Sá Coutinho, D.; Machado, A.Z.; Kessler, R.G.; Portugal, R.V.; Bernardi, A.; et al. Monoolein-based nanoparticles for drug delivery to the central nervous system: A platform for lysosomal storage disorder treatment. Eur. J. Pharm. Biopharm. 2018, 133, 96–103. [Google Scholar] [CrossRef]
- Muro, S. New biotechnological and nanomedicine strategies for treatment of lysosomal storage disorders. Wiley Interdiscip Rev. Nanomed Nanobiotechnol. 2010, 2, 189–204. [Google Scholar] [CrossRef] [Green Version]
- Matassini, C.; Vanni, C.; Goti, A.; Morrone, A.; Marradi, M.; Cardona, F. Multimerization of DAB-1 onto Au GNPs affords new potent and selective N-acetylgalactosamine-6-sulfatase (GALNS) inhibitors. Org. Biomol. Chem. 2018, 16, 8604–8612. [Google Scholar] [CrossRef]
- Gary-Bobo, M.; Nirdé, P.; Jeanjean, A.; Morère, A.; Garcia, M. Mannose 6-phosphate receptor targeting and its applications in human diseases. Curr. Med. Chem. 2007, 14, 2945–2953. [Google Scholar] [CrossRef] [Green Version]
- Shepherd, V.L.; Freeze, H.H.; Miller, A.L.; Stahl, P.D. Identification of mannose 6-phosphate receptors in rabbit alveolar macrophages. J. Biol. Chem. 1984, 259, 2257–2261. [Google Scholar] [PubMed]
- Westcott, K.R.; Searles, R.P.; Rome, L.H. Evidence for ligand- and pH-dependent conformational changes in liposome-associated mannose 6-phosphate receptor. J. Biol. Chem. 1987, 262, 6101–6107. [Google Scholar] [PubMed]
- Coutinho, M.F.; Prata, M.J.; Alves, S. Mannose-6-phosphate pathway: A review on its role in lysosomal function and dysfunction. Mol. Genet. Metab. 2012, 105, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Todkar, K.; Ilamathi, H.S.; Germain, M. Mitochondria and Lysosomes: Discovering Bonds. Front. Cell Dev. Biol. 2017, 5, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.; Nag, S.; Mason, A.B.; Barroso, M.M. Endosome mitocondria interactions are modulated by ironrelease from transferrin. J. Cell Biol. 2016, 214, 831–845. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, A.P.; Puertollano, R.; Raben, N.; Slaugenhaupt, S.; Walkley, S.U.; Ballabio, A. Autophagy in lysosomal storage disorders. Autophagy 2012, 8, 719–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraldi, A.; Annunziata, F.; Lombardi, A.; Kaiser, H.-J.; Medina, D.L.; Spampanato, C.; Fedele, A.O.; Polishchuk, R.; Sorrentino, N.C.; Simons, K.; et al. Lysosomal fusion and SNARE function are impaired by cholesterol accumulation in lysosomal storage disorders. EMBO J. 2010, 29, 3607–3620. [Google Scholar] [CrossRef] [Green Version]
- Álvarez, J.V.; Bravo, S.B.; García-Vence, M.; De Castro, M.J.; Luzardo, A.; Colón, C.; Tomatsu, S.; Otero-Espinar, F.J.; Couce, M.L. Proteomic Analysis in Morquio A Cells Treated with Immobilized Enzymatic Replacement Therapy on Nanostructured Lipid Systems. Int. J. Mol. Sci. 2019, 20, 4610. [Google Scholar] [CrossRef] [Green Version]
- Olarte-Avellaneda, S.; Rodríguez-López, A.; Alméciga-Díaz, C.J.; Barrera, L.A. Computational analysis of human N-acetylgalactosamine-6-sulfate sulfatase enzyme: An update in genotype-phenotype correlation for Morquio A. Mol. Biol. Rep. 2014, 41, 7073–7088. [Google Scholar] [CrossRef]
- Valenzano, K.J.; Khanna, R.; Powe, A.C.; Boyd, R.; Lee, G.; Flanagan, J.J.; Benjamin, E.R. Identification and characterization of pharmacological chaperones to correct enzyme deficiencies in lysosomal storage disorders. Assay. Drug. Dev. Technol. 2011, 9, 213–235. [Google Scholar] [CrossRef] [Green Version]
- Hughes, D.A.; Nicholls, K.; Shankar, S.P.; Sunder-Plassmann, G.; Koeller, D.; Nedd, K.; Vockley, G.; Hamazaki, T.; Lachmann, R.; Ohashi, T.; et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J. Med. Genet. 2017, 54, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Boyd, R.E.; Lee, G.; Rybczynski, P.; Benjamin, E.R.; Khanna, R.; Wustman, B.A.; Valenzano, K.J. Pharmacological Chaperones as Therapeutics for Lysosomal Storage Diseases. J. Med. Chem. 2013, 56, 2705–2725. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Andria, G.; Valenzano, K.J. Pharmacological Chaperone Therapy: Preclinical Development, Clinical Translation, and Prospects for the Treatment of Lysosomal Storage Disorders. Mol. Ther. 2015, 23, 1138–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshina, H.; Shimada, Y.; Higuchi, T.; Kobayashi, H.; Ida, H.; Ohashi, T. Chaperone effect of sulfated disaccharide from heparin on mutant iduronate-2-sulfatase in mucopolysaccharidosis type II. Mol. Genet. Metab. 2017, 123, 118–122. [Google Scholar] [CrossRef]
- Sawamoto, K.; Stapleton, M.; Alméciga-Díaz, C.J.; Espejo-Mojica, A.J.; Losada, J.C.; Suarez, D.A.; Tomatsu, S. Therapeutic Options for Mucopolysaccharidoses: Current and Emerging Treatments. Drugs 2019, 79, 1103–1134. [Google Scholar] [CrossRef]
- Alméciga-Diaz, C.J.; Hidalgo, O.A.; Olarte-Avellaneda, S.; Rodríguez-López, A.; Guzman, E.; Garzón, R.; Pimentel-Vera, L.N.; Puentes-Tellez, M.A.; Rojas-Rodriguez, A.F.; Gorshkov, K.; et al. Identification of Ezetimibe and Pranlukast as Pharmacological Chaperones for the Treatment of the Rare Disease Mucopolysaccharidosis Type IVA. J. Med. Chem. 2019, 62, 6175–6189. [Google Scholar] [CrossRef]





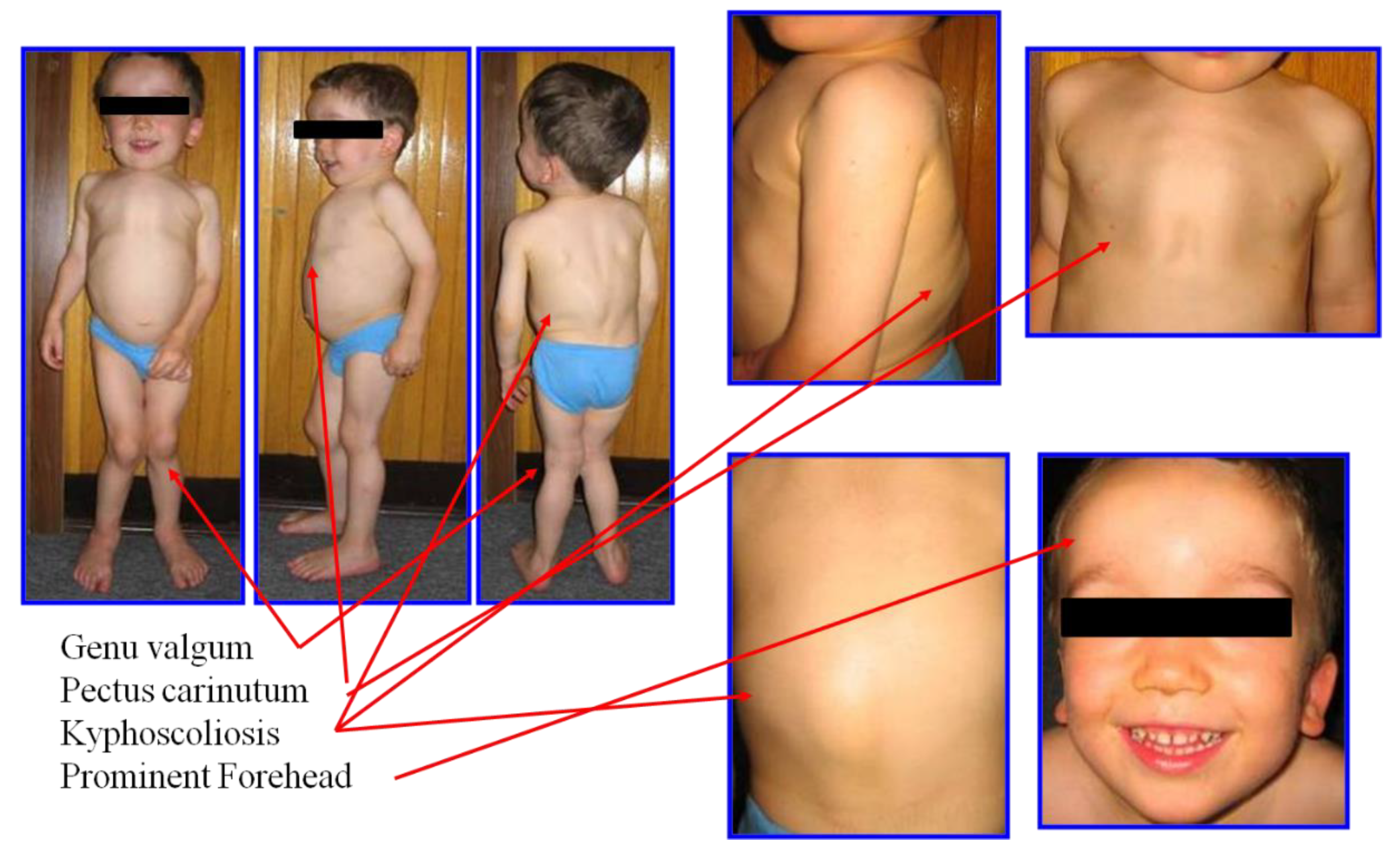{kind=link}
{kind=link}
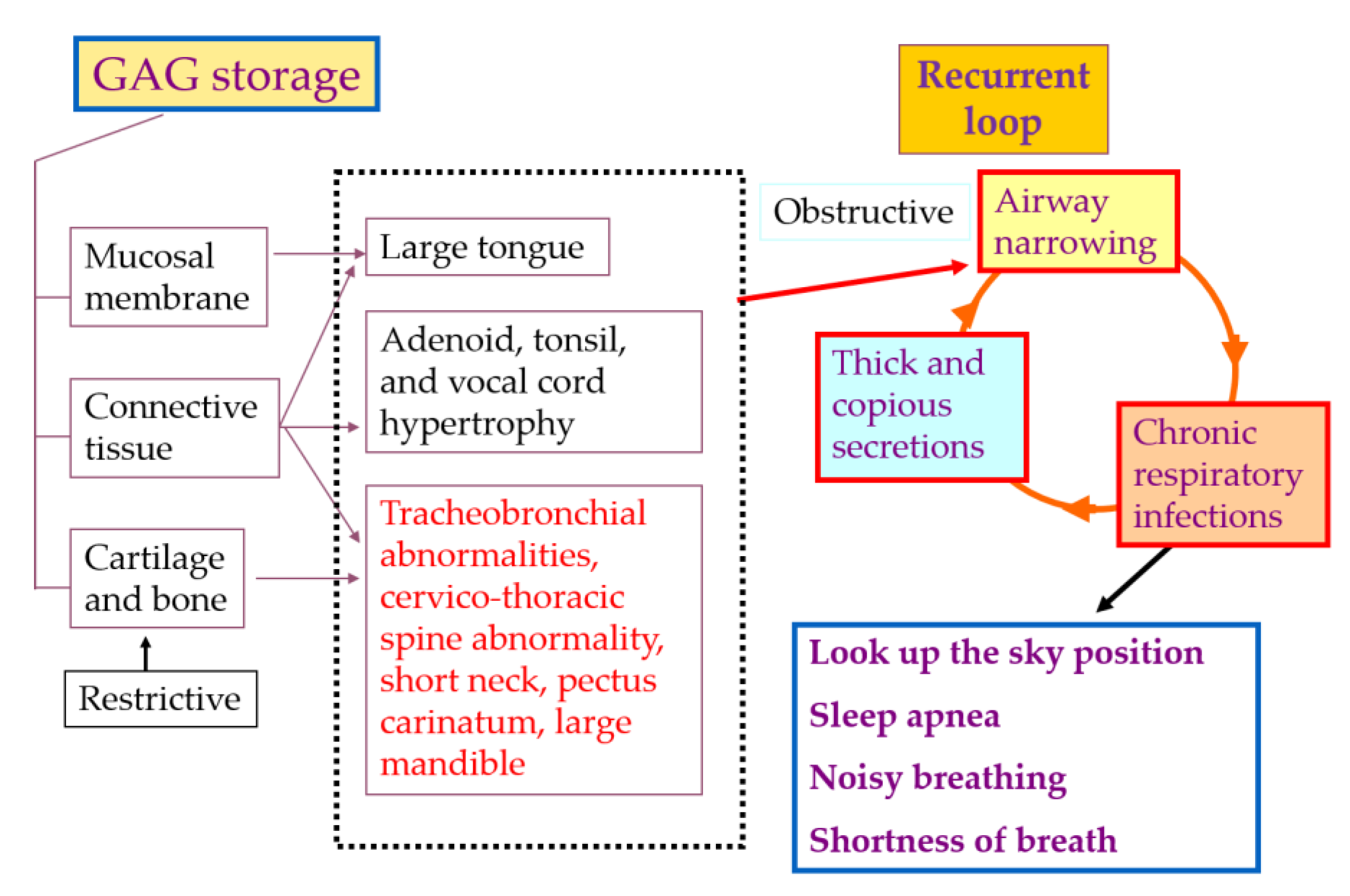{kind=link}
{kind=link}
| Advantage | Disadvantage | |
|---|---|---|
| ERT | Low risk of mortality and morbidity | High cost ($ 400,000 per year per 25 kg) |
| No limitation of age | Weekly infusion | |
| No specialized medical facility required | Short half-life time of the enzyme (40 min-human, 2 min-mouse) | |
| HSCT | Lower cost than ERT (approximately $ 100,000) | Risk of mortality and morbidity |
| One-time permanent treatment | Limitation of age | |
| Continuous activity of enzyme | Specialized medical facility required | |
| More effect in bone pathology than ERT | GVHD | |
| Availability of a donor | ||
| Advantage | Problems to overcome | |
| SDET | Enzyme is active in neutral pH (May work in circulation and ECM) | Immunogenicity to the enzyme |
| No age limitation | Optimal dose and treatment frequency | |
| Gene therapy | One-time permanent treatment | Vector selection needs to be determined |
| Continuous activity of enzyme | (optimal promoter, AAV serotype, dose etc) | |
| Does not require donor | Readministration is Not available | |
| No age limitation | Unknown duration of enzyme expression | |
| Nanomedicine | Protection of enzyme degradation | Limitation on components to make nanoparticles |
| Greater permeability through biological membranes | Optimal dose and treatment frequency | |
| Better efficacy to act on lysosomes | Unknown effects still in animal model | |
| No age limitation | ||
| Pharmacological chaperone therapy | Wide distribution in tissues | Off-target effect |
| Oral administration | Optimal dose and treatment frequency | |
| No immunogenecity | Unknown effects still in animal model |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sawamoto, K.; Álvarez González, J.V.; Piechnik, M.; Otero, F.J.; Couce, M.L.; Suzuki, Y.; Tomatsu, S. Mucopolysaccharidosis IVA: Diagnosis, Treatment, and Management. Int. J. Mol. Sci. 2020, 21, 1517. https://doi.org/10.3390/ijms21041517
Sawamoto K, Álvarez González JV, Piechnik M, Otero FJ, Couce ML, Suzuki Y, Tomatsu S. Mucopolysaccharidosis IVA: Diagnosis, Treatment, and Management. International Journal of Molecular Sciences. 2020; 21(4):1517. https://doi.org/10.3390/ijms21041517
Chicago/Turabian StyleSawamoto, Kazuki, José Víctor Álvarez González, Matthew Piechnik, Francisco J. Otero, Maria L. Couce, Yasuyuki Suzuki, and Shunji Tomatsu. 2020. "Mucopolysaccharidosis IVA: Diagnosis, Treatment, and Management" International Journal of Molecular Sciences 21, no. 4: 1517. https://doi.org/10.3390/ijms21041517
APA StyleSawamoto, K., Álvarez González, J. V., Piechnik, M., Otero, F. J., Couce, M. L., Suzuki, Y., & Tomatsu, S. (2020). Mucopolysaccharidosis IVA: Diagnosis, Treatment, and Management. International Journal of Molecular Sciences, 21(4), 1517. https://doi.org/10.3390/ijms21041517